Reviews
Genetic and Clinical Advances in Congenital Long QT Syndrome
2014 Volume 78 Issue 12 Pages 2827-2833

Details
2014 Volume 78 Issue 12 Pages 2827-2833
Congenital long QT syndrome (LQTS) is an inherited arrhythmia syndrome characterized by a prolonged QT interval on the 12-lead ECG, torsades de pointes and a higher chance of sudden cardiac death. LQTS segregates in a Mendelian fashion, which includes Romano-Ward syndrome with an autosomal dominant pattern as well as a rare autosomal recessive pattern (Jervell and Lange-Nielsen syndrome). Since 1957 when Jervell and Lange-Nielsen reported the first familial LQTS with congenital deafness, progress in understanding the genetic and electrophysiological mechanisms of LQTS has tremendously improved diagnostic methods and treatments. In the meantime, it has become evident that LQTS may not always be explained by a single gene mutation, but seems to follow a more complex genetic model intertwined with genetic common polymorphisms that have a mild to moderate effect on disease expression. In this review, we summarize the characteristics of LQTS (mainly LQT1–3) and briefly describe the most recent advances in LQTS clinical diagnostics as well as genetics. (Circ J 2014; 78: 2827–2833)
Since the first description of a LQTS family in 1957,1 tremendous progress in understanding its pathogenesis, diagnosis and treatments has been achieved. This review will describe the recent update of the diagnostic scoring system in 2011, the expert consensus statement published in 2013 and new challenges in discovering genotype-phenotype relationship in LQTS. Because of limited space, our focus is mainly on the general concept of the most frequently encountered Romano-Ward syndrome (RWS; LQTS types 1–3). Details of minor LQTS subtypes are to be found elsewhere.2–4
The prevalence of congenital LQTS is reported to be approximately 1/2,000.5 Syncope is generally the most commonly encountered first episode in LQTS patients, and aborted cardiac arrest/sudden cardiac death (ACA/SCD) is rare (1–3%).6 Of the patients who eventually become symptomatic, 50% experience their first cardiac event by the age of 12, and 90% by the age of 40.7 LQTS is also known as an etiology of sudden infant death syndrome (SIDS)8 and in approximately 10% of SIDS cases the infant carried a mutation in a LQTS-causing gene.9
