Reviews
Heart Development, Diseases, and Regeneration – New Approaches From Innervation, Fibroblasts, and Reprogramming –
2016 Volume 80 Issue 10 Pages 2081-2088

Details
2016 Volume 80 Issue 10 Pages 2081-2088
It is well known that cardiac function is tightly controlled by neural activity; however, the molecular mechanism of cardiac innervation during development and the relationship with heart disease remain undetermined. My work has revealed the molecular networks that govern cardiac innervation and its critical roles in heart diseases such as silent myocardial ischemia and arrhythmias. Cardiomyocytes proliferate during embryonic development, but lose their proliferative capacity after birth. Cardiac fibroblasts are a major source of cells during fibrosis and induce cardiac hypertrophy after myocardial injury in the adult heart. Despite the importance of fibroblasts in the adult heart, the role of fibroblasts in embryonic heart development was previously not determined. I demonstrated that cardiac fibroblasts play important roles in myocardial growth and cardiomyocyte proliferation during embryonic development, and I identified key paracrine factors and signaling pathways. In contrast to embryonic cardiomyocytes, adult cardiomyocytes have little regenerative capacity, leading to heart failure and high mortality rates after myocardial infarction. Leveraging the knowledge of developmental biology, I identified cardiac reprogramming factors that can directly convert resident cardiac fibroblasts into cardiomyocytes for heart regeneration. These findings greatly improved our understanding of heart development and diseases, and provide a new strategy for heart regenerative therapy. (Circ J 2016; 80: 2081–2088)
The heart consists of many types of cells, including cardiomyocytes, fibroblasts, neuronal cells, and vascular cells. Cardiac function is tightly controlled by interactions among these cells, but the roles and interactions of myocyte and non-myocyte cells in cardiac development, diseases, and regeneration have been unclear. I first studied the molecular mechanism of cardiac sympathetic innervation and its relationship to heart diseases in the year 2000. Endothelin-1 (ET-1), which plays a critical role in cardiac hypertrophy, was shown to induce nerve growth factor (NGF) expression in cardiomyocytes. Deletion of ET-1 or NGF in mice revealed that the ET-1-NGF pathway determines the density of sympathetic innervation in the heart.1 NGF is critical for cardiac sensory innervation, and NGF expression is reduced in heart of diabetic subjects.2 Sensory denervation and silent myocardial ischemia in diabetic rat hearts could be rescued by NGF gene transfer. The development of the cardiac sympathetic nervous system is regulated by a balance between the chemoattractant, NGF, and the chemorepellent, Sema3a, both of which are synthesized by cardiomyocytes. Sema3a, expressed abundantly in subendocardial myocytes during embryonic stages, determines the epicardial-to-endocardial innervation patterning. Misregulation of Sema3a disrupts sympathetic innervation patterning, leading to lethal arrhythmias such as sinus bradycardia and ventricular tachycardia (VT).3 From 2007, I have investigated the role of cardiac fibroblasts in heart development. Embryonic cardiac fibroblasts synthesize HB-EGF, fibronectin, and collagens, which activate myocardial proliferation through β1-integrin signaling.4 I next questioned whether resident cardiac fibroblasts could be converted into beating cardiomyocytes. In 2010, I identified cardiac reprogramming factors, Gata4, Mef2c, and Tbx5 (GMT), which can promote the generation of cardiomyocyte-like cells (iCMs) from fibroblasts without the requirement for pluripotent stem cells.5 After this discovery, I started my laboratory in Japan, and found that GMT can convert resident cardiac fibroblasts into iCMs in situ after myocardial infarction (MI) in mice.6 We also identified human cardiac reprogramming factors in 2013,7 promoted cardiac reprograming by adding miRNA to GMT,8 standardized the culture conditions for cardiac reprogramming, and revealed new molecular mechanisms of cardiac reprogramming.9 In 2016 I received the Sato Award from the Japanese Circulation Society for these achievements. Herein, I will review my research, which has changed our current understanding of cardiac development, diseases, and regeneration. The ultimate goal is to translate these findings to novel therapeutic approaches for treating heart diseases, which represent a major cause of death in Japan and other modern societies.
Cardiac tissue is richly innervated by the autonomic nervous system. Cardiac sympathetic nerves synthesize norepinephrine, which can increase heart rate and myocardial contraction via adrenergic receptors on cardiomyocytes.10,11 In contrast, NGF is synthesized by cardiomyocytes, and is a growth factor involved in maintaining sympathetic innervation in tissues.12,13 Although much attention had been given to the regulation of norepinephrine-adrenergic receptor signaling in cardiomyocytes, little was known about the regulation of NGF expression in cardiomyocytes. Among several hypertrophic hormones, ET-1 specifically increased NGF expression in cardiomyocytes.1 ET-1 was shown to induce NGF expression in cardiomyocytes though the ETA receptor, Giβγ, protein kinase C, the Src family EGRR, ERK, p38MAPK, AP-1, and C/EBP. This was one of the first studies demonstrating the molecular mechanism of NGF regulation in cardiomyocytes. My work also revealed that ET-1 regulates cardiac sympathetic innervation in the mouse heart by upregulating NGF. In ET-1 knockout (KO) mice, the stellate (sympathetic) ganglia were small and apoptotic, and cardiac innervation density was reduced. These defects were completely rescued by overexpression of NGF in cardiomyocytes, through crossing NGF transgenic (TG) mouse with ET-1 KO mice. These results demonstrated that ET-1-NGF signaling is critical for the development of the cardiac sympathetic nervous system (Figure 1).

