Reviews
Discoveries of Hydrogen Sulfide as a Novel Cardiovascular Therapeutic
2014 Volume 78 Issue 9 Pages 2111-2118

Details
2014 Volume 78 Issue 9 Pages 2111-2118
Hydrogen sulfide (H2S) is an endogenously produced gaseous signaling molecule that elicits a number of cytoprotective effects in mammalian species. H2S was originally considered toxic at elevated levels, but 15 years ago the labile molecule was discovered in mammalian tissue and termed a gasotransmitter, thus opening the door for research aimed towards understanding its physiologic nature. Since then, novel findings have depicted the beneficial aspects of H2S therapy, such as vasodilation, antioxidant upregulation, inflammation inhibition, and activation of anti-apoptotic pathways. These cytoprotective alterations effectively treat multiple forms of cardiac injury at the preclinical level of research. The field has progressed towards instituting novel H2S donors that prove more effective at activating the subsequent cardioprotective enhancements over longer time periods. As more findings explore the efficacy of H2S, research focused on detection of sulfhydrated targets is on the rise. Understanding the molecular mechanisms that stem from H2S treatment may lead the field towards powerful therapeutics in the clinical setting. This review will discuss the cytoprotective and cardioprotective effects of H2S therapy, provide analysis on the molecular alterations that lead to these enhancements, and explore recently developed therapeutics that may bring this gasotransmitter into the clinic in the near future. (Circ J 2014; 78: 2111–2118)
Hydrogen sulfide (H2S) is an endogenous, labile molecule that induces a variety of beneficial reactions in the myocardium.1 It is the latest of 3 endogenously produced molecules to be discovered and termed a gasotransmitter, along with nitric oxide (NO) and carbon monoxide (CO).2 All 3 were initially considered to be toxic, but as scientific technology has advanced over the years, researchers have discovered that these gasotransmitters are endogenously produced at low levels, and provide compensatory and protective physiological changes to the heart under times of cardiac stress and injury.3
First measured in 1989 at the neurological level, H2S was recently revealed to play a vital role in mediating physiological processes.4 Numerous studies have explored differential administration of H2S, whether through pharmacological donor therapy,5 genetic expression changes,6 or administration of H2S itself.7 The results have shown promising discoveries regarding the significance of H2S in the body. This review will delve into the cardioprotective effects of H2S and future research directions as enhanced H2S efficacy leads to more reliable therapeutic possibilities.