The most frequent LQTS subtypes are type 1 (LQT1), type 2 (LQT2) and type 3 (LQT3).10 The subdivision is based on the underlying genetic substrate, with the potassium channel genes, KCNQ1, KCNH2, and the sodium channel gene, SCN5A, as the involved genes. Specific triggers of symptoms are known in these major LQTS subtypes. For example in LQT1, cardiac events occur during physical exertion or emotional stress, typically during swimming,11 whereas auditory stimulation such as an alarm clock or a telephone bell is a typical trigger for arrhythmic events in LQT2.12 Emotional stress, as well as the postpartum period, are also frequently observed triggers of arrhythmia in LQT2.11,13 More recently, a history of epilepsy has been reported to be more common with LQT2 (39%) than with other subtypes of LQTS (10%, P<0.001),14 possibly because KCNH2 is expressed in the brain as well.14 Obviously in such cases, the differential diagnosis of LQTS and epilepsy is important for treatment, because LQTS is often mistreated as epilepsy because of generalized seizures secondary to torsades de pointes. In LQT3, symptoms are most frequently observed during rest or at night.11,15
Table 1 shows the LQTS diagnostic scoring system (updated in 2011), which includes symptoms, family history and ECG findings.16 Patients with a Schwartz score ≥3.5 points in the absence of a secondary cause for QT prolongation are diagnosed as LQTS.17 Typical LQTS cases can be readily diagnosed with this scoring system, whereas latent LQTS patients with normal QTc at rest, found in 36% of LQT1, 19% of LQT2 and 10% of LQT3, respectively,18 may show a non-diagnostic score of 1–3 points. In such cases, serial ECGs, 24-h Holter recordings, and an exercise or epinephrine test are recommended to reveal subclinical QT prolongation.19–21
| Points | |
|---|---|
| ECG findings | |
| QTc | |
| >480 ms | 3 |
| 460–470 ms | 2 |
| 450 (male) ms | 1 |
| 4-min recovery QTc after exercise test ≥480 ms | 1 |
| Torsades de pointes | 2 |
| T-wave alternans | 1 |
| Notched T wave in 3 leads | 1 |
| Low heart rate for age | 0.5 |
| Clinical history | |
| Syncope | |
| With stress | 2 |
| Without stress | 1 |
| Congenital deafness | 0.5 |
| Family history | |
| A. Family members with definite LQTS | 1 |
| B. Unexplained sudden cardiac death <age 30 among immediate family members | 0.5 |
LQTS diagnostic criteria. QTc is calculated by Bazett’s formula where QTc=QT/
With regard to the exercise test, the QT interval during the recovery phase is valuable in LQTS diagnostics.22,23 In 69 relatives of LQT1 and LQT2 probands who showed borderline to normal QTc at rest, Sy et al reported that a 4-min recovery QTc ≥445 ms discriminated LQTS gene carriers from non-carriers, and a 4-min recovery ≥480 ms showed 100% specificity.22 Horner et al reported from exercise testing of 243 patients that paradoxical QTc prolongation during the recovery phase (QTc ≥460 ms) distinguished LQTS, particularly LQT1, and ∆QTc ≥30 ms (QTc 3-min recovery minus the baseline supine QTc) showed a high sensitivity and specificity (83% and 