Abstract
Intracellular ATP is released outside cells by various stimuli and is involved in cytoprotection by activating purinergic receptors. However, it remains unclear whether targeted radionuclide therapy induces extracellular ATP release. Here, we prepared 131I-labeled trastuzumab (131I-trastuzumab) and examined extracellular ATP release and its roles in 131I-trastuzumab’s growth inhibitory effects. 131I-trastuzumab was prepared by labeling with the chloramine-T method. The binding of 131I-trastuzumab to cells was investigated using the human epidermal growth factor receptor 2 (HER2)-positive cells (SKOV3) and the HER2-negative cell (MCF7). Extracellular ATP was determined by measuring chemiluminescence using a luciferin-luciferase reagent. The growth inhibitory effects of 131I-trastuzumab were investigated by colony formation assay. 131I-trastuzumab bound exclusively to SKOV3 cells. Treatment with 131I-trastuzumab at 4 MBq/mL and higher concentrations significantly increased extracellular ATP levels, whereas non-radioactive trastuzumab didn’t. This suggested that ATP release was specifically induced by radiation derived from 131I. The growth inhibitory effects of 131I-trastuzumab were significantly enhanced by pretreatment with apyrase (ecto-ATPase) or MRS2578 (a P2Y6-selective antagonist), whereas they were significantly reduced by treatment with a P2Y6-selective agonist. In conclusion, 131I-trastuzumab induced extracellular ATP release, and the released ATP was shown to be involved in mitigating radiation-induced reduction in cell viability through P2Y6 receptor.
INTRODUCTION
Targeted radionuclide therapy (TRT) irradiates cancer cells by administering a radioisotope or a drug that combines a radioisotope with a cancer-directed drug delivery system into the body. Unlike external beam radiation therapy (EBRT), TRT is expected to be effective even for cancer that has spread throughout the body. So far, therapeutic agents using β-emitters, such as iodine-131, yttrium-90, and lutetium-177, have been developed, and strong therapeutic effects in patients have been reported.1–3) Recently, radiopharmaceuticals using α-emitters have also been developed, and strong therapeutic effects have been obtained even in patients who did not respond to treatment with β-emitters.4,5) However, some patients do not respond to treatments with α-emitters.6) Several reasons for the insufficient effects have been suggested. Due to the specific features of tumor vasculature, radiopharmaceuticals are not distributed throughout the tumor tissue and thus do not deliver sufficient radiation to all cancer cells.7) In addition, due to tumor heterogeneity, various clone populations have different drug sensitivities and target molecule expression levels within cancer tissues.8,9) In the tumor microenvironment, hypoxic conditions enhance DNA repair, increase expression of antioxidant enzymes, and facilitate immune evasion due to increased expression of PD-L1. Furthermore, radioresistant cancer-associated macrophages and cancer-associated fibroblasts are induced by crosstalk between cancer cells and surrounding cells.10) To enhance cancer therapeutic efficacy by TRT, it is important to overcome these problems.
Extracellular ATP is involved in therapeutic resistance in the tumor microenvironment. ATP is released extracellularly by various stimuli and activates ATP-selective receptors (P2 receptors) expressed on the cell membrane through autocrine and paracrine signaling.11) P2 receptors include both the ligand-gated channel-type P2X receptors (P2X1, P2X2, P2X3, P2X4, P2X5, P2X6, P2X7) and the G protein-coupled P2Y receptors (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, P2Y14). P2X3, P2X5, and P2X7 are highly expressed in cancer tissues and are involved in cell proliferation and cancer cell survival.12–14) In addition, activation of the P2Y2 or P2Y6 receptor promotes DNA repair, cancer cell growth, and metastasis.15–18) Therefore, the extracellular nucleotide signaling in the tumor microenvironment has become a new target for cancer therapy.
Previously, we clarified that γ-ray irradiation enhances intracellular antioxidant activity and DNA repair by releasing ATP outside the cell and activating P2 receptors.19–22) If extracellular ATP release occurs in TRT, the antitumor effect may be attenuated by the activation of P2 receptors. However, TRT differs from EBRT in terms of dose rate, irradiation time, and uniformity of irradiation, and it remains unclear whether TRT induces extracellular ATP release and activates P2 receptors. Therefore, in this study, we prepared 131I-labeled trastuzumab (131I-trastuzumab) and examined extracellular ATP release and its roles in TRT.
MATERIALS AND METHODS
General[131I]NaI was purchased from the Japan Radioisotope Association (Tokyo, Japan). Trastuzumab was purchased from Chugai Pharmaceutical (Tokyo, Japan). ATP, uridine 5′-triphosphate (UTP), uridine 5′-diphosphate (UDP), apyrase, and ethidium bromide (EtBr) were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Pyridoxalphosphate-6-azophenyl-2’,4’-disulfonic acid (PPADS), suramin, and MRS2578 were purchased from Tocris Bioscience (Bristol, U.K.). All other chemicals used were of the highest purity available.
Cell CultureMCF7 (product number: HTB-22) and SKOV3 (product number: HTB-77) were purchased from the American Type Culture Collection (Manassas, VA, U.S.A.). MCF7 and SKOV3 were routinely maintained in Dulbecco’s modified Eagle’s medium (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) containing 10% heat-inactivated fetal bovine serum (FBS, Serum Source International, Charlotte, NC, U.S.A.), penicillin (100 units/mL), streptomycin (100 µg/mL), and L-glutamine (2 mM) at 37 °C in 5% CO2, 95% air.