The gasotransmitters are gaseous signaling molecules that possess a unique ability to freely diffuse through cell membranes and regulate cellular signaling (Figure 1).8 This trait eliminates the need for membrane receptors and intracellular second messenger systems.9 When each of the gasotransmitters was discovered in the environment, they were all considered toxic and potentially lethal to humans, but more recently, researchers discovered that all 3 were endogenously produced, and cytoprotective.
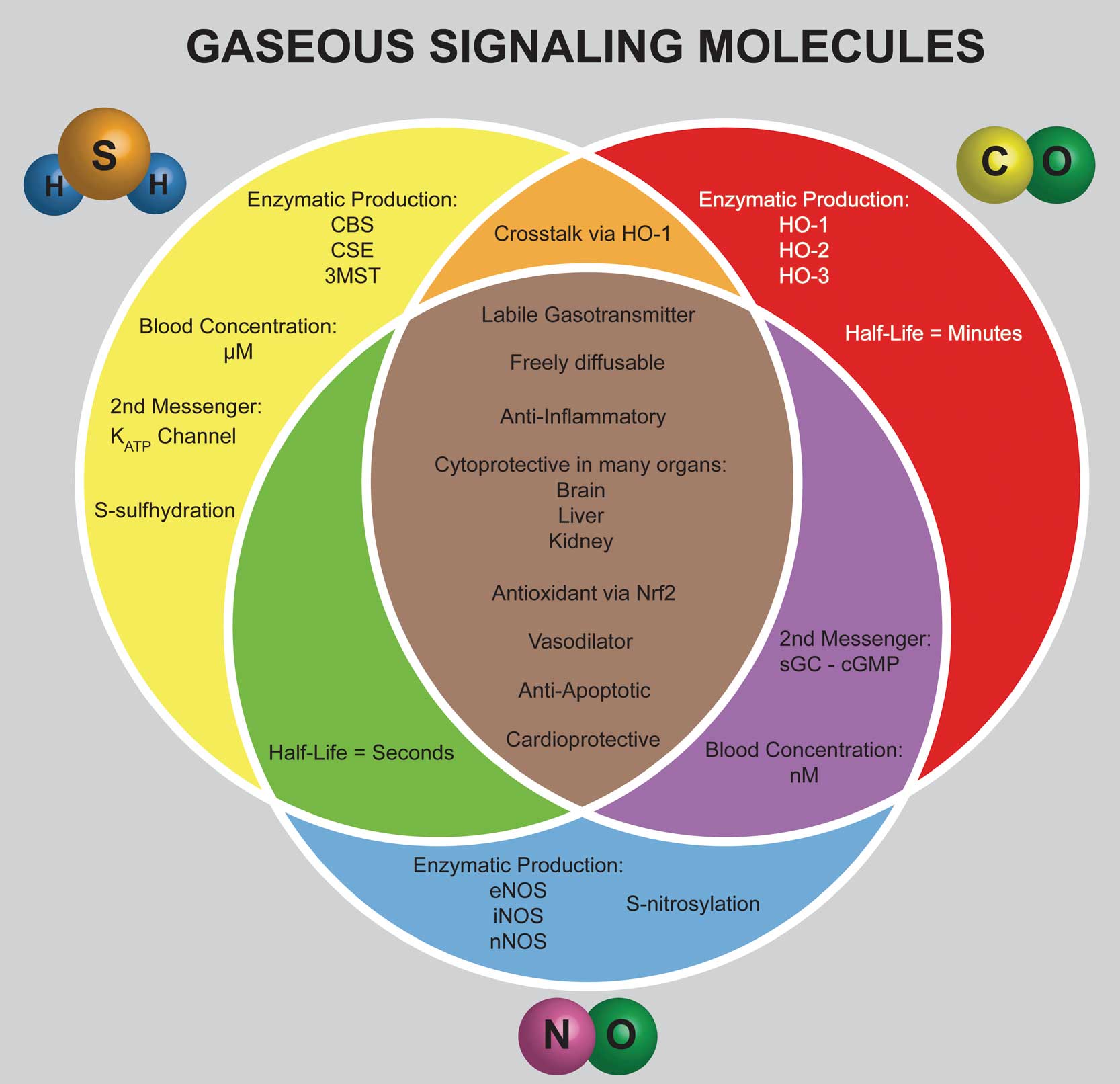
Gasotransmitters that regulate cardiovascular physiology. Hydrogen sulfide (H2S), nitric oxide (NO) and carbon monoxide (CO) make up the gasotransmitter family. These endogenous gases are produced enzymatically and provide potent cytoprotection via modulation of multiple signaling pathways. CBS, cystathionine β-synthase; cGMP, cyclic guanosine monophosphate; CSE, cystathionine γ-lyase; eNOS, endothelial nitric oxide synthase; HO-1, heme oxygenase 1; iNOS, inducible NOS; KATP, ATP-sensitive K+ channel; 3MST, 3-mercaptopyruvate sulfurtransferase; nNOS, neuronal NOS; Nrf2, nuclear-factor-E2-related factor-2; sGC, soluble guanylyl cyclase.
NO was discovered in the 18th century,10 but was found in 1987 to be an endogenous vasodilator in mammals through its generation by NO synthase (NOS).11 There are 3 isoforms of NOS: endothelial (eNOS), neuronal, and inducible. They form NO from the guanidine nitrogen of L-arginine. NO activates guanylyl cyclase in the cytosol, which subsequently causes an increase in cyclic GMP (cGMP) levels. Upregulated cGMP levels cause enhanced vasodilation and smooth muscle relaxation.12 The finding that NO mediates cGMP levels has led to numerous positive correlations in the heart, such as reduced infarct size13 and enhanced compensatory function.14
The second gasotransmitter discovered as a physiological modulator was CO, though it was initially considered an irrelevant and potentially harmful derivative of heme breakdown by the enzyme heme oxygenase (HO).15 Researchers learned in 1991 that CO is a vital cardiovascular mediator that upregulates protective mechanisms.16 The 3 known isoforms of HO are HO-1, HO-2, and HO-3. HO-1 is ubiquitously expressed and inducible, HO-2 is primarily located in the brain and testes and is constitutively active, and HO-3 is similar to HO-2 in constitutive activation but at lower efficiency.17 CO possesses similar properties to NO in its ability to elevate cGMP and augment downstream responses, such as vasodilation and vascular smooth muscle cell growth.18 Indeed, CO has been known to directly regulate NO, further evidence of their interrelationship.19
The third and latest molecule discovered as an endogenous gasotransmitter was H2S, in 2002.20 Like its fellow gasotransmitters, H2S was initially regarded as highly poisonous in the environment, but more recently this perception has changed as a growing number of studies illustrate H2S as a cytoprotective and cardioprotective agent.