93%, respectively).23 Importantly, both studies reported that exercise testing showed satisfactory results in distinguishing LQTS cases from innocent ones while on β-blockers, suggesting no need for a β-blocker washout.22,23 As another diagnostic method in LQTS, brisk movement from supine to upright has been reported.24 In that study, QTc was significantly increased by 89±47 ms in the LQTS group compared with 50±30 ms in the control group during maximal sinus tachycardia induced by standing up.24
To note, the QT interval measurement can vary according to physician and LQTS expertise.25 In addition, accurate measurement of the QTc interval at high heart rate (HR) is particularly challenging. In the case of children, by analyzing ECGs from a school-based screening program, Yoshinaga et al managed to appropriately diagnose genotype-positive LQTS cases with a QTc cut-off value (Bazett’s formula) ≥450 ms for HR <75 beats/min and ≥500 ms for HR ≥75 beats/min (94% in screened children), although it was not shown how many genotype-positive patients were missed by the use of these cut-off values.26 In any case, an expert eye on the QT interval (and ST-T morphology) remains a cornerstone of LQTS diagnostics.
In addition to the diagnostic criteria just mentioned, T wave morphology may help differentiate LQT1–3.27 Typically, a broad T wave is observed in LQT1 (Figure A), a biphasic T wave in LQT2 (Figure B) and a late-appearing T wave in LQT3, which has a narrow and tall shape, and appears at the very end of the QT interval (Figure C).27
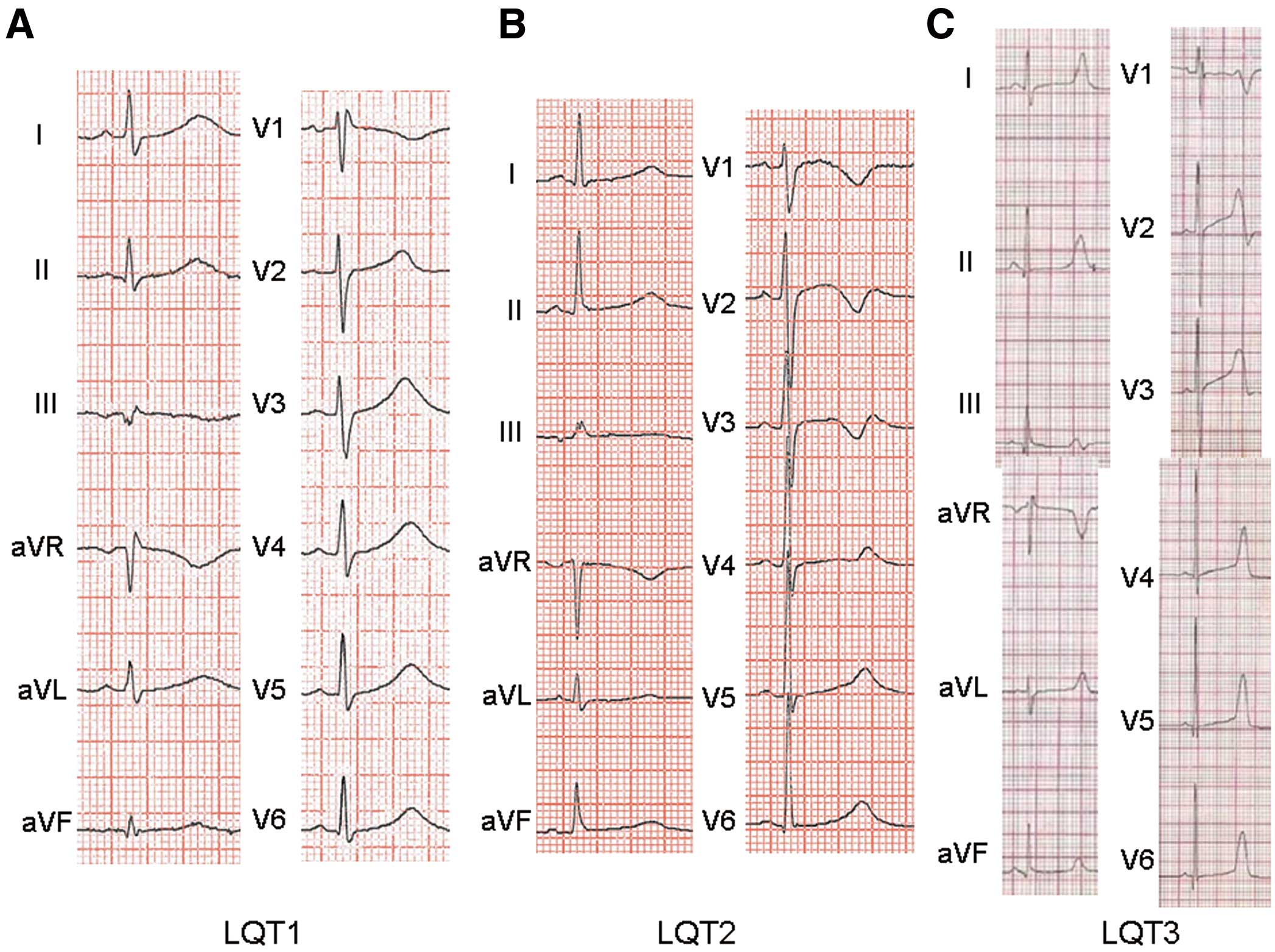
ECGs of LQTS types 1–3, with the typical T wave of each subtype. (A) LQTS type 1 with typical broad-based T wave pattern. (B) LQTS type 2 with typical bifid T wave. (C) LQTS type 3 with late-onset peaked T wave.
Furthermore, genotyping is certainly important in the diagnosis and treatment of LQTS.17 Currently in Asia, LQTS genetic testing (candidate gene approach) is reimbursed in Korea (by Korean NIH for 13 LQTS genes) and in Japan (70% covered by the government public health insurance), but it remains at research level at least in Bangladesh, China, India, Taiwan and Thailand because of either high costs or lack of facilities for broad use (personal communication). Currently, in the era of next-generation sequencing, technological advances enable us to screen many more genes through disease-associated gene panels or even whole exome sequencing. However, with these methods, there are numerous rare “variants of unknown significance” reported with unknown risk associated with disease. Such variants remain elusive and await further research to establish genotype-phenotype relationships before they can used in the clinical setting.