Cardiac innervation and heart disease. (Upper) Normal cardiac innervation. Heart rhythm is normal, and a person suffering from myocardial ischemia perceives chest pain. (Lower) Cardiac innervation is disrupted by misregulation of NGF and Sema3a. This patient might suffer fatal arrhythmias, such as VF/VT and sinus arrest, and silent myocardial ischemia because of the denervation of sensory nerves. NGF, nerve growth factor; VF, ventricular fibrillation; VT, ventricular tachycardia.
Pain perception during myocardial ischemia is transmitted by the cardiac sensory nervous system. Silent myocardial ischemia is a common clinical symptom in diabetic patients, in whom chest pain is not properly perceived during myocardial ischemia.14 It had been postulated that cardiac sensory denervation is a main cause of this disease state, but the regulatory mechanism of cardiac sensory innervation in development and in diabetes was unknown. Moreover, markers for sensory nerve endings in the heart remained unclear. I found calcitonin gene-regulated peptide (CGRP) to be a novel molecular marker for cardiac sensory innervation. Cardiac sensory nerves, immunostained as CGRP+ fibers, were increased with development from the mid-embryonic stages. My work also showed that cardiac NGF expression increased with the development of sensory innervation, and that the cardiac sensory nervous system, consisting of thoracic dorsal root ganglia (DRG), dorsal horn, and sensory nerve endings in the heart, was retarded in NGF KO mice.2,15 NGF overexpression in cardiomyocytes rescued the defects of the cardiac sensory nervous system in NGF-deficient mice, demonstrating that NGF derived from cardiomyocytes plays critical roles not only in sympathetic but also in sensory innervation. In vitro studies also revealed that NGF secreted from cardiomyocytes is required for axonal extension and CGRP expression in DRG sensory neurons. Intriguingly, the cardiac sensory nervous system was retarded and NGF expression was suppressed in streptozotocin (STZ)-induced diabetic mice, which could explain silent myocardial ischemia in diabetic patients.16 More importantly, NGF supplementation through TG cardiac expression of NGF or through NGF gene transfer was shown to rescue pain perception in diabetic mice after MI. Both the reduction in CGRP+ nerve fibers and nerve activity were rescued by NGF supplementation in STZ-induced diabetic animals. That study revealed a novel therapeutic approach using NGF gene therapy for silent myocardial ischemia in diabetic patients.
It is well known that sympathetic innervation is not homogeneous in the heart. Sympathetic nerve endings are rich in the conduction system and subepicardial myocardium, but scarce in the subendocardial region.17 This innervation patterning is well conserved among mammals, but the molecular mechanisms that govern this highly organized structure were unknown. Although we, and others, demonstrated that NGF is a critical growth factor and a strong chemoattractant for cardiac innervation, the chemorepellent that inhibits neural extension and determines innervation patterning was undetermined. I first found that Sema3a, expressed by cardiomyocytes, repelled sympathetic innervation and established cardiac innervation patterning, which is dense in epicardial sites and scarce in endocardial sites.3,18 Sema3a is highly expressed in the subendocardial trabecular layer in early embryonic stages, but is reduced with development and confined to Purkinje fibers after birth. Sympathetic epicardial-to-endocardial innervation patterning was disrupted in Sema3a KO mice. These animals exhibited protrusion of sympathetic nerve endings towards endocardial sites from the early stages of development, which resulted in similar levels of sympathetic innervation densities at the epicardial and endocardial sites after birth. Sema3a KO mice demonstrated significant reductions in heart rate variability in the low filtration band, suggesting that sympathetic nerve activity was reduced in these mice. Sema3a KO mice also showed malformation of stellate ganglia, sinus bradycardia, premature ventricular contractions, and sudden death. To determine the role of cardiac Sema3a in sympathetic innervation, I next generated cardiac-specific TG mice overexpressing Sema3a only in cardiomyocytes. Cardiac innervation patterning was disrupted in Sema3a TG mice, which exhibited a reduction in sympathetic nerve endings at the epicardial site. The expression of the β1 adrenergic receptor and action potential duration (APD) in the subepicardium were increased in Sema3a TG mice compared with wild-type controls. TG mice revealed sustained VT and ventricular fibrillation (VF) with epinephrine or electrical stimulation, and 20% of the TG mice died suddenly after 6 months with no apparent structural abnormalities in the heart, suggesting that sudden death was from lethal arrhythmias.19,20 Thus, Sema3a could be a new molecular marker for arrhythmogenesis and sudden cardiac death.