H2S activity is rapid and includes synthesis, activation of signaling pathways, and degradation. The most common method of H2S synthesis is enzymatic (Figure 2). Three different enzymes synthesize it: cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE or CGL), and 3-metacaptopyruvate sulfurtransferase (3MST). 3MST, principally localized to the mitochondria, is responsible for most neuronal H2S production and interacts with cysteine aminotransferase to produce H2S from α-ketoglutarate and L-cysteine.21 3MST–/– mice have been developed, and exhibit behavioral abnormalities driven by increased anxiety, theorized to be caused by upregulated serotonin levels or a decrease in H2S leading to reduced antioxidant protection.22 The enzyme CBS was initially discovered localized to astrocytes, through which H2S plays a key role in neuronal long-term-potentiation.23,24 CBS was later uncovered in many other tissues throughout the body. CBS homozygous and heterozygous knockouts have been developed with comprehensive follow-up studies over the years.25 CBS–/– mice exhibit poor survival with aortic endothelial dysfunction, abnormal lipid metabolism, and increased plaque formation.26 CBS+/– mice do not exhibit mortality increase, but they present with LV hypertrophy, endothelial dysfunction, and impaired angiogenesis.26 CSE, meanwhile, is predominantly found in the cardiovascular system, playing a key role in cardioprotection via H2S administration.27 Both CBS and CSE affect H2S regulation throughout body tissues by interacting with L-cysteine to produce H2S, along with the byproducts L-serine and ammonium.2 H2S is endogenously produced in other ways, such as through reduction of molecules that contain thiols, and intracellular sulfur stores that release the molecule.15
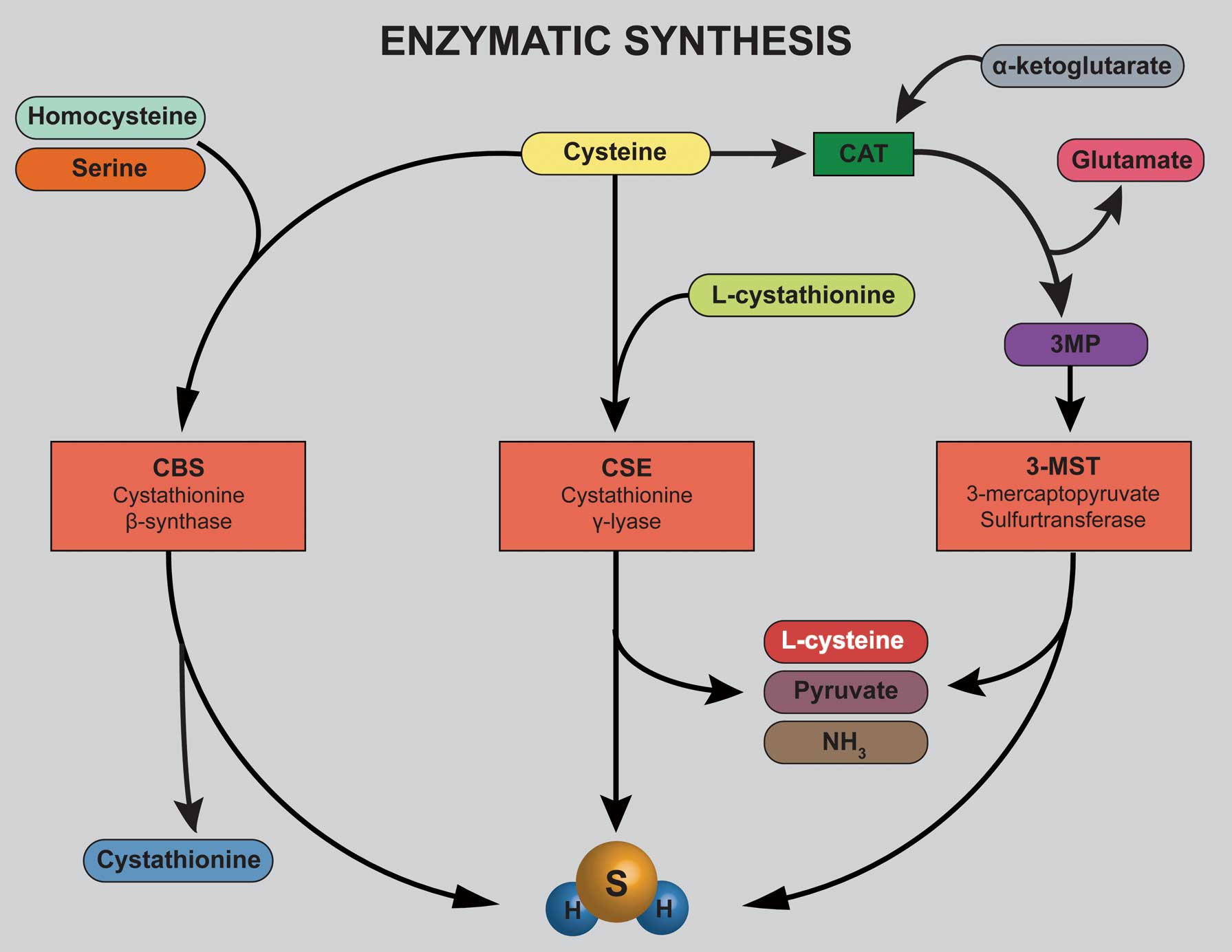
Enzymatic synthesis of hydrogen sulfide (H2S) involves 3 enzymes in the cysteine biosynthesis pathway. CBS is primarily localized to the brain, nervous system, and liver. CSE is predominantly found in the cardiovascular system and liver. 3MST is primarily found in the brain and liver. These enzymes are vital components in the regulation of H2S activity. CAT, cysteine aminotransferase; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; 3MP, 3-mercaptopyruvate; 3MST, 3-mercaptopyruvate sulfurtransferase.