The QT interval reflects repolarization of the ventricular action potentials orchestrated by various cardiac ion currents, including sodium, calcium and potassium currents.28 When a decreased potassium current (loss-of-function) or increased sodium or calcium current (gain-of-function) is caused by a mutation in an ion channel or axillary protein, a prolongation of the action potential duration occurs, which manifests on the 12-lead ECG as QT prolongation.
In 1995 and 1996, causal gene mutations of familial LQTS in 3 genes (KCNQ1, KCNH2 and SCN5A) were discovered.28–30 To date, mutations in 15 different genes have been reported in LQTS (Table 2),31–41 but KCNQ1, KCNH2 and SCN5A remain numerically the major genes in LQTS and comprise more than 90% of genotype-positive cases of LQTS.10 A causal mutation is found in approximately 75% of LQTS patients with a Schwartz score ≥4,42 but the genetic background of the other 25% of patients remain elusive.
| LQTS type | Gene | Protein | Current | Frequency (%) |
|---|---|---|---|---|
| Romano-Ward | ||||
| LQT1 | KCNQ1 | Kv7.1 | ↓ IKs | 40–55 |
| LQT2 | KCNH2 | Kv11.1 | ↓ IKr | 30–45 |
| LQT3 | SCN5A | Nav1.5 | ↑ INa | 5–10 |
| LQT4 | ANKB | Ankyrin | ↓ Coordination of Ncx, Na/K ATPase |
Rare |
| LQT5 | KCNE1 | MinK | ↓ IKs | Rare |
| LQT6 | KCNE2 | MiRP1 | ↓ IKr | Rare |
| LQT7 | KCNJ2 | Kir2.1 | ↓ IK1 | Rare |
| LQT8 | CACNA1C | Cav1.2 | ↑ ICa | Rare |
| LQT9 | CAV3 | Caveolin 3 | ↑ INa | Rare |
| LQT10 | SCN4B | Sodium channel β4-subunit |
↑ INa | Very rare |
| LQT11 | AKAP9 | Yotiao | ↓ IKs | Very rare |
| LQT12 | SNTA1 | Syntrophin-α1 | ↑ INa | Very rare |
| LQT13 | KCNJ5 | Kir3.4 | ↓ IK-Ach | Very rare |
| LQT14 | CALM1 | Calmodulin 1 | Dysfunctional Ca2+ signaling |
Rare |
| LQT15 | CALM2 | Calmodulin 2 | Rare | |
| Jervell and Lange-Nielsen | ||||
| JLN1 | KCNQ1 | Kv7.1 | ↓ IKs | Rare |
| JLN2 | KCNE1 | MinK | ↓ IKs | Rare |
In approximately 85% of genotype-positive cases of LQTS, the patient carries a mutation inherited from one of the parents and in the remaining 15% a de novo mutation is pertinent.43 In genotyped LQTS patients, approximately 50% have no lifetime symptoms, and 10–50% of such patients show no apparent QT prolongation.44–46 Compound mutations (ie, ≥2 mutations) are found in 10% of genotype-positive patients47 and the clinical manifestation of disease in such patients is often more severe.48,49
As for minor LQTS subtypes with severe clinical phenotypes, mutations in KCNQ1 or KCNE1 present as homozygous or compound heterozygous mutations in Jervell and Lange-Nielsen syndrome (J-LNS; Table 2).50,51 KCNQ1 mutations are much more prevalent (90%) than KCNE1 mutations (10%).52 Calmodulin de novo mutations were reported in infant cases of recurrent cardiac arrest by means of exome sequencing in the parents-child trio.41
Since the beginning of research into the genetic background in LQTS, linkage analysis and the candidate gene approach have been used, assuming LQTS to be a monogenic model. Later, it became evident from research into large LQTS families that LQTS actually shows incomplete penetrance with variable expressivity. In other words, different clinical phenotypes (sudden death, syncope, asymptomatic) are observed in family members carrying the same familial mutation, which may be caused by a more complex genetic model involving multiple genetic and environmental factors affecting disease development.