RadiolabelingTrastuzumab was labeled with 131I using the chloramine-T method. In brief, 2 µL of [131I]NaI (18.5 GBq/mL) was mixed with 200 µL of trastuzumab (1 mg/mL in phosphate-buffered saline (PBS)) in a reaction tube. Then, 10 µL of chloramine-T solution (10 mg/mL in PBS) was added to a reaction tube and the mixture was incubated for 10 min at room temperature. The reaction was terminated by adding 20 µL of NaHSO3 solution (20 mg/mL in PBS). The reactant was fractionated by a PD-10 column (Cytiva, Marlborough, MA, U.S.A.) equilibrated with PBS. The radioactivity was measured with a well-type γ-counter (ARC-7001, Hitachi-Aloka Medical, Tokyo, Japan), and the radiochemical yield was calculated. The fractions of 131I-trastuzumab were combined and analyzed by size exclusion radio HPLC (SE-HPLC), and the radiochemical purity of 131I-trastuzumab was calculated under the following conditions: Column: 4.6 × 300 mm TSKgel Super SW2000 (TOSOH, Tokyo, Japan); column temperature: room temperature; mobile phase: 0.1 M phosphate buffer (100 mM Na2HPO4, 100 mM NaH2PO4, pH 6.8); flow rate: 0.3 mL/min; detection: absorbance at 280 nm and radioactivity. The radiochemical yield was 77.04 ± 16.14% (N = 5) and the radiochemical purity was over 95% (Supplementary Fig. S1). For in vitro study, the fraction 131I-trastuzumab was concentrated with Amicon Ultra-0.5 mL Centrifugal Filters (100000 MWCO, Merck Millipore, Burlington, MA, U.S.A.), then 131I-trastuzumab was sterilized with a 0.22 µm pore size filter. The protein concentration of 131I-trastuzumab was determined by the Bradford method. The specific activity was 344.6 MBq/mg.
Stability of 131I-TrastuzumabFor the evaluation of in vitro stability, 20 µL of 131I-trastuzumab (10 kBq) was added to 180 µL of PBS or human plasma (Kohjin Bio, Saitama, Japan), and the solution was incubated at 37 °C for 24 h. The reactants were then fractionated by PD-10 columns equilibrated with PBS. The radioactivity of each fraction was measured with a well-type γ-counter to calculate the ratios of intact 131I-trastuzumab.
Cell Binding AssayMCF7 and SKOV3 cells (2 × 105 cells) were seeded on 12-well plates and incubated overnight. Cells were washed with Hunk’s balanced salt solution (HBSS) and treated with 40 kBq/mL of 131I-trastuzumab in HBSS for 1, 2, and 3 h. After incubation, cells were washed with HBSS and lysed with 0.1 N NaOH. Radioactivity was measured using a well-type γ-counter.
Measurement of Extracellular ATPExtracellular ATP was measured using ENLITEN® rLuciferase/Luciferin Reagent (Promega, Madison, WI, U.S.A.). SKOV3 cells (2.0 × 104 cells/well) were incubated in 100 µL of culture medium in a 96-well culture plate overnight. The medium was then replaced by 50 µL RPMI1640 medium containing 1% FBS and incubated for several hours. An aliquot (10 µL) of the conditioned medium was collected as a control sample for background ATP release. To investigate the time-course study, an aliquot (10 µL) of 131I-trastuzumab (200 kBq) or the same amount of non-radioactive trastuzumab was added and incubated for 5 to 30 min. To investigate the dose-response relationship, an aliquot (10 µL) of 131I-trastuzumab (0.5 to 16 MBq/mL) or the equivalent amount of non-radioactive trastuzumab was added and incubated for 20 min. After treatments, 20 µL of supernatant was collected and stored on ice until measurement. The ATP concentration was determined by measuring chemiluminescence with a Microplate Luminometer (Berthold, Foster City, CA, U.S.A.) 1.6 s after adding 100 µL of luciferin-luciferase reagent to 10 µL of sample solution.
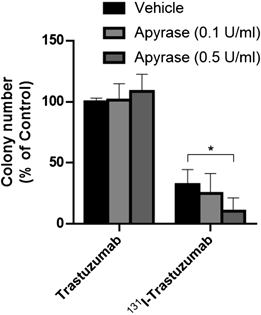
Colony Formation AssaySKOV3 cells (7.5 × 103 cells) were seeded on 96-well plates and incubated overnight. To investigate the dose-response relationship, cells were treated with 2 or 6 MBq/mL of 131I-trastuzumab or the equivalent amount of non-radioactive trastuzumab. To investigate the effect of ecto-ATPare, apyrase (0.1 or 0.5 U/mL) was added 30 min before treatment with 4 MBq/mL of 131I-trastuzumab or the equivalent amount of non-radioactive trastuzumab. To investigate the effects of P2 receptor agonists or antagonists, cells were incubated in the presence or absence of UTP (100 µM), UDP (100 µM), PPADS (10 µM), suramin (10 µM), and MRS2578 (1 µM) for 30 min. Cells were then treated with 4 MBq/mL of 131I-trastuzumab or the equivalent amount of non-radioactive trastuzumab. After incubation for 24 h, cells were detached with trypsin/PBS and seeded at 200 cells/well in a 6-well plate. After incubation for 14 d, cells were stained with crystal-violet solution for 30 min at room temperature. After the cells were washed with tap water, the colonies were counted.