H2S is a very labile molecule that quickly undergoes catabolism in numerous ways. It can interact with thiol S-methyltransferase, leading to its methylation and the formation of dimethylsulfide and methanethiol.28 It may also undergo deprotonation followed by mitochondrial oxidation leading to thiosulfate formation.29 H2S may also interact with methemoglobin, forming sulfhemoglobin.28 This interaction with heme derivatives provides a correlation with the production of the gasotransmitter CO. The growing number of studies providing novel insights regarding the synthesis and breakdown of the gasotransmitter has moved the H2S research field forward immeasurably.
Physiological PropertiesH2S activity upregulates several physiological properties that provide cardioprotection during myocardial stress states (Figure 3). For instance, H2S induces vasodilation, leading to reduced blood pressure.30 When H2S levels are reduced, such as in CSE-deficient (CSE–/–) mice, vasodilation is diminished and a hypertensive phenotype surfaces.6 In the inflammation pathology, H2S treatment in rats inhibited leukocyte adherence and reduced the inflammatory phenotype. Another inflammation study focused on CSE–/– mice, and showed enhanced leukocyte adhesion and upregulation of the inflammatory pathology. The inflamed mice were rescued by administration of H2S donors.3 These studies provide strong evidence of the function of H2S as a vasodilator and anti-inflammatory agent.
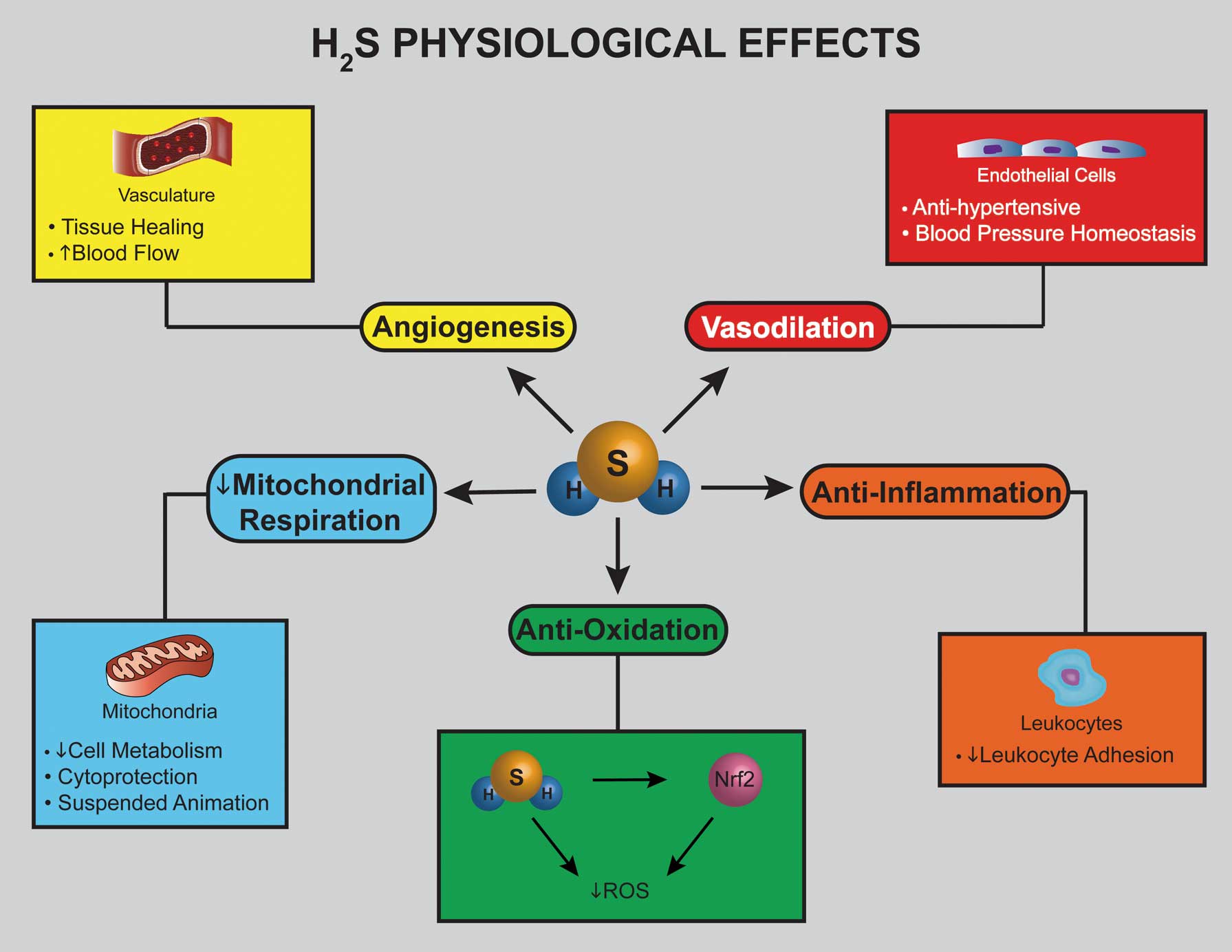
Physiological effects of hydrogen sulfide (H2S) in the cardiovascular system. Overall, cytoprotection results from these altered processes, ameliorating the adverse effects of cardiovascular disease. ARE, antioxidant response element; ROS, reactive oxygen species.
