Abstract
Macrophage apoptosis and the ability of macrophages to clean up dead cells, a process called efferocytosis, are crucial determinants of atherosclerosis lesion progression and plaque stability. Environmental stressors initiate endoplasmic reticulum (ER) stress and activate the unfolded protein response (UPR). Unresolved ER stress with activation of the UPR initiates apoptosis. Macrophages are resistant to apoptotic stimuli, because of activity of the PI3K/Akt pathway. Macrophages express 3 Akt isoforms, Akt1, Akt2 and Akt3, which are products of distinct but homologous genes. Akt displays isoform-specific effects on atherogenesis, which vary with different vascular cell types. Loss of macrophage Akt2 promotes the anti-inflammatory M2 phenotype and reduces atherosclerosis. However, Akt isoforms are redundant with regard to apoptosis. c-Jun NH2-terminal kinase (JNK) is a pro-apoptotic effector of the UPR, and the JNK1 isoform opposes anti-apoptotic Akt signaling. Loss of JNK1 in hematopoietic cells protects macrophages from apoptosis and accelerates early atherosclerosis. IκB kinase α (IKKα, a member of the serine/threonine protein kinase family) plays an important role in mTORC2-mediated Akt signaling in macrophages, and IKKα deficiency reduces macrophage survival and suppresses early atherosclerosis. Efferocytosis involves the interaction of receptors, bridging molecules, and apoptotic cell ligands. Scavenger receptor class B type I is a critical mediator of macrophage efferocytosis via the Src/PI3K/Rac1 pathway in atherosclerosis. Agonists that resolve inflammation offer promising therapeutic potential to promote efferocytosis and prevent atherosclerotic clinical events. (Circ J 2016; 80: 2259–2268)
Macrophages play crucial roles in all stages of atherosclerosis, a chronic inflammatory disease, which is the underlying cause of heart attack and stroke.1
Retention of apolipoprotein (apo) B-containing lipoproteins in the artery wall promotes endothelial dysfunction, leading to recruitment of monocytes from the blood into the intima,2
where they differentiate into macrophages. Internalization of modified low-density lipoprotein (LDL) by macrophages leads to formation of the fatty streak, the hallmark of early atherosclerosis. Mounting evidence indicates that macrophage death and the ability of macrophages to clean up dead cells, a process called efferocytosis, are crucial determinants of lesion stage and plaque stability. Accumulation of oxidized LDL (oxLDL), formation of dysfunctional high-density lipoprotein (HDL) and increased inflammation caused by peroxidase-derived oxidants and reactive oxygen species (ROS) contribute to the formation of vulnerable plaques by enhancing macrophage apoptosis and defective efferocytosis, causing increased size of the necrotic core (NC), a key feature of vulnerable plaques.3,4
Plaque rupture promotes thrombosis, blocking the blood flow in the artery and resulting in acute ischemic atherosclerotic cardiovascular events.
Macrophage Phenotypes in Atherosclerosis
Macrophages display different phenotypes and various molecular markers in the setting of atherosclerosis that influence their function in terms of promoting inflammation or its resolution and lesion progression or regression. The paradigm of designating macrophages as simply M1 or M2 is clearly an oversimplification, but has led to a framework for understanding the role of macrophage phenotype in atherogenesis. Lesional M1 and M2 macrophage phenotypes are dependent on their derivation and environment.5
These macrophages have different molecular markers because they are derived primarily from different subsets of blood monocytes, although a recent study has shown smooth muscle cells can also be the precursor of lesional macrophage-like cells.5
M1 macrophages are primarily derived from the inflammatory Ly6Chi
monocyte subset. Anti-inflammatory M2 macrophages are mainly derived from Ly6Clo
monocyte subsets, although this type of macrophage can also originate from Ly6Chi
subsets.6
M1 macrophages with monocyte chemoattractant protein (MCP)-1, interleukin (IL)-12high, IL-23high
and tumor necrosis factor (TNF)-αhigh
markers are crucial for tissue destruction. In contrast, anti-inflammatory M2 macrophages characterized by arginase I, M2 chemokines, and IL-10high
markers play important roles in the resolution of inflammation, tissue remodeling and wound healing.6
The phenotype of these macrophages is more dynamic and complex and probably undergoes conversion during the different stages of atherosclerosis, influenced by the extent of lipid uptake and retention, as well as by the presence of acceptors for cholesterol efflux, including HDL and apoE.7
The different phenotypes affect the ability of the macrophage to engulf oxLDL, efflux cholesterol to HDL, produce a large number of inflammatory or proresolving molecules for tissue remodeling and repair, and ultimately affect their survival and ability to perform efferocytosis.8
Impact of Akt Signaling on Macrophage Polarization
Environmental signals can functionally polarize macrophages, producing a large number of cell phenotypes that vary between M1, or classically activated, macrophages and M2, or alternatively activated, macrophages.9
Macrophage polarization may be induced in vitro by treatment with interferon (IFN)-γ or lipopolysaccharide (LPS) or cytokines (TNF-α, granulocyte macrophage colony-stimulating factor) to promote an inflammatory M1 phenotype. Recent reports indicate that PI3K/Akt signaling, acting through mammalian target of rapamycin (mTOR) complex 1, controls the effector responses of innate immune cells, including macrophages.10
PI3K/Akt/mTORC1 signaling regulates cellular metabolism, translation, cytokine responses, macrophage polarization and migration.10
For example, deficiency of SH2-containing inositol phosphatase (SHIP), which is a negative regulator of PI3K/Akt signaling, significantly enhances M2 programming in peritoneal and alveolar macrophages.11
This indicates that Akt levels determine the macrophage phenotype and that constitutively enhanced PI3K/Akt signaling promotes M2 macrophage differentiation. Indeed, more recent reports have confirmed that SHIP–/–
macrophages produce anti-inflammatory cytokines typical of M2-type cells, and they are skewed to the M2 phenotype.12
Similarly, loss of the phosphatase and tensin homolog in macrophages increases PI3K/Akt signaling and induces high levels of M2 macrophage markers.13
In contrast, mice with a myeloid-specific Rictor deletion, which significantly suppresses Akt signaling, are skewed to the M1 macrophage phenotype.14
Moreover, Arranz et al demonstrated that Akt1 and Akt2 play opposing roles in macrophage polarization, with Akt1 ablation generating the M1 phenotype and Akt2 deficiency skewing cells towards the M2 phenotype.15
We have recently shown that Akt2–/–
monocytes and macrophages express significantly lower levels of inflammatory genes and display the M2 phenotype with high levels of IL-10 expression in response to LPS. Furthermore, Akt2–/–
monocytes exhibit suppressed migration in response to MCP-1 and exhibited a suppressed ability for M1 polarization and C-C chemokine receptor type 2 (CCR2) induction when treated with IFN-γ.16
Importantly, LDL receptor (Ldlr)–/–
mice transplanted with Akt2–/–
bone marrow developed less atherosclerosis than control (Ldlr)–/–
mice reconstituted with wild-type (WT) bone marrow, because of the effect of Akt2 deficiency on macrophage phenotype in vivo.16
Consistent with those results, atherosclerosis was reduced in double Akt2/
Ldlr
knockout mice,17
and in a separate study of
Ldlr–/–
mice transplanted with Akt2–/–
bone marrow.18
It is important to note that the Akt1 and Akt2 isoforms exhibit distinct or opposing functions in Rac/Pak signaling and cell migration,19
in the initiation of invasion and the metastasis of tumor cancer cells.20
Together, these results highlight the critical importance of the Akt1 and Akt2 isoforms and changes in the levels of PI3K/Akt signaling in determining macrophage polarization and modulating the host’s defense. Given that the PI3K pathway is one of the most frequently altered pathways in human cancer,21
a large number of inhibitors of the PI3K/Akt/mTORC1 pathway have been designed to suppress the pathological survival and abnormal proliferation of cancer cells,22
but it is important to realize that this approach also suppresses PI3K/Akt activity and survival of non-cancer cells.