Other groups also recently reported that dysregulation of Sema3a could be a risk factor for unexplained cardiac death of patients with VF or Brugada syndrome (BrS).21,22 Nakano et al reported that a non-synonymous polymorphism (I334V) in exon 10 of the Sema3a gene was significantly associated with unexplained cardiac arrest with VF.21 VF predominantly occurred at night and sinus bradycardia was observed in these patients. Moreover, pathological examination revealed that sympathetic innervation extended to the subendocardial zone similar to that in Sema3a KO mouse, suggesting a disruption of cardiac innervation patterning in these patients. A Sema3a polymorphism (I334V) resulted in reduced chemorepellent function, and the authors concluded that this polymorphism is a new genetic marker of unexplained sudden cardiac arrest with VF in humans, and that inappropriate innervation patterning could explain this disorder. Boczek et al reported that Sema3a not only repelled sympathetic innervation but also directly altered electrophysiological properties in cardiomyocytes.22 Sema3a inhibited Kv4.3 channel activity and reduced Ito currents in cardiomyocytes, which is consistent with APD prolongation in Sema3a TG mice. In addition, 2 rare Sema3a missense gene mutations were identified in patients with BrS. BrS is characterized by conduction abnormalities, ST segment elevation on ECG, and increased risk of sudden cardiac death from VF. It is well known that not only dysregulation of the Na and Ito currents, but also the activity of the autonomic nervous system can cause arrhythmia. These Sema3a missense mutations disrupted the ability of SEMA3A to inhibit Kv4.3 channels, resulting in significantly increased Ito currents, which could be the pathogenic basis of BrS.23 These results clearly demonstrated that Sema3a is a new genetic marker for sudden cardiac death in unknown genetic disorders.24
Cardiac fibroblasts, which are abundant in the adult heart, are quiescent and provide a mechanical scaffold for myocardial contractions under physiological conditions. These fibroblasts are activated, proliferate, and synthesize extracellular matrix (ECM) and growth factors after myocardial injury.25 The extrinsic factors secreted from cardiac fibroblasts affect neighboring cardiomyocytes, leading to cardiac hypertrophy and remodeling. Although much attention has been given to the function and roles of cardiac fibroblasts in the adult heart, little was known about embryonic cardiac fibroblasts. It was unclear when and how fibroblasts migrate from the epicardium and proliferate during embryogenesis. During development, cardiomyocytes proliferate actively; however, the function of cardiac fibroblasts at this stage was unclear.26 I hypothesized that embryonic fibroblasts resembled activated fibroblasts in adult hearts after injury, synthesizing various factors and promoting cardiac hypertrophy.27 I first discovered that embryonic cardiac fibroblasts appeared in mouse hearts from embryonic day 12.5, and increased in number through myocardial proliferation. Next, I investigated whether cardiac fibroblasts promote cardiomyocyte proliferation in coculture experiments. Intriguingly, embryonic cardiac fibroblasts promoted cardiomyocyte proliferation, but adult fibroblasts promoted cardiac hypertrophy. Embryonic fibroblasts specifically expressed several molecules, including Fn1, Col3a1, and HB-EGF, which could collaboratively promote cardiomyocyte proliferation through the β1 integrin, ERK, and PI3K/Akt pathways. To test the role of β1 integrin signaling in cardiac development in vivo, I generated cardiomyocyte-specific β1 integrin KO mice; 50% of the mutant mice died before birth, and demonstrated reduced myocardial growth and proliferation, leading to cardiac fibrosis.28 Mechanistically, the expression of cell cycle regulators, such as Ccnd1, Ccne1, Ccng2, and Cdkn1a, were dysregulated in β1 integrin KO mice. Thus, embryonic cardiac fibroblasts synthesized specific ECM components and growth factors to promote myocardial proliferation through β1 integrin, which was a previously unrecognized function of cardiac fibroblasts during development (Figure 2).4
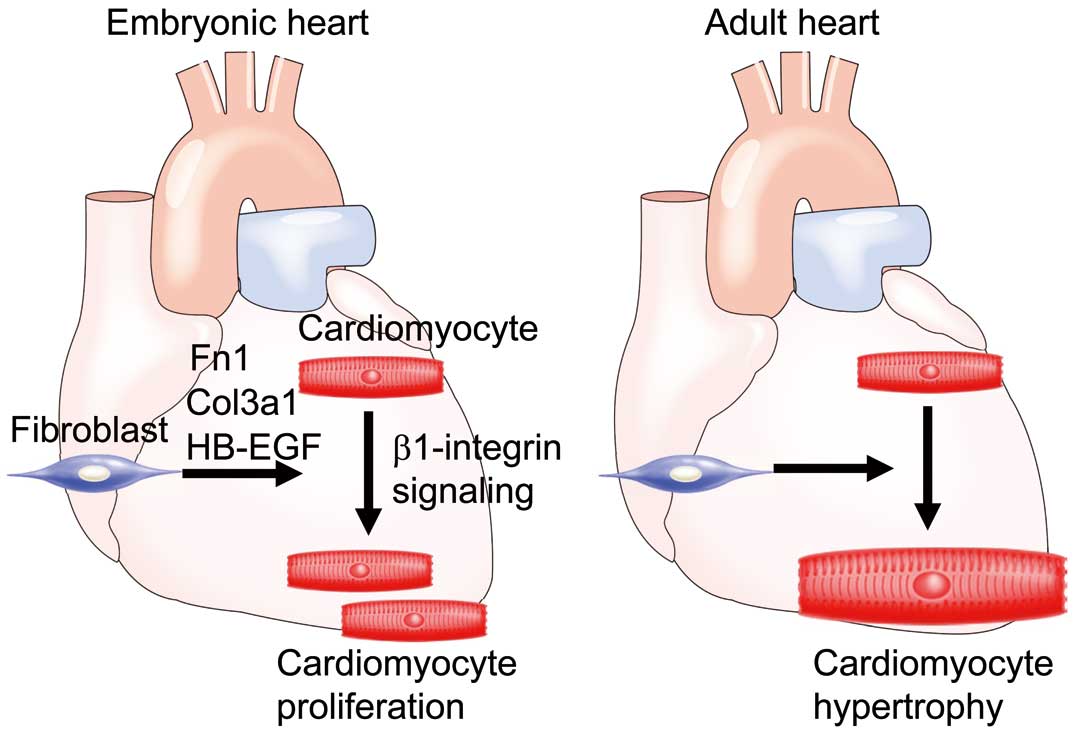
Embryonic cardiac fibroblasts promote cardiomyocyte proliferation. Embryonic cardiac fibroblasts synthesize Fn1, Col3a1, and HB-EGF, which collaboratively promote cardiomyocyte proliferation through β1-integrin (Left). In contrast, adult cardiac fibroblasts induce cardiac hypertrophy (Right).