The inflammatory leukocyte pathology, in tandem with other deleterious factors such as dysfunctional endothelium and foam cell formation, may lead to atherosclerosis, and H2S could be a potent treatment. Macrophages are able to produce H2S via CSE activity in the wake of the inflammatory endotoxin lipopolysaccharide, providing an endogenous atherosclerotic treatment.31 Furthermore, administration of the H2S donor, sodium hydrosulfide (NaHS), can inhibit atherosclerotic precursors in macrophages, such as proatherogenic-oxidized low-density lipoprotein-induced foam cell formation.32 In another study, the H2S donor, sodium sulfide (Na2S), and NaHS were both able to inhibit leukocyte adherence and the resultant atherosclerotic pathology.33 This is further evidence of the protective nature of H2S therapy during pathological stress states.
H2S also provides antioxidant effects in a number of tissues, including vascular smooth muscle cells,34 neurons,7 and cardiomyocytes.35 As reactive oxygen species (ROS) are produced from electron transfer reactions in the cell, they are scavenged by antioxidant molecules.36 Oxidative stress states, such as ischemia-reperfusion (IR), cause increased ROS production, a demand that the antioxidant defense system cannot meet.37 The roaming ROS can cause a number of cellular maladaptations, such as lipid peroxidation, protein oxidation to inactive states, and DNA strand breaks.38 H2S reduces this ROS derangement by upregulating antioxidant properties. It can scavenge the ROS superoxide35 and protect neuronal death by upregulating levels of glutathione, another antioxidant.7 These findings suggest that H2S is an antioxidant itself, but can also upregulate antioxidant protective mechanisms. One such mechanism is through the nuclear-factor-E2-related factor-2 (Nrf2) dependent signaling pathway. Nrf2 is a member of the NF-E2 family of nuclear basic leucine zipper transcription factors, and activates enzymes that scavenge pro-oxidative stressors.39
Moreover, H2S has been shown to play a role as an angiogenesis mediator. Knockout of CSE leads to downregulation of the Akt pathway, preventing activation of pro-angiogenic factors.40 Furthermore, H2S upregulates the mitogen-activated protein kinase (MAPK) pathway, primarily the ERK (extracellular signal-regulated kinase) and p38 proteins, leading to enhanced cell proliferation. In a model of hypertension-induced heart failure, H2S ameliorated the LV remodeling process, while upregulating angiogenesis.41 Additionally, administration of NaHS in drinking water increased VEGF expression and inhibited angiostatin and endostatin, 2 antiangiogenic factors.42 Also, the upregulation of both Akt and VEGF may cause this evident angiogenesis phenotype after administration with a H2S donor molecule (SG-1002).5
H2S also modulates cellular homeostasis by affecting mitochondria. It inhibits complex IV of the mitochondrial electron transport chain, cytochrome c oxidase,43 leading to downregulated mitochondrial respiration and subsequent cardioprotective effects during myocardial IR injury.44,45 The ability of H2S to inhibit mitochondrial respiration can cause a state of suspended animation in mice.46 This novel finding may lead to H2S therapies aimed at mitigating pathologies stemming from IR injury or hypoxia.
There have been many studies of the beneficial aspects of H2S therapy in several organ systems. The molecule was found to be neuroprotective in vivo, reducing cerebral IR injury and oxidative stress effects via administration of NaHS.47 Another study found that H2S, administered through NaHS, protects neurons in vitro from oxidative stress resulting from IR.7 Additionally, in vivo models show that NaHS is able to reduce inflammation after cerebral IR,48 and that the H2S donor diallyl sulfide, the primary organosulfur component of garlic, can reduce cerebral infarct volume and caspase-3 activation, a hallmark of apoptosis.49
The gasotransmitter also shows cytoprotective effects in the liver; Na2S was administered to mice in a hepatic IR injury model and mitigated elevations of serum alanine aminotransferase and aspartate aminotransferase levels.50 In the pulmonary system, exogenous H2S solubilized in Krebs-Henseleit Buffer has been shown to protect lungs from IR injury in an ex vivo rat model.51 H2S also plays a protective role in the renal system. In a rat kidney IR model, the injury yielded a reduction in endogenous H2S production through CBS synthesis. This was ameliorated by exogenous H2S administration and led to improved renal function.52 These studies illustrate the protective actions of H2S throughout the body.