Modifier genes are common genetic variants found in more than 1% of affected individuals, which have an influence on susceptibility to disease. They have less effect on disease compared with a causal gene mutation, but by summing up the effects of such modifier genes, the disease severity varies. In LQTS, variants in the coding regions of LQTS-related genes such as KCNE1 D85N,53 KCNH2 K897T,54 and SCN5A H558R55 are known to influence the QT interval. Single nucleotide polymorphisms (SNPs) in non-coding regions (intron, 3’UTR) of KCNQ1 are also reported as an important QT-interval modifier.56,57 Furthermore, genome-wide association studies have revealed SNPs in NOS1AP as a modulator of QT interval in LQTS patients,58,59 as well as in the general population.60 An effort to find any SNPs affecting QT interval of the Caucasian general population continues by larger international consortia.61 This field is expected to expand to further understand the complexity of genotype-phenotype relationship in LQTS. However, it remains currently at the research level, which requires careful interpretation by experts.62
The well-established parameters of risk stratification in LQTS patients are prolonged QTc interval ≥500 ms and a history of syncope.18 More detailed studies have revealed that the risk of a first cardiac event in males is higher in childhood before puberty, whereas in females, cardiac events occur during adolescence and the postpartum period.63,64 In adults between 18 and 40 years of age, the risks of experiencing any cardiac event are a history of ≥1 cardiac events before age 18 years, female sex, longer QTc interval (≥440 ms) and LQT2.65 For patients older than 40 years, LQT3 patients have significantly more cumulative lethal arrhythmic events (35%) than LQT1 (14%), LQT2 (24%) and genotype-negative patients (10%).66,67 Importantly, a family history of SCD in a first-degree relative is not a significant risk for ACA/SCD.68
LQTS patients with a normal QTc interval (≤440 ms) carry a lower risk of ACA/SCD compared with LQTS patients with QT prolongation (QTc >440 ms), but still show >10-fold risk of fatal arrhythmia compared with genotype- and phenotype-negative family members.69 Such “latent” cases of LQTS can be recapitulated by cellular electrophysiological studies and can give further insights to the mechanisms. For example, Wu et al used a heterozygous construct of KCNQ1-G269S in CHO and HEK cells and showed that this mutation located in segment 5 of IKs channel significantly disrupted upregulation of IKs currents in response to protein kinase A (PKA) stimulation, which fits with the clinical characteristics of patients carrying KCNQ1-G269S (borderline QT interval at rest and QT prolongation and syncope after exercise).70
More recent studies have shown that mutation location and type influence the variability of symptoms. In patients with LQT1, missense cytoplasmic-loop mutations have been reported to cause a longer QT interval at enrolment and an increased risk of ACA/SCD.71 As a specific mutation in KCNQ1, A341V, more than other KCNQ1 mutations (transmembrane or dominant-negative mutations), harbors the most severe phenotype.72–74 In LQT2 patients, the risk of ACA/SCD is higher in females regardless of mutation site compared with males.75 Among male subjects, patients with pore-loop mutations,75 especially missense mutations,76 show a higher incidence of ACA/SCD compared with non-pore-loop mutations.