Role of FABP4-Mediated ER Stress in Atherosclerosis
The adipocyte fatty acid-binding protein, also known as fatty acid binding protein 4 (FABP4), is expressed by adipocytes and macrophages.23
FABP4 plays an important role in intracellular fatty acid transportation and regulating lipid metabolism, oxidation and endoplasmic reticulum (ER) stress. The effect of FABP4 on ER stress is via liver X receptor (LXR)-mediated de novo lipogenesis, as well as integrating inflammatory and metabolic responses (Figure 1).24
FABP4 expression is regulated by phorbol 12-myristate 13-acetate, peroxisome proliferator-activated receptor-γ (PPAR-γ) agonists, LPS and oxidized LDL.25
Studies in FABP4-deficient mice have demonstrated its essential role in many aspects of metabolic syndrome. FABP4 has been shown to affect insulin sensitivity, lipid metabolism and lipolysis in diabetes and atherosclerosis.26
FABP4 deficiency decreases the atherosclerotic lesions of apoE-deficient (Apoe–/–) mice.27
Interestingly, loss of FABP4 in macrophages affects formation of inflammatory cytokines and cholesterol loading, and protects against atherogenesis in
Apoe–/–
mice.25
Combined adipocyte-macrophage fatty acid-binding protein deficiency (FABP4 and mal1 knockout) in
Apoe–/–
mice leads to them displaying significantly lower levels of serum cholesterol and triglycerides, and reduced atherosclerotic lesions in both early and late-stage atherosclerosis compared with control
Apoe–/–
mice. A FABP4 inhibitor decreases production of MCP-1 and reduces atherosclerosis in
Apoe–/–
mice on a western diet in both early and late intervention studies.25
Therefore, pharmacological inhibition of FABP4 has been shown to be a potential therapeutic approach to the treatment of atherosclerosis.28
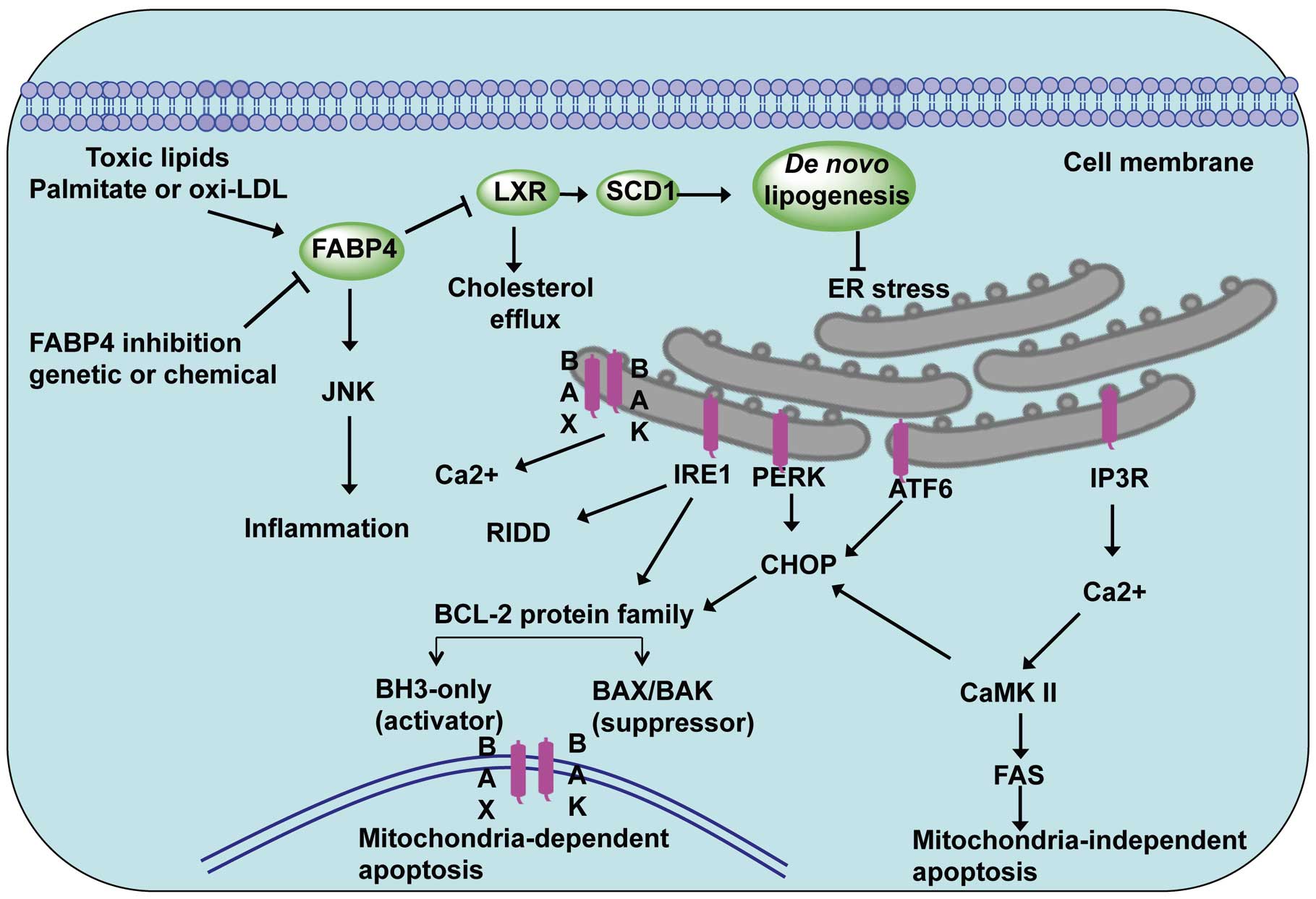
Role of the UPR Signaling Pathway in ER Stress in Macrophages
Both oxidative stress- and ER stress-induced apoptosis are associated with the initiation and progression of atherosclerosis. ER homeostasis is maintained by a series of checks and balances between the production of the protective unfolded protein response (UPR) and other ER stress sensors. Factors contributing to ER stress include increased protein synthesis, a redox imbalance in the ER lumen, and formation of misfolded protein exceeding the cellular protein quality control system, which leads to disorder of the ER homeostasis.29
Three major ER sensors, protein kinase RNA-like ER kinase (PERK), inositol requiring protein 1α (IRE1α), and activating transcription factor 6 (ATF6), have been implicated in initiating the UPR (Figure 1).30
In response to an imbalance of unfolded proteins and chaperones, the ER stress sensors IRE1and PERK are activated.29
PERK phosphorylates the α-subunit of the translation initiation factor eIF2α (eukaryotic translation initiation factor 2α), which increases C/EBP-homologous protein (CHOP, also known as GADD153).29
CHOP activation leads to increases in both the BCL-2 family of proteins that promote mitochondria-dependent apoptosis and activation of CaMKII-FAS in mitochondria-independent apoptosis. Similarly, the transcription factor ATF6 is involved in chaperone induction and also transcriptionally promotes CHOP activation. Interestingly, growth arrest and DNA damage-inducible protein-34 (GADD34), a crucial transcriptional target of CHOP and ATF4, is able to dephosphorylate phosphorylated eIF2α and thus reinstitute protein translation and bring ATF4 translation back to basal levels.31
Studies in CHOP-deficient mice have demonstrated its essential role in the ER stress-induced apoptosis of atherosclerosis. There is reduced apoptosis and necrosis in the atherosclerotic lesions of
Ldlr–/–
and
Apoe–/–
mice lacking CHOP32
and ablation of CHOP decreases ER stress-induced apoptosis and cardiac dysfunction in the mice.33
These UPR signaling pathways play crucial roles in ER stress-induced apoptosis in atherosclerosis,34
and attenuation of ER stress by a macrophage lipid chaperone could be a therapeutic approach to the treatment of atherosclerosis.28
Macrophage Apoptosis and Atherosclerosis
Macrophage apoptosis occurs during all stages of atherosclerosis and influences early lesion formation, plaque progression, and plaque stability.35
Chronic or unresolved ER stress with the activation of different branches of the UPR eventually initiates apoptosis.30
Deletion of the pro-apoptotic factor Bax in hematopoietic cells accelerates early atherosclerosis in
Ldlr–/–
mice.36
In agreement with those findings, when the apoptotic inhibitor AIM (Spα/Api6) is deleted,
Ldlr–/–
mice show increased macrophage apoptosis and suppressed early atherosclerosis.37
These results support the concept increased macrophage apoptosis is associated with diminished lesion cellularity and decreased lesion progression in the early stages of atherosclerosis. In contrast, the suppression of apoptosis generates bigger atherosclerotic lesions and increases the area of the NC, with possible plaque expansion.38
Several studies have verified that decreased levels of macrophage apoptosis result in dramatically increased size of early atherosclerotic lesions but are associated with reduced plaque burden in more advanced lesions.39
For example, c-Jun NH2-terminal kinases (JNK) is a pro-apoptotic effector of the UPR40
and the JNK1 isoform directly opposes anti-apoptotic Akt signaling41
(Figure 2). Recently, we showed that loss of JNK1, but not JNK2, in hematopoietic cells protects macrophages from apoptosis and this accelerates early atherosclerosis42