CardioprotectionCardiovascular H2S studies are growing at the greatest rate among those regarding gasotransmitters, as mounting evidence shows its potent cardioprotection. Ex vivo administration of NaHS before IR reduced myocardial injury.53 An in vivo murine model of IR injury showed that Na2S reduced infarct size at a dose-dependent cardioprotective rate.54 In another IR porcine model, subjects received Na2S treatment 10 min prior to reperfusion. At the end of the reperfusion period, treated pigs exhibited enhanced LV function compared with control pigs.55
In the setting of heart failure, overexpression of the enzyme CSE has uncovered interesting findings. This transgenic alteration leads to cardioprotective effects after either IR-induced heart failure or pressure-overload induced heart failure.5 Furthermore, knockout of CSE in murine models of heart failure showed worsened myocardial function and greater infarct size.6,20 In another model of pressure-overload induced heart failure, mice administered Na2S exhibited enhanced pro-angiogenesis factors, such as matrix metalloproteinase (MMP)-2, and suppressed anti-angiogenesis factors, including MMP-9.42 These studies illuminate the multifaceted capacity for cardioprotection of H2S through various signaling modifications.
Cellular TargetsH2S interacts with numerous signaling targets that activate various cardioprotective pathways (Figure 4). One such target is the ATP-sensitive K+ (KATP) channel.56 These channels are located on the surface of cell membranes and mitochondria in many cell types, including neurons, smooth muscle cells, and cardiac myocytes.57 Studies have shown that KATP channels are affected by dose-dependent H2S administration,30 and that endogenous H2S is a key regulator of KATP channel activity.35 KATP channels activate protein kinase C (PKC), which in turn activates the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA). This leads to augmented resequestration of Ca2+ from the cytosol to the sarcoplasmic reticulum,58 causing enhanced cardiomyocyte metabolism and compensatory Ca2+ handling during myocardial stress states. In isolated cardiomyocytes, investigators discovered that IR injury and causative Ca2+ overload are ameliorated by H2S-induced activation of PKC, leading to upregulated SERCA activity, providing strong evidence of H2S protection via KATP and Ca2+ regulation.
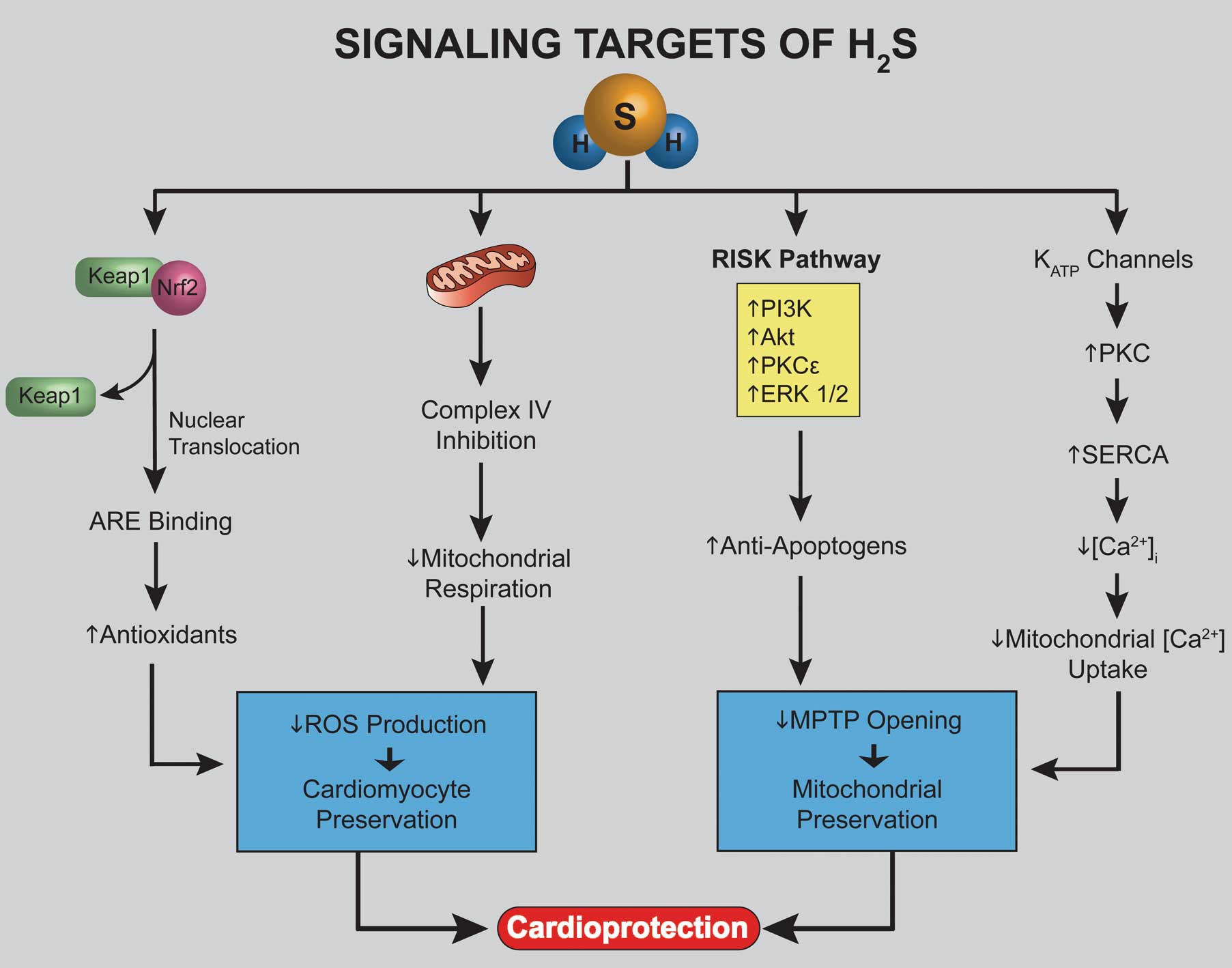
Signaling pathways mediated by hydrogen sulfide (H2S) that provide similar cardioprotective results. These protective pathways strengthen myocardial rescue from various cardiomyopathies. ERK, extracellular signal-regulated kinase; ARE, antioxidant response element; Keap1, Kelch-like ECH-associated protein 1; MPTP, mitochondrial permeability transition pore; PI3K, phosphatidylinositide 3-kinase; PKC, protein kinase C; RISK, reperfusion injury salvage kinase; ROS, reactive oxygen species; SERCA, sarco-endoplasmic reticulum Ca2+-ATPase.
In the setting of diabetes, H2S pretreatment has proven cardioprotective in an IR injury mouse model. This protection resulted from activation of the aforementioned Nrf2, an antioxidant signaling molecule.59 The Nrf2 protein can modulate mitochondrial oxidative stress by promoting the expression of antioxidant genes.59 H2S activation causes Nrf2 to separate itself from its adherent inhibitor, Kelch-like ECH-associated protein 1 in the cytosol,60 then translocate to the nucleus and bind to a specific enhancer sequence, known as the antioxidant responsive element, in the promoter region of antioxidant genes. This leads to upregulation of antioxidant proteins, including HO-1 and thioredoxin 1.59 Chronic administration of H2S for 7 days caused increased Nrf2 and antioxidant levels,3 as did CSE overexpression in mice.59