During my study of cardiac fibroblasts and heart development, I identified molecules that are specifically expressed in cardiomyocytes but not in fibroblasts.4 These include cardiac-specific transcription factors, known to regulate cardiac development and with roles in cell fate decisions. Although there were reports demonstrating that the deletion of a single gene can cause embryonic lethality and cardiac abnormality, no single ‘‘master regulator’’ of cardiomyocytes, similar to MyoD for skeletal muscle, was identified. In contrast, it was well known that a network of cardiac transcription factors, namely “core transcription factors”, synergistically activate the cardiac program to differentiate stem/progenitor cells into cardiomyocytes. Takahashi and Yamanaka also demonstrated that a combination of core stem cell-specific transcription factors, Oct4, Sox2, Klf4, and c-myc, could reprogram fibroblasts into pluripotent stem cells.29 Thus, I hypothesized that not a single factor, but a combination of core cardiac transcription factors might be the master regulator of cardiomyocytes, functioning to determine cardiomyocyte cell fate. I suggested that if cardiac fibroblasts could be directly converted into beating cardiomyocytes, new approaches could be developed for heart regeneration, as this strategy would not require cell transplantation. Identification of cardiac reprogramming factors that convert fibroblasts into cardiomyocytes, without reversion to stem cells, seemed to be a high-risk project, but I initiated such research based on its attractiveness. To avoid bias in the selection of reprogramming factors and to increase the probability of success, I first developed a screening system to identify these factors. I generated an αMHC-GFP TG mouse, in which green fluorescent protein (GFP) is driven by the αMHC promoter, and quantitatively determined the efficiency of cardiac induction. The next important question was which genes should be selected as candidate factors. I used 2 criteria to select candidate factors: (1) genes that are expressed specifically in cardiomyocytes but not in fibroblasts and (2) genes for which genetic deletion resulted in embryonic lethality and cardiac malformation; this was to ensure that the reprogramming factors were functionally important, with crucial roles in cardiogenesis. To select cardiac-specific factors, I utilized microarray data and the differential gene expression profiles between cardiomyocytes and fibroblasts, based on my previous analysis.4 Based on bioinformatics publications, I selected 14 candidate genes for cardiac reprogramming: Baf60c, Gata4, Hand2, Hopx, Hrt2, Isl1, Mef2c, Mesp1, Myocd, Nkx2.5, Pitx2c, Smyd1, Srf, and Tbx5. These genes are known to play important roles in cardiac differentiation and form a core transcriptional network during development. However, single deletions of these genes have failed to inhibit heart tube formation (nascent heart) during development, suggesting that the gene network is important for cardiogenesis.