RT-PCRTotal RNA was isolated from cells using a NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany). The first-strand cDNA was synthesized from 0.5 µg of total RNA with PrimeScript Reverse Transcriptase (TaKaRa Bio, Shiga, Japan). The sequences of specific primers for P2 receptors are shown in Supplementary Table S1. PCR was carried out by incubating each cDNA sample with the primers (0.5 µM each), Tks Gflex DNA polymerase (1.25 U: Toyobo, Osaka, Japan), and 2x Gflex PCR Buffer (25 µL, Toyobo). Amplification was carried out for 35 cycles (98 °C for 10 s, 60 °C for 15 s, 72 °C for 30 s). The products were then subjected to 2% agarose gel electrophoresis. Bands were stained with EtBr and photographed.
Statistical AnalysisResults are expressed as means ± standard deviation (S.D.). The statistical significance of differences between two groups was calculated using the unpaired Student’s t-test. The significance of differences was determined by a one-way ANOVA followed by Tukey’s multiple comparisons. The criterion of significance was p < 0.05, as determined with GraphPad Prism 6 software.
RESULTS
Cell Binding AssayThe ratios of intact 131I-trastuzumab in PBS and human plasma were 88.69 and 87.32%, respectively. To investigate the HER2 binding activity, 131I-trastuzumab was incubated with SKOV3, an HER2-positive cell line, or MCF7, an HER2-negative cell line. As shown in Fig. 1, the binding of 131I-trastuzumab to SKOV3 cells increased time-dependently, reaching 14.29 ± 0.12%dose/105 cells at 3 h after addition. On the other hand, the binding of 131I-trastuzumab to MCF7 cells did not increase in a time-dependent manner and was less than 0.44 ± 0.02%dose/105 cells even 3 h after addition. These results demonstrate the binding activity of 131I-trastuzumab to HER2-expressing cells.
Growth Inhibition with 131I-TrastuzumabUsing SKOV3 cells, the growth inhibitory effects of 131I-trastuzumab or equal amounts of non-radioactive trastuzumab were examined by colony formation assay. 131I-trastuzumab treatment significantly reduced the colony numbers, with the ratios of colony number being 39.6 ± 3.7% of the control at 2 MBq/mL and 10.7 ± 4.5% at 6 MBq/mL (Fig. 2). No reduction in SKOV3 colony numbers was observed after treatment with an equivalent dose of non-radioactive trastuzumab. These results indicate that irradiation with 131I suppressed SKOV3 cell proliferation.
ATP Release after Treatment with 131I-TrastuzumabTo investigate whether 131I-trastuzumab induces extracellular ATP release, ATP concentrations in cell supernatants were quantified after treatment with 131I-trastuzumab (4 MBq/mL) or an equal dose of non-radioactive trastuzumab. As shown in Fig. 3A, the concentration of extracellular ATP was significantly increased at 20 min after the addition of 131I-trastuzumab. In contrast, non-radioactive trastuzumab did not alter extracellular ATP levels (Fig. 3B). Examination of extracellular ATP release using various doses of 131I-trastuzumab and non-radioactive trastuzumab revealed that 131I-trastuzumab significantly increased extracellular ATP concentration from 4 MBq/mL compared with non-radioactive trastuzumab (Fig. 3C). These results indicate that irradiation with 131I-trastuzumab causes extracellular release of ATP from SKOV3 cells.
Effects of Ecto-ATPase on the Growth Inhibitory Effects of 131I-trastuzumabTo investigate how the extracellular ATP released by 131I-trastuzumab affects 131I-trastuzumab’s growth inhibitory effects, SKOV3 cells were treated with 131I-trastuzumab or an equivalent dose of non-radioactive trastuzumab in the presence of an ecto-ATPase, Apyrase. The results showed that, after the addition of 0.5 U/mL of Apyrase, the number of colonies after treatment with 131I-trastuzumab was significantly reduced (Fig. 4). Apyrase-induced changes in colony numbers were not observed with equal doses of non-radioactive trastuzumab.
Effects of P2 Agonists or Antagonists on the Growth Inhibitory Effects of 131I-TrastuzumabWe investigated the involvement of P2 receptors in the biological effects of extracellular ATP released by 131I-trastuzumab. As shown in Fig. 5A, P2X4, P2Y2, and P2Y6 were expressed in SKOV3 cells among P2 receptors. In the presence of each of the P2 receptor agonists UTP (P2Y2 and P2Y4 agonists) and UDP (P2Y6 agonist), UDP significantly inhibited the growth inhibitory effects of 131I-trastuzumab (Fig. 5B). In contrast, the growth inhibitory effects of 131I-trastuzumab were significantly enhanced by the P2 receptor inhibitor PPADS and the P2Y6 receptor selective inhibitor MRS2578 (Fig. 5C). No effects of P2 receptor agonists or antagonists were observed on cells treated with non-radioactive trastuzumab.