(Figure 2). Therefore, the ability of macrophages to resist pro-apoptotic stimuli, which are abundant in atherosclerotic lesions, is a crucial determinant of macrophage survival and plaque cellularity.

Macrophages are remarkably resistant to apoptotic stimuli and this ability to survive is mainly based on activity of the PI3K/Akt pathway. Akt signaling is vital for cell proliferation, migration and survival.43
In human and mouse macrophages Akt phosphorylation is constitutively active44
and inhibition of Akt signaling induces cell apoptosis.44,45
Macrophages express 3 Akt isoforms, Akt1, Akt2 and Akt3, which are products of distinct genes with similar structural organization,43
exhibiting high homology with substantial differences between the isoforms only in the last 130 amino acids.46
Recent in vivo studies using knockout mouse models have provided evidence for isotype-specific functions of Akt. For example, mice lacking Akt1 demonstrate increased perinatal death and reductions in body weight.47,48
In contrast, Akt2-deficient mice display normal growth, but develop a diabetes-like syndrome with hyperglycemia and insulin resistance.48
Akt3 knockout mice exhibit a reduction in brain weight resulting from decreases in both cell size and cell number, but maintain normal glucose homeostasis and body weight.49
These data indicate that all 3 Akt isoforms have differential or non-redundant physiological functions.50
It is important to note that loss of either the Akt1 or Akt2 isoform in embryonic fibroblasts51
and mouse macrophages,16
as well as Akt3 cell deficiency,52
has no effect on apoptosis. In contrast, macrophages that are double deficient for Akt1 and apoE have been reported to have increased cell sensitivity to apoptosis.53
Interestingly, loss of Akt1 or Akt2 expression in the macrophages of
Ldlr–/–
mice did not affect macrophage apoptosis,16
suggesting that apoE may interact with Akt to promote cell survival. Furthermore, loss of 2 Akt isoforms, Akt2/3 or and Akt1/2, in macrophages dramatically increased their sensitivity to apoptosis, yet suppression of JNK1 signaling by the JNK inhibitor, SP600125, completely reversed the effect of loss of 2 Akt isoforms on macrophage viability.42
Thus, several lines of evidence indicate that Akt isoforms are redundant with regard to apoptosis.
IκB kinase α (IKKα, a member of the serine/threonine protein kinase family) is implicated in the non-canonical NF-κB pathway.54
IKKα is also associated with 2 major prosurvival pathways, PI3K/Akt and NF-κB, and Akt signaling mediated through IKKα may activate the anti-apoptotic activity of the NF-κB pathway.55
Therefore, we recently investigated the role of IKKα in macrophage survival and atherosclerosis (Figure 2). Interestingly, both genetic deficiency of IKKα and pharmacological inhibition of IKKα suppressed Akt phosphorylation in macrophages.56
Transplantation studies revealed that loss of IKKα in the hematopoietic cells of
Ldlr–/–
mice was associated with reduced atherosclerotic lesion size, and the lesions displayed increased numbers of apoptotic macrophages compared with control mice transplanted with WT cells.56
Tilstam et al have reported that knock-in of a non-activatable IKKα kinase (IkkαAA/AA) mutant in the bone marrow of
Apoe–/–
mice influenced hematopoiesis but not atherosclerosis.57
However, it is important to note that IKKα activity was not abolished in these IkkαAA/AA mice,57
as evidenced by a lack of morphogenic defects and downstream signaling effects specific to loss of IKKα in the original descriptions. In contrast, our data represent the first report of the effect of complete loss of expression and activity of IKKα in hematopoietic cells on atherogenesis. Thus, IKKα plays an important role in Akt phosphorylation and IKKα deficiency reduces macrophage survival and suppresses early atherosclerosis. Interestingly, IKKα is required for activity of mTOR,58
and we found that IKKα plays an important role in mTORC2-mediated Akt signaling of mouse macrophages. Therefore, future studies to elucidate the effect of individual components of mTORC2 on Akt activity of macrophages, including Rictor, will be crucial for understanding the role of mTORC2 in atherosclerosis.
Akt promotes cell survival through either direct phosphorylation of anti-apoptotic molecules (Bad, Caspase) or transcriptional activation of anti-apoptotic genes (MDM2, IKK, Yap) that regulate cell survival (Figure 3).59
Phosphorylation of Bad inactivates Bad at Ser 136 by promoting its binding with cytosolic 14-3-3 proteins, which prevents Bad from inhibiting the anti-apoptotic factor BCL-XL.60
Previously, we have shown that the loss of the EP4 receptor in mouse macrophages suppresses Akt and BAD phosphorylation, compromising macrophage survival.45
Akt also inhibits the evolutionarily conserved Forkhead family of transcription factors FoxO, which induces very diverse responses, including apoptosis initiated by activation of the Bcl-2 family member Bim.61
In addition, Akt promotes cell survival by inhibiting murine double minute-2 (MDM2), which triggers p53 degradation.62
Acting through glycogen synthase kinase, Akt may inhibit an anti-apoptotic member of the Bcl family, Mcl-1.63
In addition, Akt directly inhibits phosphorylation of the pro-apoptotic protein Bax, which is a key regulator of mitochondrial permeability.64
Finally, Akt phosphorylates GSK3 isoforms at a highly conserved N-terminal regulatory site, resulting in inactivation of the apoptotic mitochondrial pathway that includes caspase-9.65
Together, these data demonstrate that constitutive activity of Akt signaling is critical for survival of macrophages.