For cardiac induction, I used cardiac fibroblasts from the αMHC-GFP TG mice. Immunofluorescence and mRNA expression analyses revealed that there was no contamination of cardiomyocytes in the initial fibroblast population. I generated 14 retroviral vectors to express the candidate factors. Transduction of single vectors did not induce cardiac induction in fibroblasts, as expected. In contrast, transduction of all 14 factors resulted in the generation of αMHC-GFP cells after 1 week, suggesting that reprogramming factors were included in the set of 14 genes. I next quantified the reprogramming efficiency by FACS, and determined that approximately 1.7% of fibroblasts expressed αMHC-GFP after 1 week. Removal of Gata4 reduced αMHC-GFP induction to 0.5%, whereas removal of Baf60c, Hand2, Hopx, Hrt2, and Pitx2c did not change or increase αMHC-GFP induction after 1 week, suggesting that these 5 factors were not needed for (or inhibited) cardiac induction. I narrowed down the factors from 14 to 9, and continued to remove individual factors to define the minimal set of required factors for cardiac induction. I finally found that only 3 factors, Gata4, Mef2c, and Tbx5 (GMT), were sufficient for cardiac reprogramming, and approximately 20% of the fibroblasts expressed αMHC-GFP reporter 1 week after transduction. Transduction of 1–2 factors of Gata4, Mef2c, and Tbx5 did not result in the generation of cardiomyocytes from fibroblasts, suggesting that all 3 factors are minimally required for cardiac reprogramming. Next, I analyzed protein expression and the structure of cardiomyocytes by immunostaining. The iCMs expressed a panel of cardiac proteins, including cardiac troponin T (cTnT), α-actinin, and atrial natriuretic peptide (ANP), and the cells displayed sarcomeric structures. The extent of cardiac maturation and differentiation increased after 4 weeks in culture, as did the levels of cTnT protein and mRNA, and the expression of cardiac genes, Actc1, Myh6, Ryr2, and Gja1, increased in a time-dependent manner. In contrast, the fibroblast signature, including Col1a2 expression, was downregulated starting from 1 week after transduction, suggesting that the fibroblast program was suppressed at an early stage of reprogramming. Microarray analyses revealed that the global gene expression profiles of the iCMs at 4 weeks were completely different from those of the original fibroblasts, and similar, but not identical, to those of endogenous cardiomyocytes. ChIP-qPCR, assessing the enrichment of H3K27 me3 and H3K4 me3 at the promoters of cardiac-specific genes including Actn2, Ryr2, and Tnnt2, revealed that the iCMs derived from cardiac fibroblasts altered their chromatin status from that of a fibroblast state to that of a cardiac-like state. Bisulfite genomic sequencing in the Nppa and Myh6 promoter regions demonstrated that the DNA methylation state of iCMs was similar to that of cardiomyocytes but different from that of fibroblasts. Although I did not perform whole-genome sequencing of the epigenetic status, these results suggested that GMT, at least in part, epigenetically reprogrammed fibroblasts to a cardiomyocyte-like state. Next, I investigated whether fibroblasts could be stably reprogrammed into iCMs without the requirement of exogenous transgenes. I found that 2 weeks of transgene expression was sufficient for cardiac reprogramming using a doxycycline-mediated temporal gene induction system. GMT resulted in direct reprogramming to iCMs, without mediating an intermediate cardiac progenitor cell or mesodermal cell state, as GMT-transduced fibroblasts did not express Mesp1 or isl1 during reprogramming. Finally, I found that iCMs exhibited spontaneous calcium oscillations, and that a small subset of iCMs (0.1–0.01% of starting fibroblasts) started to contract spontaneously in culture. The beating iCMs revealed action potentials similar to those of mouse cardiomyocytes, suggesting that GMT can reprogram neonatal cardiac fibroblasts into functional cardiomyocytes in vitro. Adult cardiac fibroblasts and tail-tip fibroblasts could also be converted into iCMs by GMT, but the reprogramming efficiency was lower than that of neonatal cardiac fibroblasts, suggesting that some epigenetic barriers must be overcome and that other factors are needed to improve reprogramming efficiency.30 Importantly, this is a first study demonstrating that somatic cells can be directly reprogrammed into functional cardiomyocytes by defined factors (Figure 3).5
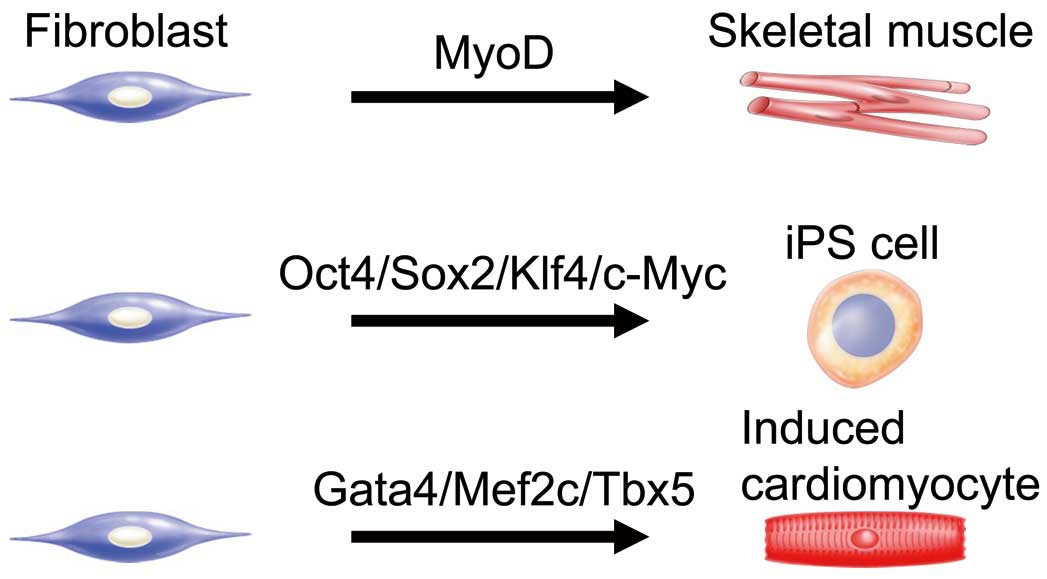
Direct reprogramming into various cell types by defined factors. Fibroblasts can be reprogrammed (transdifferentiated) into skeletal muscle, induced pluripotent stem (iPS) cells, and induced cardiomyocytes using combinations of tissue-specific transcription factors.