DISCUSSION
After ionizing radiation, cells in cancer tissue release various transmitters and engage in intercellular communication.23) Radiation-induced bystander effects refer to widely recognized intercellular communication observed after ionizing radiation. These effects signify that radiation’s impact extends to unirradiated cells surrounding the directly irradiated cells. This phenomenon occurs through the release of reactive oxygen species (ROS), nitric oxide, and cytokines. Radiation-induced bystander effects reduce the viability of unirradiated cells but induce radioresistance, activation of metastasis, tumor-promoting inflammation, and genomic instability.24–27) In addition, the activation of immune responses by radiation-induced immunogenic cell death after radiation therapy is widely known as abscopal effects. The activation is expected to have an antitumor effect not only on irradiated sites but also on distantly metastasized cancers.28) In TRT, cancer cells are subjected to heterogeneous long-term irradiation at a low dose rate, in contrast to EBRT, which involves high-dose-rate, short-time uniform irradiation. However, similar to the case with EBRT, TRT also induces immune activation.29) In a report by Zhang et al., immunosuppressive cells (T helper 17, T cytotoxic 17, regulatory T cells) and cytokines (interleukin (IL)-17, IL-23, IL-10, transforming growth factor-β1) in patients with differentiated thyroid cancer were reduced to the same levels as in healthy individuals by 131I therapy.30) In addition, [177Lu]oxodotreotide treatment in NCI-H727 cell xenograft model increased the infiltration of CD86+ cells and CD49b+/FasL+ NK cells in tumor tissue, and was shown to be able to kill the tumor.31) In TRT using α-emitters, the release of danger-associated molecular patterns (DAMPs) outside the cells has been reported, as well as the induction of immune activation. Gorin et al. reported that high-mobility group box 1 (HMGB1) and heat shock protein 70 (HSP70), known as DAMPs, were released from 213Bi-BSA-treated cancer cell line MC-38 and that the addition of the culture supernatant to dendritic cells induced the activation of those cells. Furthermore, they reported that 213Bi-BSA-treated MC-38 inoculation activates T-cell-dependent antitumor immunity.32) Hagemann et al. reported that treatment with 227Th-labeled compounds induces not only apoptosis in cancer cells through DNA damage but also promotes antitumor immune activation via the extracellular release of immunostimulatory chemokines and DAMPs such as calreticulin, HSP70, HSP90, and HMGB1. They also showed that the combination of 227Th-labeled compounds and immune checkpoint inhibitors enhanced the antitumor effects.33) Thus, it is possible to enhance the antitumor effect of TRT by appropriately controlling intercellular communication in the cancer microenvironment with specific inhibitors. We previously demonstrated cell-to-cell communication through P2 receptor activation by radiation-induced extracellular ATP release.19–22) Recently, we also showed that adenosine contributes to the activation of DNA damage repair by activating the adenosine receptor A2BR; we also found that treatments using selective inhibitors for the adenosine receptor A2BR enhance the antitumor effect of radiation.34–36) If we can elucidate extracellular ATP release and P2 receptor-mediated cellular responses in TRT, it may lead to new ways to improve antitumor efficacy.
In this study, we investigated the extracellular release of ATP and its roles in TRT using 131I-trastuzumab in ovarian cancer cell line SKOV3. TRT is one of the promising treatments for ovarian cancer, and various compounds labeled with α- and β-emitters have been developed.37,38) 131I-trastuzumab has also been evaluated preclinically as a candidate for the treatment of ovarian cancer.39) Clinical trials of TRT for ovarian cancer are already underway, but as with other types of cancer, the effectiveness of TRT varies from patient to patient.40) Therefore, elucidating the extracellular ATP release and its biological effects on TRT may contribute to improving the therapeutic efficacy of TRT for ovarian cancer. 131I-trastuzumab was synthesized by directly labeling the antibody with iodine-131 using chloramine T. Over 85% of 131I-trastuzumab was intact at 24 h after incubation with human plasma, indicating that 131I-trastuzumab remained almost stable during exposure to cells. 131I-trastuzumab bound only to HER2-positive cells, and the amount of binding increased with time, indicating that 131I-trastuzumab retained HER2 binding activity even after radiolabeling. The growth inhibitory effect was observed only with 131I-trastuzumab and not with non-radioactive trastuzumab. These results suggest that 131I-trastuzumab binds to SKOV3 cells in a HER2-dependent manner and induces growth inhibition by emitted radiation.
Treatment with 131I-trastuzumab induced the extracellular release of ATP, which exhibited a significant increase at 20 min. Non-radioactive trastuzumab did not induce extracellular ATP release, indicating that ATP release was induced by 131I-derived radiation; the antibody itself did not affect ATP release. Although the manner of irradiation is physically different between TRT and EBRT, the time course of ATP release after treatment with 131I-trastuzumab was similar to previous studies. The radiation-induced ATP release reached a peak within 30 min after γ-irradiation, then gradually returned to baseline.20–22) Connexin 43 (Cx43) gap junction hemichannel mainly involves the radiation-induced ATP release. An opening of Cx43 gap junction hemichannel is mediated by an increase in intracellular Ca2+ concentration caused by ROS generated by radiation.41) Intracellular ATP is released outside the cell and extracellular Ca2+ flows into the cell through Cx43 gap junction hemichannel simultaneously.42) When the intracellular Ca2+ concentration exceeds a certain value, the Cx43 hemichannel closes.43) Based on these mechanisms, ATP levels possibly peaked at 20 min after treatment with 131I-trastuzumab. In the future, we will deepen our understanding of the difference between EBRT and TRT by examining the extracellular ATP release mechanism by 131I-trastuzumab. After treatment with 131I-trastuzumab, extracellular ATP was released when the radioactivity was 4 MBq/mL or higher, and there was no radioactivity-dependent change up to 16 MBg/mL. According to the mechanism of radiation-induced ATP release, a radioactivity concentration of 4 MBq/mL may have been the threshold above which ROS is generated in an amount sufficient to induce ATP release. Compared with radiation-induced ATP release, hypotonic stress induced a large amount of ATP release but did not result in cell death (Supplementary Figs. S2, S3). Furthermore, the extracellular ATP concentration after 131I-trastuzumab treatment was similar to the values for radiation-induced ATP release in previous reports. Therefore, ATP was possibly released by radiation rather than leakage due to cell membrane damage. The difference in the ATP release mechanism of hypotonic stress and radiation might affect the amount of extracellular ATP. Various pathways for ATP release by hypotonic stress have been elucidated, e.g. Gap junction hemichannels, volume-regulated anion channels, maxi-anion channels,44) and exocytosis.45) In addition, it has been reported that hypotonic stress induces a hypotonic stress-induced signaling cascade via the activation of P2Y receptors by released ATP, inducing a feed-forward cycle of long-term ATP release.46) Based on these reports, hypotonic stress could release a larger amount of ATP outside the cells than radiation exposure.