Mechanisms of Efferocytosis in the Atherosclerotic Lesion
Studies of human atherosclerotic lesions and genetic manipulations in experimental mice show that efficient efferocytosis is critical to limiting the progression and vulnerability of atherosclerotic plaque. Apoptotic cells produce “find me” molecules (ie, lysophosphatidylcholine, sphingosine-1-phosphate) to attract phagocytes. Efferocytosis is then initiated by phagocytic receptors interacting directly with apoptotic cell “eat me” ligands (ie, phosphatidylserine, oxidized phospholipids)66
or indirectly via extracellular bridging molecules.67
Efferocyte receptor engagement stimulates various signaling cascades that lead to Rac1 activation, cytoskeletal rearrangement, and apoptotic cell engulfment.67
Macrophages are the main efferocyte present in the intima and studies suggest that a number of macrophage receptor pathways are critical to efficient efferocytosis in atherosclerotic lesions (Figure 4).

Groundbreaking studies demonstrated that Mer tyrosine kinase (MERTK) mediates efferocytosis in atherosclerotic lesions (Figure 4). MERTK engulfs apoptotic cells by interacting with the bridging molecules, Gas6 or Protein S, which are phosphatidylserine-binding proteins.67
MERTK-mediated efferocytosis is enhanced by its co-receptor, αvβ5 integrin, which interacts with milk fat globule epidermal growth factor (MFGE) 8, which serves as a bridging molecule and binds phosphatidylserine on the apoptotic cell.67
MERTK engagement of Gas6 promotes autophosphorylation,68
leading to activation of Akt and PLCγ2 followed by protein kinase C activation and phosphorylation of FAK, and p130cas
upstream of Rac1 activation and cytoskeletal rearrangement. Studies have demonstrated that in contrast to CD36, MERTK engulfs cells made apoptotic by free cholesterol enrichment, which is likely a death stimulus in lesions.69
Importantly, Thorp and colleagues70
demonstrated that lesions of
Apoe–/–
mice with a tyrosine kinase-defective MERTK receptor (MertkKD) had enlarged NCs, and a higher number of apoptotic cells that were not associated with macrophages, compared with the lesions in control
Apoe–/–
mice, indicating defective efferocytosis. Similarly, Mallat and colleagues71
demonstrated that lesions of
Ldlr
knockout mice transplanted with MERTK-deficient vs. WT bone marrow had increased numbers of apoptotic cells and NC area. Consistent with studies showing that MERTK signaling suppresses macrophage toll-like receptor 4 (TLR4) inflammatory signaling,72
these same studies demonstrated increased inflammation in the spleens of mice transplanted with MERTK–/–
vs. WT bone marrow.71
In concert with the integrin, αvβ3, transglutaminase-2 (TG2) mediates the engulfment of apoptotic cells (Figure 4) by interacting with the phosphatidylserine bridging molecule, MFGE8.73
A role for the TG2-αvβ3 complex in atherosclerotic lesion efferocytosis has been suggested by studies showing that deletion of TG2 increases lesion size.74
In addition, transplantation of
Ldlr–/–
mice with
MFGE8–/–
vs. WT bone marrow increases lesion necrosis and the number of apoptotic cells.75
Studies have shown that macrophage LDLr-related protein 1 (LRP1) is critical to atherosclerotic lesion efferocytosis (Figure 4).76
Transplantation of
Ldlr–/–
mice with bone marrow from WT mice vs. bone marrow from macrophage LRP1-deficient mice resulted in accelerated atherosclerosis development,77
enhanced necrosis, and markedly increased numbers of apoptotic cells that were not associated with macrophages.76,78
With its co-receptor, calreticulin, LRP1 interacts with phosphatidylserine-binding complement factor C1q to engulf apoptotic cells.79
LRP1 signals via the adapter protein, GULP,80
to activate Rac1. Other studies have suggested that ABCA7 is associated with the LRP1/calreticulin complex and facilitates engulfment by activating ERK.81
Similar to the MERTK pathway, LRP1 signaling reduces production of inflammatory cytokines and promotes phagocyte Akt phosphorylation to increase cell survival.76
In addition, it is likely that apoE facilitates efferocytosis (Figure 4) via the LRP1 complex by acting as a bridging molecule and/or as an apoptotic cell ligand for LRP1, as apoE binds phosphatidylserine82
and is an avid ligand of LRP1. In addition, the synthesis of apoE is increased in macrophages undergoing apoptosis,83
and apoE on both apoptotic cells and phagocytes enhances efferocytosis.76,84
Furthermore, the efferocytosis of LRP1–/–
vs. WT apoptotic macrophages, which express more apoE than WT cells, is enhanced in WT phagocytes, but not in LRP1–/–
phagocytes.76
Studies have shown that deletion of macrophage apoE markedly accelerates atherosclerosis,85–87
and tissues of
Apoe–/–
mice show marked accumulation of apoptotic cells.84
Importantly, lesions of
Ldlr–/–
mice transplanted with
Apoe–/–
vs. WT bone marrow have increased necrosis and a higher percentage of macrophage-free apoptotic cells.87
Interestingly, the human apoE4 isoform is less efficient at mediating efferocytosis than apoE3.88
Consistent with the compromised efferocytotic function of apoE4, human carriers are at increased risk of coronary artery disease compared with humans expressing apoE3 or apoE2 (heterozygous).89
Our recent studies demonstrated that the HDL receptor, Scavenger receptor class B type I (SR-BI), is a critical mediator of efferocytosis (Figure 4).87
Using in vitro and in vivo assays, we demonstrated that deletion of macrophage SR-BI results in impaired efferocytosis of apoptotic cells.87
SR-BI is localized to phagosomes and directly interacts with apoptotic cell phosphatidylserine.87
SR-BI engagement with apoptotic cell phosphatidylserine promotes Src membrane recruitment and phosphorylation, leading to downstream activation of phosphoinositide 3-kinase and Rac1.87
As apoE is a ligand for SR-BI, apoE may facilitate SR-BI-mediated efferocytosis. Defective efferocytosis by SR-BI–/–
macrophages results in enhanced expression of inflammatory cytokines, including IL-1β, IL-6 and TNF-α,
and reduced expression of anti-inflammatory transforming growth factor (TGF) β.87
Furthermore, Akt is activated in SR-BI-expressing cells compared with SR-BI–/–
macrophages, thereby promoting efferocyte survival.87,90
Deletion of macrophage SR-BI greatly accelerates atherosclerotic lesion formation in
Apoe–/–
mice91
and
Ldlr–/–
mice.87,92
Our studies demonstrated that, in both mouse models, atherosclerotic lesions containing SR-BI-deficient macrophages had marked accumulation of apoptotic cells, increased NC, and thinner fibrous cap.87
Importantly, plaque containing SR-BI–/–
macrophages had a higher percentage of apoptotic cells that were not associated with macrophages, suggesting defective lesion efferocytosis.87
In addition, in vitro pharmacological activation of Rac1 corrected the defective efferocytosis in SR-BI-deficient macrophages,87
raising the possibility that impaired SR-BI signaling affects other efferocytosis pathways, making SR-BI a novel therapeutic target.