I next investigated whether resident cardiac fibroblasts could be reprogrammed into iCMs in situ by gene transfer of retroviral GMT after MI.6 First, it was confirmed that the GFP retrovirus infected only proliferating non-myocytes, but not terminally differentiated cardiomyocytes, after coronary artery ligation in mice. The infective efficiency of GFP retrovirus in cardiac fibroblasts could be increased by concentrating the viral solution by 100-fold. The number of GFP+ cells was reduced after 2 weeks in wild-type immunocompetent mice, but not in immunosuppressed mice such as nude mice. GFP expression was maintained for up to 3 months in nude mice, suggesting that retroviral transgene expression was epigenetically silenced and/or that GFP-infected fibroblasts were eliminated by an immune response in wild-type mice. To increase the in vivo survival of iCMs, nude mice were used for cardiac reprogramming experiments in this study. Mixtures of GMT and GFP viruses, or GFP alone, were directly injected into the myocardium after MI to trace the cell fate of retroviral-transduced cells using GFP. Injection of GFP alone marked non-myocytes (mainly fibroblasts), which did not display altered cell fate after 1 month. In contrast, approximately 1% of the GFP+ cells expressed cardiac proteins after GMT+GFP injection. However, most cells remained smaller than endogenous cardiomyocytes and only 15% of iCMs had sarcomeric structures, suggesting that most cells were partially reprogrammed iCMs. Next, to transduce GMT genes more efficiently in vivo, a single polycistronic vector encoding GMT was generated. Injection of polycistronic GMT retroviral vector generated more mature-looking iCMs (2-fold compared with separate GMT retroviruses), suggesting that the appropriate expression of transgenes is critical for cardiac reprogramming. It was also found that some iCMs in the infarct area had clear sarcomeric structures and were similar to mature ventricular cardiomyocytes. These results suggested that GMT gene transfer into infarcted mouse hearts could reprogram resident cardiac fibroblasts into iCMs in situ. Two other groups also independently reported that gene transfer of GMT or GMT plus Hand2 into mouse infarcted hearts could improve cardiac function and regenerate myocardium by reprogramming endogenous cardiac fibroblasts into iCMs in vivo.31,32
I next asked whether human fibroblasts could be similarly reprogrammed into iCMs by defined factors.7 Human cardiac fibroblasts were used as the cell source for cardiac reprogramming. GMT transduction resulted in the induction of very few cells that weakly expressed α-actinin, suggesting that mouse-reprogramming factors were insufficient for human reprogramming. Human cardiac reprogramming factors were screened by adding 11 other factors to GMT. The addition of Myocd to GMT resulted in marked upregulation of cTnT expression, whereas the addition of Mesp1 increased functional gene expression and calcium oscillation. The addition of both Myocd and Mesp1 to GMT (GMTMM) greatly increased the expression of multiple cardiac genes such as Actc1, Myh6, Myl2, Pln, Scn5a, endogenous Mef2c, and Tbx20, compared with GMT alone. Moreover, GMTMM-transduced cells expressed the cardiac proteins, α-actinin, cTnT, and ANP, and exhibited clear sarcomeric structures. Approximately 5% of cells expressed cTnT and α-actinin based on FACS analysis. Importantly, global gene expression analyses (by microarray) revealed that GMTMM upregulated a cardiac transcriptional program and concomitantly downregulated a fibroblast program in human fibroblasts during reprogramming. Human iCMs exhibited spontaneous calcium oscillations, but they did not beat spontaneously. Coculture of human iCMs with rat cardiomyocytes differentiated the iCMs into beating myocytes, which had action potentials similar to those of human embryonic stem cell-derived cardiomyocytes. GMTMM was also shown to induce iCM reprogramming from human dermal fibroblasts, suggesting that GMTMM are universal human cardiac reprogramming factors. Thus, it has been demonstrated that human fibroblasts can be directly converted to iCMs through overexpression of defined factors. Other groups also report that overexpression of GMT is not sufficient for human cardiac reprogramming, but that addition of other factors, including miRNAs, to GMT could result in reprogramming of human fibroblasts into iCMs.33,34