In relation to the impact of P2 receptors on the growth inhibitory effects of 131I-trastuzumab, UDP, a specific agonist of the P2Y6 receptor, led to a significant increase in the number of colonies formed following treatment with 131I-trastuzumab. These results suggest that P2Y6 receptors contribute to the mitigation of 131I-trastuzumab-induced radiation damage in SKOV3 cells. Furthermore, the number of colonies formed following treatment with 131I-trastuzumab was significantly decreased when cells were pre-treated with apyrase, PPADS, and MRS2578. This suggests that extracellularly released ATP by 131I-trastuzumab played a role in the reduction of cell damage through the P2Y6 receptor. In this study, PPADS and Suramin, broad P2 receptor inhibitors, were used. PPADS has been reported to have an inhibitory effect on the P2X4 and P2Y6 receptors.47,48) Suramin has also been reported to have an inhibitory effect on the P2X4 and P2Y2 receptors.49,50) Since the colony formation of SKOV3 after 131I-trastuzumab treatment was not affected by UTP, a P2Y2/4 receptor agonist, nor by Suramin, the P2Y2 receptor is unlikely involved in the suppression of the growth inhibitory effect of 131I-trastuzumab. In addition, colony proliferation of SKOV3 after 131I-trastuzumab treatment was significantly reduced by PPADS, but was comparable to that of MRS2578, suggesting that the involvement of P2X4 receptors is low and that P2Y6 receptors are predominant. UTP is degraded to UMP by CD39 expressed on the cell membrane and degraded to uridine by CD73. Gentile et al. have reported that CD39 expression is low and CD73 expression is high in SKOV3.51) Therefore, regarding the possibility that UDP generated by dephosphorylation of UTP acts on the P2Y6 receptor, the UDP concentration might not be sufficient to demonstrate an inhibitory response to the cytotoxic effect of 131I-trastuzumab because of the slow production of UDP from UTP as well as parallel dephosphorylation to UMP.
In the γ-irradiated cells, P2Y6 receptor activation by radiation-induced ATP induces epidermal growth factor receptor (EGFR) activation and extracellular signal-regulated kinase 1/2 (ERK1/2) phosphorylation, as well as the DNA repair response.15,22,52) In addition, P2Y6 receptor activation in cancer is known to be involved in cell death inhibition.53,54) P2Y6 receptor activation by 131I-trastuzumab-induced ATP release may attenuate the cytotoxic effects by promoting DNA repair activation and cell death inhibition. In addition, it has been reported that P2Y6 receptor activation promotes the migration, invasion, and metastasis of cancer cells.17,55,56) Therefore, the P2Y6 receptor may be an important biological marker in TRT, and the inhibition of the P2Y6 receptor may be useful not only to enhance antitumor efficacy but also to prevent ATP release-mediated cancer malignant transformation.
The limitation of this study is that we used only the SKOV3 cell line. Because it has been reported that P2Y6 receptor-mediated cellular responses vary depending on the type of cancer,54,57) it is necessary to verify further whether our findings apply to other types of cancer in order to utilize radiation-induced purinergic signaling in the development of TRT in the future. As far as we have searched, there has been no literature on P2Y6 receptor-mediated biological responses in ovarian cancer cell lines. Therefore, although the results of this study are based only on the SKOV3, our findings will help in understanding P2 receptor-mediated biological responses in ovarian cancer.
In conclusion, we found that 131I-trastuzumab induced extracellular ATP release and that the released ATP was involved in mitigating the reduction of cell viability through P2Y6 receptor. Since selective inhibition of the P2Y6 receptor enhanced 131I-trastuzumab’s growth inhibitory effects, a combination of selective inhibitors for the P2Y6 receptor may be useful to enhance the therapeutic efficacy of TRT.
Acknowledgments
We would like to thank the staff of the Medical Radioisotope Application Project at the National Institute for Quantum Science and Technology as well as the Departments of Diagnostic Radiology and Nuclear Medicine at Gunma University Graduate School of Medicine for their cooperation and helpful input. This work was partially supported by Japan Society for the Promotion of Science (JSPS) KAKENHI Grant 22K07780 to Mitsutoshi Tsukimoto.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Beierwaltes WH. Treatment of neuroblastoma with 131I-MIBG: dosimetric problems and perspectives. Med. Pediatr. Oncol., 15, 188–191 (1987).