Beta-blocker therapy is the first choice for LQTS.17 It dramatically decreases event rates, from 0.97 to 0.31 events per patient per year.77 Recurrent events were often seen in patients with a history of ACA or in patients who are non-compliant with β-blocker therapy.77 In LQT1 and LQT2, propranolol (2–4 mg·kg–1·day–1) and nadolol (1–1.5 mg·kg–1·day–1) have been shown to be much more effective than metoprolol in suppressing recurrent cardiac events.78 Therefore, metoprolol should probably not be prescribed in symptomatic LQTS patients.78,79 Atenolol, not included in the aforementioned study, appears to be less effective, according to a study performed in (only) 28 genotyped patients with a median follow-up of 46 months.80
Of the 3 major genotypes of LQTS, β-blocker therapy is extremely effective in LQT1 because of the prominent involvement of adrenergic stimulation in its pathogenesis.11,70 Pure β-blockers are less effective in LQT2 than LQT1, possibly because of α1A adrenoreceptor-mediated IKr reduction.81 Preliminary data recently extracted from >400 LQT3 patients showed that β-blocker therapy is also protective in LQT3.82
As adjunctive therapy, some drugs are used in a genotype-specific manner. For example, oral K+ supplements are considered especially in LQT2 patients because the underlying genetic defect is very sensitive to the serum potassium level.83–85 This holds true particularly when the serum potassium level is low (eg, with diarrhea). In LQT3, mexiletine in addition to β-blocker therapy may be considered in patients with a specific mutation.86 Of note, worsening of QT prolongation by mexiletine was reported in 1 individual with LQT3.87 Therefore, it is important in LQT3 patients to re-check the QT interval after mexiletine administration when the serum mexiletine level reaches the therapeutic level.79 Another drug, ranolazine, a FDA-approved anti-anginal agent, has recently drawn attention as an alternative choice to treat patients with LQTS because it blocks the late sodium current, INa late, (and to a minor extent the IKr and ICa,L without proarrhythmic effects).88 There is so far only 1 clinical report, which includes a small number of patients, on the short-term effectiveness of ranolazine in LQT3.89
Cardiac events in LQTS patients are generally well controlled with β-blocker therapy. However, patients with a history of ACA, symptomatic patients in the first year of life, and patients with J-LNS carrying KCNQ1 mutations are at particularly high risk and therefore, special aids, including implantable cardioverter defibrillator (ICD) and left cardiac sympathetic denervation (LCSD) on top of β-blocker therapy may be deemed necessary because of the high recurrence rate of fatal arrhythmia.77,90 Besides medical treatment, life-style advice for patients is important.17
LCSDLCSD is a surgical procedure to ablate the lower two-thirds of the left stellate ganglion together with the thoracic ganglia T2–T4 to denervate cardiac sympathetic innervation to the heart.91 It is currently used as an adjunctive therapy in symptomatic patients who are refractory to β-blocker therapy. LCSD has been shown to reduce cardiac events significantly in LQTS with a rather high long-term cardiac-event-free survival (46%/5 years,91 59%/2 years92). In patients with a history of syncope, post-LCSD QTc <500 ms predicts efficacy of LCSD.91 However, those with persistent QTc prolongation (≥500 ms) have a high chance of SCD and need to be protected with an ICD. A known complication of this procedure is Horner’s syndrome, but in most cases, it is only transiently observed after the surgery and the patient recovers afterwards.91
ICDUse of an ICD should be regarded as adjunctive therapy in LQTS. An ICD is recommended only for patients who have frequent syncopal episodes despite being on maximal doses of β-blocker (and eventually other additional pharmacological therapies) or at high risk of recurrent ACA/SCD,17 such as patients who have a history of ACA, symptomatic infant cases (<1 year of age) or those with J-LNS.52
Research on LQTS in the past 2 decades has broadened our knowledge of the mechanism as well as genotype-specific therapeutic options. An ongoing challenge is LQTS with normal to borderline QTc at rest. In such cases, diagnostic tests to detect maladaptation of the QT interval to HR are clinically important. Indeed, the updated scoring system in 2011 now includes QT interval of a recovery phase after exercise. In LQTS genetics, next-generation sequencing enables us to detect SNPs in coding/non-coding regions of (LQTS-related) genes that modify the QT interval. Currently, analysis and interpretation of results by next-generation sequencing remains at the research level while advances in the understanding of genotype-phenotype relationships, including LQTS causal genes as well as SNPs, are expected to guide us further to genetically-guided personalized treatment in the future.
We thank Dr Api Khong (Thailand), Dr Jie Wu (China), Dr Jyh-Ming Juang (Taiwan), Dr Laila Banu (Bangladesh), Dr Priya Chockalingam (India) and Dr Seil Oh (Korea) for providing information on LQTS genetic testing in their countries.