miRNAs play important roles in heart development and cardiac function. However, in my first study, the focus was on transcription factors and miRNAs were not screened as candidates for cardiac reprogramming factors. Moreover, the molecular mechanisms of direct reprogramming from fibroblasts to iCMs remained unknown. It was next investigated whether the cardiac miRNAs, miR-1, miR-133, miR-208, and miR-499, played a role in cardiac reprogramming. miRNA alone did not induce cardiac reprogramming in mouse embryonic fibroblasts, but the addition of miR-133 to GMT greatly improved cardiac induction. miR-133 increased the number of cTnT+ cells by 8-fold, and the GMT/miR-133-transduced cells were converted into iCMs more rapidly than those transduced with GMT alone. Surprisingly, cell contraction, which generally takes 30 days with GMT alone, started from 10 days after GMT/miR-133 transduction. Thus, miR-133 rapidly and efficiently induced cardiac reprogramming from mouse fibroblasts in combination with GMT transduction. Next, the molecular mechanisms of miR-133-mediated cardiac reprogramming were investigated and it was found that miR-133 directly binds to Snai1, a master regulator of epithelial-to-mesenchymal transition, and that miR-133-mediated Snai1 suppression globally silenced the fibroblast program to promote cardiac reprogramming. MiR-133 downregulated fibroblast gene expression after only 3 days of transduction, leading to rapid conversion of fibroblasts to cardiomyocytes. Knockdown of Snai1 by siRNA promoted cardiac reprogramming by suppressing the fibroblast program, recapitulating the effect of miR-133, although the effects were not as strong as those mediated by miR-133. In contrast, overexpression of Snai1 in conjunction with miR-133 resulted in the maintenance of fibroblast signatures and mitigated the effects of miR-133, suggesting that suppression of Snai1 is crucial for miR-133-mediated cardiac reprogramming. These results suggested that miR-133 promotes cardiac reprogramming by targeting Snai1 and suppressing a fibroblast signature. The addition of miR-133 to GMTMM also increased induction of cTnT+ cells from human fibroblasts by 10-fold (from 2% to 27%), which was suppressed by Snai1 overexpression. Thus, miR-133 promoted cardiac reprogramming in human fibroblasts by suppressing Snai1. This is a first study demonstrating a molecular mechanism for direct reprogramming, in which silencing of the transcriptional program of the initial cell population was critical for cell fate switch.8 Recently, it was reported that chemical compounds, including transforming growth factor β inhibitors, also promote cardiac reprogramming by suppressing fibroblast signatures.35,36
A major issue in direct reprogramming is that the efficiency of generating functional iCMs from fibroblasts was low. Although a relatively large proportion of GMT-transduced fibroblasts express cardiac markers and proteins (~20% of fibroblasts), only a few of them differentiated into beating iCMs (~0.1% of fibroblasts) under original serum-containing conditions.5 Moreover, standardized protocols or defined culture conditions for cardiac reprogramming remain undetermined, hindering the reproducibility of this new technology in many laboratories. Thus, I sought to identify efficient, reproducible, and defined culture conditions for cardiac reprogramming. To identify the culture conditions that efficiently induced functional iCMs, GMT-transduced cells were cultured in serum-free medium with 8 cardiogenic compounds. Among them, fibroblast growth factor (FGF) 2, FGF10, and vascular endothelial growth factor increased the number of beating iCMs after 4 weeks, and the addition of all 3 growth factors (FFV) further increased the reprogramming efficiency by 40-fold.9 Beating cells cultured under defined conditions expressed multiple cardiac proteins, had sarcomeric structures, and exhibited more calcium oscillations. Intriguingly, FFV did not increase the number of cTnT+ partially reprogrammed iCMs, but converted them into fully reprogrammed functional iCMs during the late stage of reprogramming. Mechanistically, FFV did not promote cell proliferation or generate other types of myocytes, such as pacemaker cells, to increase the proportion of beating iCMs. Instead, FFV upregulated the expression of multiple cardiac transcription factors, including Gata6, Nkx2.5, Hand2, Ppargc1a, and Srf, and upregulated global cardiac gene expression related to sarcomeric genes and ion channel genes. Activation of the cardiac program by FFV was mediated by ERK and PI3k/Akt, but not p38MAPK. The next question was whether it was possible to reduce the number of transcription factors needed to reprogram fibroblasts into iCMs using FFV. In fact, Mef2c and Tbx5 were able to promote the generation of beating iCMs from MEFs without Gata4 under defined culture conditions; therein, FFV induced endogenous Gata4 expression. It was also found that the addition of the Wnt inhibitor, IWP4 (with FFV), further increased the proportion of beating iCMs, and this defined condition was effective not only in MEFs but also in tail-tip fibroblasts with other reprogramming factors. Almost 9% of MEFs could be induced to form beating iCMs with GMT+Hand2, which was 100-fold more than the original reprogramming efficiency (~0.1%). Importantly, this defined condition is efficient, reproducible, and available in any laboratory worldwide, which should greatly promote the widespread use of this new technique (Figure 4).