- 2) Knox SJ, Goris ML, Trisler K, Negrin R, Davis T, Liles TM, Grillo-López A, Chinn P, Varns C, Ning SC, Fowler S, Deb N, Becker M, Marquez C, Levy R. Yttrium-90-labeled anti-CD20 monoclonal antibody therapy of recurrent B-cell lymphoma. Clin. Cancer Res., 2, 457–470 (1996).
- 3) Kwekkeboom DJ, Bakker WH, Kam BL, Teunissen JJ, Kooij PP, de Herder WW, Feelders RA, van Eijck CH, de Jong M, Srinivasan A, Erion JL, Krenning EP. Treatment of patients with gastro-entero-pancreatic (GEP) tumours with the novel radiolabelled somatostatin analogue [177Lu-DOTA(0),Tyr3]octreotate. Eur. J. Nucl. Med. Mol. Imaging, 30, 417–422 (2003).
- 4) Wissing MD, van Leeuwen FW, van der Pluijm G, Gelderblom H. Radium-223 chloride: extending life in prostate cancer patients by treating bone metastases. Clin. Cancer Res., 19, 5822–5827 (2013).
- 5) Kratochwil C, Bruchertseifer F, Giesel FL, Weis M, Verburg FA, Mottaghy F, Kopka K, Apostolidis C, Haberkorn U, Morgenstern A. 225Ac-PSMA-617 for PSMA-targeted α-radiation therapy of metastatic castration-resistant prostate cancer. J. Nucl. Med., 57, 1941–1944 (2016).
- 6) Kratochwil C, Bruchertseifer F, Rathke H, Hohenfellner M, Giesel FL, Haberkorn U, Morgenstern A. Targeted α-therapy of metastatic castration-resistant prostate cancer with 225Ac-PSMA-617: swimmer-plot analysis suggests efficacy regarding duration of tumor control. J. Nucl. Med., 59, 795–802 (2018).
- 7) Haeck JC, Bol K, de Ridder CM, Brunel L, Fehrentz JA, Martinez J, van Weerden WM, Bernsen MR, de Jong M, Veenland JF. Imaging heterogeneity of peptide delivery and binding in solid tumors using SPECT imaging and MRI. EJNMMI Res., 6, 3 (2016).
- 8) McAbee JH, Degorre-Kerbaul C, Valdez K, Wendler A, Shankavaram UT, Watts C, Camphausen K, Tofilon PJ. Detection of glioblastoma intratumor heterogeneity in radiosensitivity using patient-derived neurosphere cultures. J. Neurooncol., 149, 383–390 (2020).
- 9) Puranik AD, Dromain C, Fleshner N, Sathekge M, Pavel M, Eberhardt N, Zengerling F, Marienfeld R, Grunert M, Prasad V. Target heterogeneity in oncology: the best predictor for differential response to radioligand therapy in neuroendocrine tumors and prostate cancer. Cancers (Basel), 13, 3607 (2021).
- 10) Suwa T, Kobayashi M, Nam JM, Harada H. Tumor microenvironment and radioresistance. Exp. Mol. Med., 53, 1029–1035 (2021).
- 11) Linden J, Koch-Nolte F, Dahl G. Purine release, metabolism, and signaling in the inflammatory response. Annu. Rev. Immunol., 37, 325–347 (2019).
- 12) Greig AVH, Linge C, Healy V, Lim P, Clayton E, Rustin MHA, McGrouther DA, Burnstock G. Expression of purinergic receptors in non-melanoma skin cancers and their functional roles in A431 cells. J. Invest. Dermatol., 121, 315–327 (2003).
- 13) Raffaghello L, Chiozzi P, Falzoni S, Di Virgilio F, Pistoia V. The P2X7 receptor sustains the growth of human neuroblastoma cells through a substance P-dependent mechanism. Cancer Res., 66, 907–914 (2006).
- 14) Maynard JP, Lee JS, Sohn BH, Yu X, Lopez-Terrada D, Finegold MJ, Goss JA, Thevananther S. P2X3 purinergic receptor overexpression is associated with poor recurrence-free survival in hepatocellular carcinoma patients. Oncotarget, 6, 41162–41179 (2015).
- 15) Ide S, Nishimaki N, Tsukimoto M, Kojima S. Purine receptor P2Y6 mediates cellular response to γ-ray-induced DNA damage. J. Toxicol. Sci., 39, 15–23 (2014).
- 16) Schulien I, Hockenjos B, van Marck V, Ayata CK, Follo M, Thimme R, Hasselblatt P. Extracellular ATP and purinergic P2Y2 receptor signaling promote liver tumorigenesis in mice by exacerbating DNA damage. Cancer Res., 80, 699–708 (2020).
- 17) Ma X, Pan X, Wei Y, Tan B, Yang L, Ren H, Qian M, Du B. Chemotherapy-induced uridine diphosphate release promotes breast cancer metastasis through P2Y6 activation. Oncotarget, 7, 29036–29050 (2016).
- 18) Zhang JL, Liu Y, Yang H, Zhang HQ, Tian XX, Fang WG. ATP-P2Y2-β-catenin axis promotes cell invasion in breast cancer cells. Cancer Sci., 108, 1318–1327 (2017).
- 19) Tsukimoto M, Homma T, Ohshima Y, Kojima S. Involvement of purinergic signaling in cellular response to gamma radiation. Radiat. Res., 173, 298–309 (2010).