H2S can also affect mitochondrial targets via upregulation of the reperfusion injury salvage kinase pathway, which is able to inhibit the opening of mitochondria permeability transition pores (MPTP).61 Downregulation of MPTP leads to reduced mitochondrial membrane potential depolarization, and consequent inhibition of pro-apoptotic protein activation.62 Furthermore, H2S is able to downregulate ROS at the mitochondria, providing cardioprotection through reduced respiration.44 Coupled with the ability of H2S to enhance anti-apoptogen activation, such as HSP90 and Bcl-2,63 the gasotransmitter is a potent regulator of cellular death through mitochondrial regulation.59
H2S can elicit further cytoprotective effects via crosstalk with the other gasotransmitters. Most researched has been the interaction between H2S and NO, primarily through H2S activation of eNOS.5 Similarly, NO has been found to upregulate the H2S enzyme CSE.64 It has been theorized that the vasodilatory effects of these molecules, though produced through different pathways, work concomitantly to accentuate this beneficial response in the body. The interactions between H2S and CO have been less researched, but one study found that H2S is able to upregulate the CO/HO pathway during hypertensive stress states.65 Furthermore, H2S can upregulate HO-1 expression via the Nrf-2 signaling pathway.66
The significance of H2S has grown with the discovery that it modifies a diverse array of proteins. Measuring H2S levels accurately is paramount in properly characterizing this molecule. Previously established methods only provide partial results of H2S processes. In one such process, gas chromatography-chemiluminescence, fresh tissue homogenate is incubated in buffer, allowing for sulfide gas to be released. The released gas is measured in the headspace and quantified, but the incubation period takes time, removing the possibility for real-time sulfide release analysis.67 H2S levels are also measured with the methylene blue assay, which indicates sulfide concentrations through colorimetry. However, results may not be wholly accurate, because of interference and artifact.68 Another method is fluorescent probe detection, but this may read thiol interference, leading to inaccurate results.29
Sulfhydration diversity has been recently studied by Mustafa et al,69 using a modification of the biotin switch assay that traditionally detects S-nitrosylation (SNO). The biotin switch assay is normally able to detect nitrosylated thiols by selectively exposing them via ascorbate treatment, followed by labeling of the sulfhydryl groups with biotin-HPDP.70 The sulfhydration study found that biotin labeling still occurs without the ascorbate-induced exposure of nitrosylated thiols. These newly discovered thiols may be S-sulfhydrated (cysteine –SH groups are converted to –SSH), providing potential evidence of direct H2S modification of a number of key proteins.69 The group examined murine hepatic sulfhydration and found 39 sulfhydrated proteins, which may lead to fascinating insights regarding the sulfhydration pathway. For example, CSE may directly bind to target proteins, leading to H2S production at the site where it upregulates physiological changes. This would be analogous to NOS binding to SNO targets. Or perhaps CSE produces H2S from elsewhere in the cell, and the labile gasotransmitter itself quickly travels to the target proteins. This novel method of identifying S-sulfhydrated proteins shows that H2S is a key factor in posttranslational modification, and must be further characterized to understand its protective abilities.