Mechanisms of Defective Efferocytosis in the Atherosclerotic Lesion
Histological examination of advanced atherosclerotic lesions in humans and mice suggests that efferocytosis is defective.93
Consistent with defective efferocytosis is significant NC formation and thinning of the fibrous cap. Efficient efferocytosis prevents secondary necrosis (Figure 4) and leakage of inflammatory and toxic molecules from dying cells.69
Many of the efferocytotic pathways, including LRP1, SR-BI, and MERTK,68,76,87
also limit inflammation via anti-inflammatory and phagocyte prosurvival signaling. Thus, impaired engulfment of apoptotic cells leads to secondary necrosis, promoting unresolved inflammation and autoimmune responses.69
It is clear from the mouse atherosclerosis studies that disruption of one efferocytotic pathway is sufficient to promote lesion necrosis, and it is likely that several mechanisms conspire to promote defective lesion efferocytosis by impairing multiple pathways.
As lesions progress, macrophages produce more ROS and oxidation of extracellular molecules such as LDL increases. OxLDL and oxidized phospholipids can impair efferocytosis by competing with apoptotic cells for interaction with phagocytic receptors (ie, SR-BI)94
and bridging molecules (ie, MFGE8).95
As apoptotic cells and oxidized phospholipids have similar epitopes, antibodies recognizing oxidized phospholipids can also prevent efferocytosis.96
In addition, oxLDL increases TLR4 expression and signaling, causing increased proinflammatory cytokine (ie, TNF-α, IL-1β) secretion and reduced anti-inflammatory cytokine production (ie, TGFβ, IL-10).97
Studies have shown that activation of TLR4 impairs efferocytosis98
and the increased inflammatory environment could affect efferocytosis via a number of processes. Indeed, studies have shown that TLR4 activation reduces both SR-BI and LRP1 expression.99
In addition, increased inflammatory cytokine production decreases LXR activation, promoting decreased MERTK expression.100
Furthermore, production of inflammatory proteases (ie, ADAM17) likely causes shedding of MERTK101
and LRP1,102
leading to decreased functional efferocytosis receptors and competition of soluble forms with bridging molecules.
The proinflammatory signaling molecule, high mobility group box 1 (HMGB1), increases during human atherosclerotic plaque formation103
and can impair efferocytosis via a number of mechanisms. Our studies demonstrated that SR-BI regulates expression of HMGB1, suggesting that factors that reduce SR-BI expression and signaling will compromise other efferocytotic pathways by increasing HMGB1 expression.87
Intracellular HMGB1 interacts with Src, preventing its activation and recruitment to the plasma membrane, resulting in decreased Rac1 activation.104
Secreted HMGB1 interacts with both integrin αvβ3105
and apoptotic cell phosphatidylserine106
to effectively block efferocytotic pathways. Importantly, neutralization of HMGB1 reduces atherosclerosis107
and our studies have shown that downregulation of HMGB1 expression partially restores defective efferocytosis in SR-BI–/–
macrophages.87
Recent studies have demonstrated that activation of ERK5 prevents atherosclerosis development,108
and ERK5 activation may be compromised in advanced atherosclerotic lesions because ERK5 is activated via anti-inflammatory TGFβ signaling.109
Those same studies demonstrated increased necrosis and apoptotic cell accumulation in the lesions of
Ldlr–/–
mice transplanted with ERK5–/–
vs. WT bone marrow, indicating defective efferocytosis.108
Indeed, ERK5 activation has been shown to upregulate the expression of a number of efferocytotic players, including MERTK, C1q, Gas6 and MFGE8.108
In addition, ERK5 activation promotes an anti-inflammatory macrophage M2 phenotype. Taken together, the findings indicate that ERK5 activation is critical to maintaining efficient macrophage efferocytosis and thus limiting inflammation.
More recent studies have identified CD47 as a target to prevent defective macrophage efferocytosis in atherosclerotic lesions.110
CD47 is normally present on live cells and is a “do not eat me” signal for efferocytes. CD47 signals through the phagocyte SIRPα receptor protein, thereby preventing engulfment. The expression of CD47 increases as atherosclerotic lesions advance in both humans and
Apoe–/–
mice. Plaque CD47 is localized to dying macrophages and smooth muscle cells and to the NC.110
In addition, administration of antibodies against CD47 in several mouse models greatly reduced atherosclerotic lesion formation compared with controls, and there was less NC formation and fewer apoptotic cells not associated with macrophages.110
The increased expression of CD47 in cells made apoptotic by proatherosclerotic oxidized phospholipids was found to be related to TNF-α signaling of TNFR1, leading to enhanced transcription via activation of NF-κB.110
Furthermore, other studies have shown that enzyme-triggered cell primary necrotic death promotes inefficient efferocytosis of these dying cells, which also was likely caused by abnormal CD47 expression in the cells killed via this mechanism.111
Interestingly, compared with treatment with anti-CD47 antibody alone, combined anti-TNF-α treatment additionally improved efferocytosis of apoptotic cells, likely resulting from decreased CD47 expression.
Future Directions
Evidence has mounted that defective efferocytosis of dying cells is a critical event in the formation of vulnerable atherosclerotic plaque. Therefore, therapeutic treatments that effectively maintain efficient efferocytosis are necessary to prevent thrombus formation and development of clinical events. As lesions progress, the oxidative and inflammatory environment likely results in impairment of one or more efferocytotic pathways, which then promotes unresolved inflammation and toxicity, culminating in rendering other efferocytotic pathways inoperable. Even with lipid-lowering statins, there is still substantial risk for humans to develop coronary artery disease, and in recent years, therapies using antagonists to target individual inflammatory molecules or pathways have had limited success.112
This is likely because of the redundancy of multiple inflammatory pathways that operate in atherosclerosis. Interesting studies recently revealed the “do not eat me” signal, CD47, as an attractive therapeutic target.110
Although anti-CD47 antibody and/or anti-TNF-α treatment may provide some benefit to increasing lesional efferocytosis, it should be realized that any benefit would likely be limited by compromised efferocytotic machinery. Thus, the therapy that would be most beneficial is one that simultaneously decreases the “do not eat me” signals in dying cells and maximizes expression of the efferocytotic machinery. Such therapy is feasible using agonists that induce alternative macrophage development (ie, PPARγ, PPARδ, LXR) or promote conversion of macrophages to an anti-inflammatory proresolution state (ie, lipoxins, resolvins).113
Agonists that promote an overall anti-inflammatory, proresolving phenotype would be expected to maximize expression of the efferocytotic machinery while limiting TNF-α production, inflammatory NF-κB activation, and expression of CD47 in dying cells. The lipid-resolving mediators, lipopoxin and resolvins, enhance macrophage efferocytosis,113
and administration of a fish oil diet to enhance in vivo production of lipid-resolving mediators prevents defective efferocytosis in atherosclerotic lesions in mice.114
In addition, both PPAR and LXR activation of macrophages to an anti-inflammatory phenotype have been shown to increase efferocytosis and expression of critical efferocytotic players.115
Recent studies demonstrated that treatment with the selective LXR activator, nagilactone B, reduced atherosclerotic lesion area, NC, and macrophage content.116
Studies showing that ERK5 regulates macrophage inflammatory status, efferocytotic machinery, and atherosclerosis108
have identified ERK5 activation as a viable proresolving target. Interestingly, Fredman and colleagues showed that administration of an inflammatory-resolving Annexin 1 agonist peptide that was enclosed in collagen IV nanoparticles to specifically target existing atherosclerotic lesions in
Apoe–/–
mice decreased the atherosclerotic lesion area and necrosis and increased the fibrous cap thickness.117
Taken together, these studies highlight the possibility that agonists that resolve inflammation and induce wound healing offer promising therapeutic potential to prevent atherosclerotic clinical events.