- 20) Ohshima Y, Tsukimoto M, Takenouchi T, Harada H, Suzuki A, Sato M, Kitani H, Kojima S. γ-Irradiation induces P2X7 receptor-dependent ATP release from B16 melanoma cells. Biochim. Biophys. Acta, Gen. Subj., 1800, 40–46 (2010).
- 21) Ohshima Y, Kitami A, Kawano A, Tsukimoto M, Kojima S. Induction of extracellular ATP mediates increase in intracellular thioredoxin in RAW264.7 cells exposed to low-dose γ-rays. Free Radic. Biol. Med., 51, 1240–1248 (2011).
- 22) Nishimaki N, Tsukimoto M, Kitami A, Kojima S. Autocrine regulation of γ-irradiation-induced DNA damage response via extracellular nucleotides-mediated activation of P2Y6 and P2Y12 receptors. DNA Repair (Amst.), 11, 657–665 (2012).
- 23) Daguenet E, Louati S, Wozny AS, Vial N, Gras M, Guy JB, Vallard A, Rodriguez-Lafrasse C, Magné N. Radiation-induced bystander and abscopal effects: important lessons from preclinical models. Br. J. Cancer, 123, 339–348 (2020).
- 24) Lyng FM, Seymour CB, Mothersill C. Production of a signal by irradiated cells which leads to a response in unirradiated cells characteristic of initiation of apoptosis. Br. J. Cancer, 83, 1223–1230 (2000).
- 25) Mothersill C, Seymour CB. Cell–cell contact during gamma irradiation is not required to induce a bystander effect in normal human keratinocytes: evidence for release during irradiation of a signal controlling survival into the medium. Radiat. Res., 149, 256–262 (1998).
- 26) Seymour CB, Mothersill C. Delayed expression of lethal mutations and genomic instability in the progeny of human epithelial cells that survived in a bystander-killing environment. Radiat. Oncol. Investig., 5, 106–110 (1997).
- 27) Heeran AB, Berrigan HP, O’Sullivan J. The radiation-induced bystander effect (RIBE) and its connections with the hallmarks of cancer. Radiat. Res., 192, 668–679 (2019).
- 28) Zhao X, Shao C. Radiotherapy-mediated immunomodulation and anti-tumor abscopal effect combining immune checkpoint blockade. Cancers (Basel), 12, 2762 (2020).
- 29) Sun Q, Li J, Ding Z, Liu Z. Radiopharmaceuticals heat anti-tumor immunity. Theranostics, 13, 767–786 (2023).
- 30) Zhang L, Chen J, Xu C, Qi L, Ren Y. Effects of iodine-131 radiotherapy on Th17/Tc17 and Treg/Th17 cells of patients with differentiated thyroid carcinoma. Exp. Ther. Med., 15, 2661–2666 (2018).
- 31) Wu Y, Pfeifer AK, Myschetzky R, Garbyal RS, Rasmussen P, Knigge U, Bzorek M, Kristensen MH, Kjaer A. Induction of anti-tumor immune responses by peptide receptor radionuclide therapy with 177Lu-DOTATATE in a murine model of a human neuroendocrine tumor. Diagnostics (Basel), 3, 344–355 (2013).
- 32) Gorin JB, Ménager J, Gouard S, Maurel C, Guilloux Y, Faivre-Chauvet A, Morgenstern A, Bruchertseifer F, Chérel M, Davodeau F, Gaschet J. Antitumor immunity induced after α irradiation. Neoplasia, 16, 319–328 (2014).
- 33) Hagemann UB, Ellingsen C, Schuhmacher J, Kristian A, Mobergslien A, Cruciani V, Wickstroem K, Schatz CA, Kneip C, Golfier S, Smeets R, Uran S, Hennekes H, Karlsson J, Bjerke RM, Ryan OB, Mumberg D, Ziegelbauer K, Cuthbertson AS. Mesothelin-targeted thorium-227 conjugate (MSLN-TTC): preclinical evaluation of a new targeted alpha therapy for mesothelin-positive cancers. Clin. Cancer Res., 25, 4723–4734 (2019).
- 34) Tanaka Y, Kitabatake K, Abe R, Tsukimoto M. Involvement of A2B receptor in DNA damage response and radiosensitizing effect of A2B receptor antagonists on mouse B16 melanoma. Biol. Pharm. Bull., 43, 516–525 (2020).
- 35) Kitabatake K, Yoshida E, Kaji T, Tsukimoto M. Involvement of adenosine A2B receptor in radiation-induced translocation of epidermal growth factor receptor and DNA damage response leading to radioresistance in human lung cancer cells. Biochim. Biophys. Acta, Gen. Subj., 1864, 129457 (2020).
- 36) Kitabatake K, Kaji T, Tsukimoto M. Involvement of CD73 and A2B receptor in radiation-induced DNA damage response and cell migration in human glioblastoma A172 cells. Biol. Pharm. Bull., 44, 197–210 (2021).
- 37) Alvarez RD, Partridge EE, Khazaeli MB, Plott G, Austin M, Kilgore L, Russell CD, Liu T, Grizzle WE, Schlom J, LoBuglio AF, Meredith RF. Intraperitoneal radioimmunotherapy of ovarian cancer with 177Lu-CC49: a phase I/II study. Gynecol. Oncol., 65, 94–101 (1997).