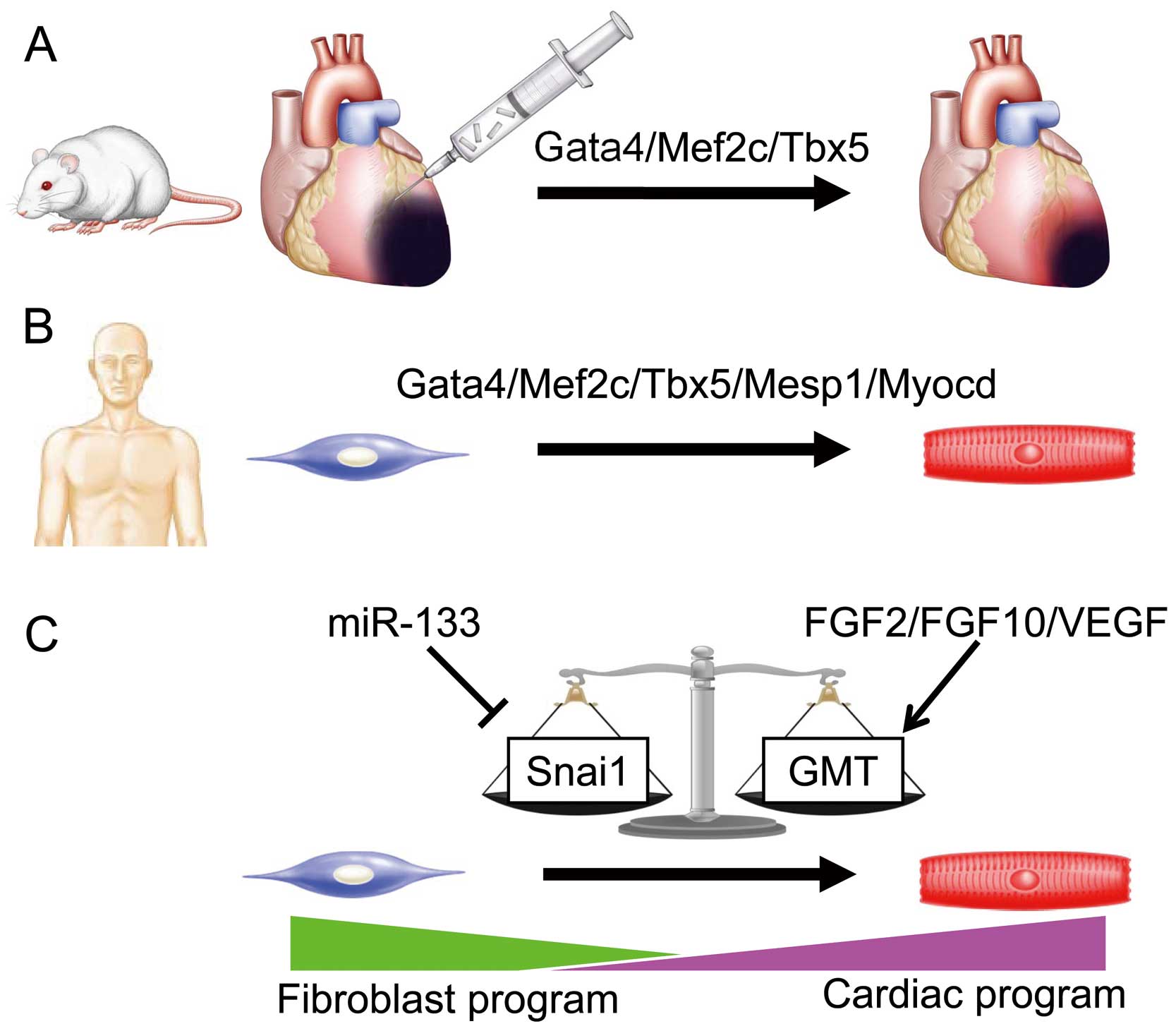
Progress of cardiac reprogramming research. (A) In vivo cardiac reprogramming by Gata4/Mef2c/Tbx5 regenerates myocardial tissue in mice. (B) Gata4/Mef2c/Tbx5/Mesp1/Myocd are human cardiac reprogramming factors. (C) Molecular mechanism of cardiac reprogramming. The balance of master regulators, Snai1 and Gata4/Mef2c/Tbx5 (GMT), modified by miR133 and FGF2/FGF10/VEGF, determines cardiac reprogramming. FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor.
Here I have reviewed 16 years of my work. Starting from the identification of the molecular mechanisms of cardiac innervation, I realized that developmental biology is a very powerful model to understand the pathophysiology of heart diseases in adults. I also applied the knowledge of cardiac fibrosis and hypertrophy in the adult heart to developmental biology to elucidate the mechanism of cardiac growth and myocyte proliferation in the embryo. I believe that the application of knowledge from one field to another can be a cue for important discoveries, which can lead to breakthroughs in basic science. In fact, I applied the theory of iPSC generation to the cardiac field, and found that combinations of cardiac transcription factors can directly reprogram fibroblasts into cardiomyocytes. This finding opened a new avenue for regenerative medicine and personalized medicine. However, to transfer the reprogramming technology to the clinic, there are still big hurdles to overcome. Safety issues for in vivo cardiac reprogramming, optimal cocktails of reprogramming factors, and the efficacy of regeneration in large animals and humans need to be addressed. As the demand is high for regenerative therapies, future efforts and advances in basic science will overcome these issues.37–39 I believe we will advance this new technology to the clinic in the near future, and it will be a game changer for the treatment of numerous heart diseases.
It is my great honor and privilege to receive the Sato Award. I express my enormous gratitude and appreciation to my mentors: Satoshi Ogawa (Professor Emeritus at Keio University School of Medicine), Keiichi Fukuda (Professor at Keio University School of Medicine), and Deepak Srivastava (Professor at the Gladstone Institute of Cardiovascular Disease) for their strong support. I also acknowledge my colleagues, collaborators, and lab members for their collaboration and discussion. I thank my family for their immense support of my work and life. I was supported by research grants from AMED-PRIME, JSPS, Keio University Program for the Advancement of Next Generation Research Projects, Banyu Life Science, Senshin Medical Research Foundation, Takeda Science Foundation, and a Novartis Research Grant.