Acknowledgments
This work was supported by National Institutes of Health grants HL116263, HL127173, HL105375.
None of the authors of this paper have a financial interest related to these studies.
References
- 1.
Linton MF, Yancey PG, Davies SS, Jerome WGJ, Linton EF, Vickers KC. The role of lipids and lipoproteins in atherosclerosis. [Updated 2015 December 24]. In: De Groot LJ, Chrousos G, Dungan K, et al, editors. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc; 2000– (pages: 1–91). Available from: https://www.ncbi.nlm.nih.gov/books/NBK343489/ [PMID: 26844337].
- 2.
Tabas I. Heart disease: Death-defying plaque cells. Nature 2016; 536: 32–33.
- 3.
Shao B, Pennathur S, Heinecke JW. Myeloperoxidase targets apolipoprotein A-I, the major high density lipoprotein protein, for site-specific oxidation in human atherosclerotic lesions. J Biol Chem 2012; 287: 6375–6386.
- 4.
Huang J, Milton A, Arnold RD, Huang H, Smith F, Panizzi JR, et al. Methods for measuring myeloperoxidase activity toward assessing inhibitor efficacy in living systems. J Leukoc Biol 2016; 99: 541–548.
- 5.
Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 2016; 118: 653–667.
- 6.
Fisher EA. Regression of atherosclerosis: The journey from the liver to the plaque and back. Arterioscler Thromb Vasc Biol 2016; 36: 226–235.
- 7.
Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev 2014; 262: 153–166.
- 8.
da Rocha RF, De Bastiani MA, Klamt F. Bioinformatics approach to evaluate differential gene expression of M1/M2 macrophage phenotypes and antioxidant genes in atherosclerosis. Cell Biochem Biophys 2014; 70: 831–839.
- 9.
Gordon S, Martinez FO. Alternative activation of macrophages: Mechanism and functions. Immunity 2010; 32: 593–604.
- 10.
Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol 2015; 25: 545–555.
- 11.
Rauh MJ. SHIP represses the generation of alternatively activated macrophages. Immunity 2005; 23: 361–374.
- 12.
Weisser SB, McLarren KW, Voglmaier N, van Netten-Thomas CJ, Antov A, Flavell RA, et al. Alternative activation of macrophages by IL-4 requires SHIP degradation. Eur J Immun 2011; 41: 1742–1753.
- 13.
Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AB, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol 2014; 193: 1717–1727.
- 14.
Festuccia WT, Pouliot P, Bakan I, Sabatini DM, Laplante M. Myeloid-specific rictor deletion induces M1 macrophage polarization and potentiates in vivo pro-inflammatory response to lipopolysaccharide. PLoS One 2014; 9: e95432, doi:10.1371/journal.pone.0095432.
- 15.
Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Nat Acad Sci USA 2012; 109: 9517–9522.
- 16.
Babaev VR, Hebron KE, Wiese CB, Toth CL, Ding L, Zhang Y, et al. Macrophage deficiency of Akt2 reduces atherosclerosis in Ldlr null mice. J Lipid Res 2014; 55: 2296–2308.
- 17.
Rensing KL, de Jager SCA, Stroes ES, Vos M, Twickler MTB, Dallinga-Thie GM, et al. Akt2/LDLr double knockout mice display impaired glucose tolerance and develop more complex atherosclerotic plaques than LDLr knockout mice. Cardiovasc Res 2014; 101: 277–287.
- 18.
Rotllan N, Chamorro-Jorganes A, Araldi E, Wanschel AC, Aryal B, Aranda JF, et al. Hematopoietic Akt2 deficiency attenuates the progression of atherosclerosis. FASEB J 2015; 29: 597–610.
- 19.
Zhou GL, Tucker DF, Bae SS, Bhatheja K, Birnbaum MJ, Field J. Opposing roles for Akt1 and Akt2 in Rac/Pak signaling and cell migration. J Biol Chem 2006; 281: 36443–36453.
- 20.
Chin YR, Toker A. Akt isoform-specific signaling in breast cancer: Uncovering an anti-migratory role for palladin. Cell Adh Migr 2011; 5: 211–214.
- 21.
Samuels Y, Waldman T. Oncogenic mutations of PIK3CA in human cancers. Curr Topics Microbiol Immunol 2010; 347: 21–41.
- 22.
Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells 2008; 26: 3027–3036.
- 23.
Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006; 444: 860–867.
- 24.
Coe NR, Bernlohr DA. Physiological properties and functions of intracellular fatty acid-binding proteins. Biochim Biophys Acta 1998; 1391: 287–306.
- 25.
Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med 2001; 7: 699–705.
- 26.
Uysal KT, Scheja L, Wiesbrock SM, Bonner-Weir S, Hotamisligil GS. Improved glucose and lipid metabolism in genetically obese mice lacking aP2. Endocrinology 2000; 141: 3388–3396.
- 27.
Boord JB, Maeda K, Makowski L, Babaev VR, Fazio S, Linton MF, et al. Adipocyte fatty acid-binding protein, aP2, alters late atherosclerotic lesion formation in severe hypercholesterolemia. Arterioscler Thromb Vasc Biol 2002; 22: 1686–1691.
- 28.
Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 2009; 15: 1383–1391.
- 29.
Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 2007; 8: 519–529.
- 30.
Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol 2011; 13: 184–190.
- 31.
Yagi A, Hasegawa Y, Xiao H, Haneda M, Kojima E, Nishikimi A, et al. GADD34 induces p53 phosphorylation and p21/WAF1 transcription. J Cell Biochem 2003; 90: 1242–1249.
- 32.
Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe–/– and Ldlr–/– mice lacking CHOP. Cell Metab 2009; 9: 474–481.
- 33.
Fu HY, Okada K, Liao Y, Tsukamoto O, Isomura T, Asai M, et al. Ablation of C/EBP homologous protein attenuates endoplasmic reticulum-mediated apoptosis and cardiac dysfunction induced by pressure overload. Circulation 2010; 122: 361–369.
- 34.
Tabas I. The role of endoplasmic reticulum stress in the progression of atherosclerosis. Circ Res 2010; 107: 839–850.
- 35.
Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol 2015; 209: 13–22.
- 36.
Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol 2005; 25: 174–179.
- 37.
Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, et al. A role for the apoptosis inhibitory factor AIM/Spα/Api6 in atherosclerosis development. Cell Metab 2005; 1: 201–213.
- 38.
Seimon T, Tabas I. Mechanisms and consequences of macrophage apoptosis in atherosclerosis. J Lipid Res 2009; 50: S382–S387.
- 39.