- 38) Minnix M, Li L, Yazaki PJ, Miller AD, Chea J, Poku E, Liu A, Wong JYC, Rockne RC, Colcher D, Shively JE. TAG-72-targeted α-radionuclide therapy of ovarian cancer using 225Ac-labeled DOTAylated-huCC49 antibody. J. Nucl. Med., 62, 55–61 (2021).
- 39) Deng H, Liu W, Yang X, Li K, Liao W, Zhao P, Yang Y, Wei H, Wang J, Chen Y. Preliminary evaluation and in vitro cytotoxicity studies of [131I]I-trastuzumab in HER2 expressing ovarian cancer cells. J. Radioanal. Nucl. Chem., 331, 2451–2460 (2022).
- 40) Hallqvist A, Bergmark K, Bäck T, Andersson H, Dahm-Kähler P, Johansson M, Lindegren S, Jensen H, Jacobsson L, Hultborn R, Palm S, Albertsson P. Intraperitoneal α-emitting radioimmunotherapy with 211At in relapsed ovarian cancer: long-term follow-up with individual absorbed dose estimations. J. Nucl. Med., 60, 1073–1079 (2019).
- 41) Tsukimoto M. Purinergic signaling is a novel mechanism of the cellular response to ionizing radiation. Biol. Pharm. Bull., 38, 951–959 (2015).
- 42) Schalper KA, Sánchez HA, Lee SC, Altenberg GA, Nathanson MH, Sáez JC. Connexin 43 hemichannels mediate the Ca2+ influx induced by extracellular alkalinization. Am. J. Physiol. Cell Physiol., 299, C1504–C1515 (2010).
- 43) Lurtz MM, Louis CF. Intracellular calcium regulation of connexin43. Am. J. Physiol. Cell Physiol., 293, C1806–C1813 (2007).
- 44) Taruno A. ATP release channels. Int. J. Mol. Sci., 19, 808 (2018).
- 45) Koyama T, Oike M, Ito Y. Involvement of Rho-kinase and tyrosine kinase in hypotonic stress-induced ATP release in bovine aortic endothelial cells. J. Physiol., 532, 759–769 (2001).
- 46) Nandigama R, Padmasekar M, Wartenberg M, Sauer H. Feed forward cycle of hypotonic stress-induced ATP release, purinergic receptor activation, and growth stimulation of prostate cancer cells. J. Biol. Chem., 281, 5686–5693 (2006).
- 47) Garcia-Guzman M, Soto F, Gomez-Hernandez JM, Lund PE, Stühmer W. Characterization of recombinant human P2X4 receptor reveals pharmacological differences to the rat homologue. Mol. Pharmacol., 51, 109–118 (1997).
- 48) Robaye B, Boeynaems JM, Communi D. Slow desensitization of the human P2Y6 receptor. Eur. J. Pharmacol., 329, 231–236 (1997).
- 49) Jones CA, Chessell IP, Simon J, Barnard EA, Miller KJ, Michel AD, Humphrey PP. Functional characterization of the P2X4 receptor orthologues. Br. J. Pharmacol., 129, 388–394 (2000).
- 50) Charlton SJ, Brown CA, Weisman GA, Turner JT, Erb L, Boarder MR. PPADS and suramin as antagonists at cloned P2Y- and P2U-purinoceptors. Br. J. Pharmacol., 118, 704–710 (1996).
- 51) Gentile C, Finizio A, Froechlich G, D’Alise AM, Cotugno G, Amiranda S, Nicosia A, Scarselli E, Zambrano N, Sasso E. Generation of a retargeted oncolytic herpes virus encoding adenosine deaminase for tumor adenosine clearance. Int. J. Mol. Sci., 22, 13521 (2021).
- 52) Tamaishi N, Tsukimoto M, Kitami A, Kojima S. P2Y6 receptors and ADAM17 mediate low-dose gamma-ray-induced focus formation (activation) of EGF receptor. Radiat. Res., 175, 193–200 (2011).
- 53) Kim SG, Soltysiak KA, Gao ZG, Chang TS, Chung E, Jacobson KA. Tumor necrosis factor alpha-induced apoptosis in astrocytes is prevented by the activation of P2Y6, but not P2Y4 nucleotide receptors. Biochem. Pharmacol., 65, 923–931 (2003).
- 54) Placet M, Arguin G, Molle CM, Babeu JP, Jones C, Carrier JC, Robaye B, Geha S, Boudreau F, Gendron FP. The G protein-coupled P2Y6 receptor promotes colorectal cancer tumorigenesis by inhibiting apoptosis. Biochim. Biophys. Acta Mol. Basis Dis., 1864 (5 Pt. A), 1539–1551 (2018).
- 55) Sharma S, Kalra H, Akundi RS. Extracellular ATP mediates cancer cell migration and invasion through increased expression of cyclooxygenase 2. Front. Pharmacol., 11, 617211 (2021).
- 56) Qin J, Zhang Z, Fu Z, Ren H, Liu M, Qian M, Du B. The UDP/P2y6 axis promotes lung metastasis of melanoma by remodeling the premetastatic niche. Cell. Mol. Immunol., 17, 1269–1271 (2020).
- 57) Wan H, Xie R, Xu J, He J, Tang B, Liu Q, Wang S, Guo Y, Yang X, Dong TX, Carethers JM, Yang S, Dong H. Anti-proliferative effects of nucleotides on gastric cancer via a novel P2Y6/SOCE/Ca2+/β-catenin pathway. Sci. Rep., 7, 2459 (2017).