An innovative method of labeling SNO has been developed by Kohr et al,71 providing for the first time an in-depth look at SNO occupancy in the myocardium. Whole heart homogenates were used with a cysteine-reactive tandem mass tag labeling and enrichment kit. Ischemic preconditioning was administered to show upregulated SNO in various proteins, exhibiting the protective trait of NO. As discussed, H2S shares many traits with NO, so a modified version of this protocol may be an effective method for determining sulfhydration of pertinent proteins.
H2S donors are on the rise, providing more insights into instituting the molecule as an efficacious therapeutic. The most frequently utilized H2S donors are NaHS and Na2S, salts that can quickly release H2S in vivo. As beneficial as they are in providing H2S at appropriately high levels to elicit physiologic responses, problems lie in their subsequent short half-life.72 Donors that remain stable and long lasting in the body would be better suited in the clinic. One such stable donor, diallyl trisulfide, is a derivative of garlic that is able to decrease infarct size and improve mitochondrial coupling in mice after acute myocardial ischemia.73
Another H2S donor has recently been synthetically developed, termed SG-1002, and was orally administered in a murine model of TAC.5 The subsequent upregulation of H2S levels led to enhanced cardiac function and mitigation of LV remodeling. This donor was also able to upregulate eNOS activity and resultant NO production, leading to ameliorative vasodilation that contributed to cardiac rescue.5 A clinical trial has just begun to assess the efficacy of the SG-1002 compound in patients (clinicaltrials.gov). Furthermore, a water-soluble H2S donor has recently been developed, called GYY4137.74 This molecule slowly releases H2S over the course of hours, and provides the hallmark beneficial effects of H2S, such as vasodilation and antihypertensive activity. Moving forward, it is imperative that researchers investigate the ability of these stable donors to provide the least disruptive therapeutic effect in the clinic. Novel therapeutic development is not the only goal of H2S clinical studies. A new clinical trial is underway to understand how cardiovascular disease affects H2S levels, specifically in women (clinicaltrials.gov). The study will distinguish H2S levels in the cardiovascular system in women with and without peripheral or coronary artery disease, with the aim of defining novel differences in H2S during these pathological disease states.
Therapeutic uses of H2S are growing outside of the cardiovascular system, as well. There is a recently developed H2S-releasing derivative of naproxen (ATV-346) that reduces the gastrointestinal damage induced by the toxicity of standard naproxen.75 This novel drug compound was also found to augment the chemotherapeutic effects of standard naproxen in a murine model of colon cancer. Not only did smaller doses of ATV-346 reduce cancerous lesions at a greater rate than standard naproxen, but the comorbidity of gastrointestinal injury was abrogated as well.76 This drug is an example of the varied benefits that H2S can provide in the therapeutic setting.
This review has explored the groundwork discoveries that elucidate the newfound protective ability of H2S in the heart. Future experiments will shed light on the molecular mechanisms that are altered with endogenous or exogenous H2S administration. There are a growing number of studies making use of the stable donors of H2S, providing insights into the development of effective pharmacological agents in patients. Characterization of sulfhydrated proteins is a novel topic that may guide the field towards previously unknown molecular findings. As the physiologic benefit of H2S is further understood, its cardioprotective effects can be utilized more efficaciously at the clinical level.
Funding Support: This study was supported by a grant from the National Institutes of Health National Heart Lung and Blood Institute (NHLBI) (5R01HL098481-05) to J.W.C. The work was also supported by funding from the Carlyle Fraser Heart Center of Emory University Hospital Midtown.