Gautier EL, Huby T, Witztum JL, Ouzilleau B, Miller ER, Saint-Charles F, et al. Macrophage apoptosis exerts divergent effects on atherogenesis as a function of lesion stage. Circulation 2009; 119: 1795–1804.
- 40.
Scull CM, Tabas I. Mechanisms of ER stress-induced apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol 2011; 31: 2792–2797.
- 41.
Sunayama J, Tsuruta F, Masuyama N, Gotoh Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J Cell Biol 2005; 170: 295–304.
- 42.
Babaev VR, Yeung M, Erbay E, Ding L, Zhang Y, May JM, et al. Jnk1 deficiency in hematopoietic cells suppresses macrophage apoptosis and increases atherosclerosis in low-density lipoprotein receptor null mice. Arterioscler Thromb Vasc Biol 2016; 36: 1122–1131.
- 43.
Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell 2007; 129: 1261–1274.
- 44.
Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: Role of Mcl-1, independent of nuclear factor (NF)-{kappa}B, Bad, or caspase activation. J Exp Med 2001; 194: 113–126.
- 45.
Babaev VR, Chew JD, Ding L, Davis S, Breyer MD, Breyer RM, et al. Macrophage EP4 deficiency increases apoptosis and suppresses early atherosclerosis. Cell Metab 2008; 8: 492.
- 46.
Heron-Milhavet L, Khouya N, Fernandez A, Lamb NJ. Akt1 and Akt2: Differentiating the aktion. Histol Histopathol 2011; 26: 651–662.
- 47.
Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood 2010; 115: 4237–4246.
- 48.
Cho H, Thorvaldsen JL, Chu Q, Feng F, Birnbaum MJ. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 2001; 276: 38349–38352.
- 49.
Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, et al. Role for Akt3/protein kinase Bγ in attainment of normal brain size. Mol Cell Biol 2005; 25: 1869–1878.
- 50.
Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, et al. Regulation of PTEN transcription by p53. Mol Cell 2001; 8: 317–325.
- 51.
Liu X, Shi Y, Birnbaum MJ, Ye K, De Jong R, Oltersdorf T, et al. Quantitative analysis of anti-apoptotic function of Akt in Akt1 and Akt2 double knock-out mouse embryonic fibroblast cells under normal and stressed conditions. J Biol Chem 2006; 281: 31380–31388.
- 52.
Ding L, Biswas S, Morton RE, Smith JD, Hay N, Byzova TV, et al. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab 2012; 15: 861–872.
- 53.
Fernandez-Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, et al. Loss of Akt1 leads to severe atherosclerosis and occlusive coronary artery disease. Cell Metab 2007; 6: 446–457.
- 54.
Karin M. The IκB kinase: A bridge between inflammation and cancer. Cell Res 2008; 18: 334–342.
- 55.
Romashkova JA, Makarov SS. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature 1999; 401: 86.
- 56.
Babaev VR, Ding L, Zhang Y, May JM, Lin PC, Fazio S, et al. Macrophage IKKα deficiency suppresses Akt phosphorylation, reduces cell survival, and decreases early atherosclerosis. Arterioscler Thromb Vasc Biol 2016; 36: 598–607.
- 57.
Tilstam PV, Gijbels MJ, Habbeddine M, Cudejko C, Asare Y, Theelen W, et al. Bone marrow-specific knock-in of a non-activatable Ikkalpha kinase mutant influences haematopoiesis but not atherosclerosis in Apoe-deficient mice. PLoS One 2014; 9: e87452, doi:10.1371/journal.pone.0087452.
- 58.
Dan HC, Baldwin AS. Differential involvement of IkappaB kinases alpha and beta in cytokine- and insulin-induced mammalian target of rapamycin activation determined by Akt. J Immunol 2008; 180: 7582–7589.
- 59.
Duronio V. The life of a cell: Apoptosis regulation by the PI3K/PKB pathway. Biochem J 2008; 415: 333–344.
- 60.
Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, et al. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 2002; 3: 631–643.
- 61.
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999; 96: 857–868.
- 62.
Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 2001; 98: 11598–11603.
- 63.
Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21: 749.
- 64.
Gardai SJ, Hildeman DA, Frankel SK, Whitlock BB, Frasch SC, Borregaard N, et al. Phosphorylation of Bax Ser184 by Akt regulates its activity and apoptosis in neutrophils. J Biol Chem 2004; 279: 21085–21095.
- 65.
Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 2001; 359: 1–16.
- 66.
Kadl A, Bochkov VN, Huber J, Leitinger N. Apoptotic cells as sources for biologically active oxidized phospholipids. Antioxid Redox Signal 2004; 6: 311–320.
- 67.
Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognition by phagocytes: A view to a kill. Trends Cell Biol 2006; 16: 189–197.
- 68.
Tibrewal N, Wu Y, D’Mello V, Akakura R, George TC, Varnum B, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem 2008; 283: 3618–3627.
- 69.
Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010; 10: 36–46.
- 70.
Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoE–/– mice. Arterioscler Thromb Vasc Biol 2008; 28: 1421–1428.
- 71.
Ait-Oufella H, Pouresmail V, Simon T, Blanc-Brude O, Kinugawa K, Merval R, et al. Defective mer receptor tyrosine kinase signaling in bone marrow cells promotes apoptotic cell accumulation and accelerates atherosclerosis. Arterioscler Thromb Vasc Biol 2008; 28: 1429–1431.
- 72.
Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol 1999; 162: 3498–3503.
- 73.
Toth B, Garabuczi E, Sarang Z, Vereb G, Vamosi G, Aeschlimann D, et al. Transglutaminase 2 is needed for the formation of an efficient phagocyte portal in macrophages engulfing apoptotic cells. J Immunol 2009; 182: 2084–2092.
- 74.
Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, et al. Leukocyte transglutaminase 2 expression limits atherosclerotic lesion size. Arterioscler Thromb Vasc Biol 2006; 26: 563–569.
- 75.
Ait-Oufella H, Kinugawa K, Zoll J, Simon T, Boddaert J, Heeneman S, et al. Lactadherin deficiency leads to apoptotic cell accumulation and accelerated atherosclerosis in mice. Circulation 2007; 115: 2168–2177.
- 76.
Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, et al. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler Thromb Vasc Biol 2010; 30: 787–795.
- 77.
Overton CD, Yancey PG, Major AS, Linton MF, Fazio S. Deletion of macrophage LDL receptor-related protein increases atherogenesis in the mouse. Circ Res 2007; 100: 670–677.
- 78.
Yancey PG, Ding Y, Fan D, Blakemore JL, Zhang Y, Ding L, et al. Low-density lipoprotein receptor-related protein 1 prevents early atherosclerosis by limiting lesional apoptosis and inflammatory Ly-6Chigh monocytosis: Evidence that the effects are not apolipoprotein E dependent. Circulation 2011; 124: 454–464.
- 79.
Orr AW, Pedraza CE, Pallero MA, Elzie CA, Goicoechea S, Strickland DK, et al. Low density lipoprotein receptor-related protein is a calreticulin coreceptor that signals focal adhesion disassembly. J Cell Biol 2003; 161: 1179–1189.
- 80.
Su HP, Nakada-Tsukui K, Tosello-Trampont AC, Li Y, Bu G, Henson PM, et al. Interaction of CED-6/GULP, an adapter protein involved in engulfment of apoptotic cells with CED-1 and CD91/low density lipoprotein receptor-related protein (LRP). J Biol Chem 2002; 277: 11772–11779.
- 81.
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke P, Morimoto K, Janssen WJ, et al. ATP-binding cassette transporter A7 enhances phagocytosis of apoptotic cells and associated ERK signaling in macrophages. J Cell Biol 2006; 174: 547–556.
- 82.
Lee G, Pollard HB, Arispe N. Annexin 5 and apolipoprotein E2 protect against Alzheimer’s amyloid-beta-peptide cytotoxicity by competitive inhibition at a common phosphatidylserine interaction site. Peptides 2002; 23: 1249–1263.
- 83.
Tedla N, Glaros E, Brunk U, Jessup W, Garner B. Heterogeneous expression of apolipoprotein-E by human macrophages. Immunology 2004; 113: 338–347.
- 84.
Grainger DJ, Reckless J, McKilligin E. Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J Immunol 2004; 173: 6366–6375.
- 85.
Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E-deficient mice by bone marrow transplantation. Science 1995; 267: 1034–1037.
- 86.
Fazio S, Babaev VR, Murray AB, Hasty AH, Carter KJ, Gleaves LA, et al. Increased atherosclerosis in C57BL/6 mice reconstituted with apolipoprotein E null macrophages. Proc Natl Acad Sci USA 1997; 94: 4647–4652.
- 87.
Tao H, Yancey PG, Babaev VR, Blakemore JL, Zhang Y, Ding L, et al. Macrophage SR-BI mediates efferocytosis via Src/PI3K/Rac1 signaling and reduces atherosclerotic lesion necrosis. J Lipid Res 2015; 56: 1449–1460.
- 88.
Cash JG, Kuhel DG, Basford JE, Jaeschke A, Chatterjee TK, Weintraub NL, et al. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem 2012; 287: 27876–27884.
- 89.
Song Y, Stampfer MJ, Liu S. Meta-analysis: Apolipoprotein E genotypes and risk for coronary heart disease. Ann Intern Med 2004; 141: 137–147.
- 90.
Reddy SM, Hsiao KH, Abernethy VE, Fan H, Longacre A, Lieberthal W, et al. Phagocytosis of apoptotic cells by macrophages induces novel signaling events leading to cytokine-independent survival and inhibition of proliferation: Activation of Akt and inhibition of extracellular signal-regulated kinases 1 and 2. J Immunol 2002; 169: 702–713.
- 91.
Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, et al. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation 2003; 108: 2258–2263.
- 92.
Covey SD, Krieger M, Wang W, Penman M, Trigatti BL. Scavenger receptor class B type I-mediated protection against atherosclerosis in LDL receptor-negative mice involves its expression in bone marrow-derived cells. Arterioscler Thromb Vasc Biol 2003; 23: 1589–1594.
- 93.
Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 2005; 25: 1256–1261.
- 94.
Gillotte-Taylor K, Boullier A, Witztum JL, Steinberg D, Quehenberger O. Scavenger receptor class B type I as a receptor for oxidized low density lipoprotein. J Lipid Res 2001; 42: 1474–1482.
- 95.
Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: Implications for macrophage clearance of apoptotic cells. Cell Death Differ 2004; 11: 943–945.
- 96.
Chang MK, Bergmark C, Laurila A, Horkko S, Han KH, Friedman P, et al. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc Natl Acad Sci USA 1999; 96: 6353–6358.
- 97.
Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, et al. Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: Toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ Res 2009; 104: 210–218.
- 98.
Miller YI, Viriyakosol S, Binder CJ, Feramisco JR, Kirkland TN, Witztum JL. Minimally modified LDL binds to CD14, induces macrophage spreading via TLR4/MD-2, and inhibits phagocytosis of apoptotic cells. J Biol Chem 2003; 278: 1561–1568.
- 99.
Costales P, Castellano J, Revuelta-Lopez E, Cal R, Aledo R, Llampayas O, et al. Lipopolysaccharide downregulates CD91/low-density lipoprotein receptor-related protein 1 expression through SREBP-1 overexpression in human macrophages. Atherosclerosis 2013; 227: 79–88.
- 100.
A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009; 31: 245–258.
- 101.
Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). J Biol Chem 2011; 286: 33335–33344.
- 102.
Gorovoy M, Gaultier A, Campana WM, Firestein GS, Gonias SL. Inflammatory mediators promote production of shed LRP1/CD91, which regulates cell signaling and cytokine expression by macrophages. J Leukoc Biol 2010; 88: 769–778.
- 103.
de Souza AW, Westra J, Limburg PC, Bijl M, Kallenberg CG. HMGB1 in vascular diseases: Its role in vascular inflammation and atherosclerosis. Autoimmun Rev 2012; 11: 909–917.
- 104.
Banerjee S, de Freitas A, Friggeri A, Zmijewski JW, Liu G, Abraham E. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol 2011; 187: 4686–4694.
- 105.
Friggeri A, Yang Y, Banerjee S, Park YJ, Liu G, Abraham E. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Physiol Cell Physiol 2010; 299: C1267–C1276.
- 106.
Liu G, Wang J, Park YJ, Tsuruta Y, Lorne EF, Zhao X, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol 2008; 181: 4240–4246.
- 107.
Kanellakis P, Agrotis A, Kyaw TS, Koulis C, Ahrens I, Mori S, et al. High-mobility group box protein 1 neutralization reduces development of diet-induced atherosclerosis in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol 2011; 31: 313–319.
- 108.
Heo KS, Cushman HJ, Akaike M, Woo CH, Wang X, Qiu X, et al. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation 2014; 130: 180–191.
- 109.
Marchetti A, Colletti M, Cozzolino AM, Steindler C, Lunadei M, Mancone C, et al. ERK5/MAPK is activated by TGFbeta in hepatocytes and required for the GSK-3beta-mediated Snail protein stabilization. Cell Signal 2008; 20: 2113–2118.
- 110.
Kojima Y, Volkmer JP, McKenna K, Civelek M, Lusis AJ, Miller CL, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature 2016; 536: 86–90.
- 111.
Greenlee-Wacker MC, Rigby KM, Kobayashi SD, Porter AR, DeLeo FR, Nauseef WM. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J Immunol 2014; 192: 4709–4717.
- 112.
Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature 2011; 473: 317–325.
- 113.
Mitchell S, Thomas G, Harvey K, Cottell D, Reville K, Berlasconi G, et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: Stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J Am Soc Nephrol 2002; 13: 2497–2507.
- 114.
Li S, Sun Y, Liang CP, Thorp EB, Han S, Jehle AW, et al. Defective phagocytosis of apoptotic cells by macrophages in atherosclerotic lesions of ob/ob mice and reversal by a fish oil diet. Circ Res 2009; 105: 1072–1082.
- 115.
Korns D, Frasch SC, Fernandez-Boyanapalli R, Henson PM, Bratton DL. Modulation of macrophage efferocytosis in inflammation. Front Immunol 2011; 2: 57.
- 116.
Gui Y, Yao S, Yan H, Hu L, Yu C, Gao F, et al. A novel small molecule liver X receptor transcriptional regulator, nagilactone B, suppresses atherosclerosis in apoE-deficient mice. Cardiovasc Res 2016; 112: 502–514.
- 117.
Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, et al. Targeted nanoparticles containing the proresolving peptide Ac2-26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med 2015; 7: 275ra220.
