Abstract
It is almost a quarter of century that a pioneering work of 2 researchers named Brugada brought the entire scientific community to understanding the molecular, clinical, and electrophysiological aspects of a distinctive syndrome. It affects mainly young adults with syncope and/or sudden cardiac death caused by polymorphic ventricular tachycardia or ventricular fibrillation in the absence of any sign of cardiac degeneration or alteration. Although the involvement of the epicardial layer of the right ventricular outflow tract, and the requirement of pharmacologic challenge for unveiling concealed forms, have been fully characterized, many areas of uncertainties remain to be elucidated, such as the unpredictable usefulness of programmed ventricular stimulation, the role of radiofrequency catheter ablation for reducing ST-segment elevation, and the value of risk stratification in patients diagnosed with upper displacement of right precordial leads. How much Brugada syndrome is an intense field of research is witnessed by 4 different consensus committees that took place in a relatively short period of time considering the recent discovery of this intricate arrhythmogenic disease. The main focus of this review is to describe the milestones in Brugada syndrome from its first phenotypic and genotypic appraisals to recent achievements in electrical therapies proposed for the management of this fascinating rhythm disturbance that, despite new diagnostic and therapeutic learnings, still predisposes to sudden cardiac death.
The Brugada syndrome (BrS) is characterized on ECG by right bundle branch block (RBBB) and an unusual form of ST-T wave elevation (unrelated to ischemia, electrolyte abnormalities or structural heart disease) in the anterior right precordial leads (RPL).1
As circumstantiated by local traditions and cultural myths that attributed some fantastic names (Lai-tai, Bangungut, Pokkuri) to a syndrome that could induce sudden unexpected nocturnal death or simply sudden unexplained death, BrS is more diffused in Japan (0.15–0.27%) and Philippines (0.18%),2
compared with Western countries (Europe: 0–0.017%; North America: 0.005–0.1%),3
although an intermittent ECG pattern often conceals the true prevalence. Despite risk distribution not being uniform among patients, and most not experiencing adverse cardiac events during their lives, the “Brugada sign” usually qualifies patients with syncope and a family history of sudden cardiac death (SCD) for an implantable cardioverter-defibrillator (ICD).
The aims of this review are to summarize the recent appraisals in BrS and to provide updates in the diagnostic and therapeutic paths of a disease that bears a non-negligible risk of SCD.
History
Provided the ECG abnormalities of the right ventricle (RV) were all associated to SCD,4
Joseph and Pedro Brugada5
reported the full phenotypic characterization of the RBBB in the prognosis of a syndrome in which altered autonomic tone and antiarrhythmic drugs could modulate the extent of ST-segment elevation (Figure 1).6

In 1997, a series initially consisting of 47 patients,7
and subsequently extended to 63, demonstrated the usefulness of the ICD in preventing SCD in patients with RBBB and ST-segment elevation in leads V1–3.1
A few years later, Priori et al analyzed 52 BrS families with 44 asymptomatic family members (FM) displaying positive genetic screening but negative ECGs, hence documenting the effect of family history in the prognosis of the disease.8
Following these studies, a first consensus report was elaborated.9
Subsequently, a second consensus was drafted,10
which introduced the concept of changing eating habits, because cardiac arrest (CA) caused by polymorphic ventricular tachycardia (pVT) or ventricular fibrillation (VF) can occur after a large meal, especially with foods rich in carbohydrates and glutinous rice, which are believed to shift potassium from free circulation into cells.11
Etiopathogenesis
In the majority of genetically diagnosed cases, BrS displays an autosomal dominant mode of transmission with low penetrance8,12
(i.e., the abnormal gene is inherited by 50% of the offspring, and both males and females equally inherit the defective gene, but not all will develop the disease). The identification of a loss-of-function mutation affecting the SCN5A (sodium voltage-gated channel α subunit 5) gene on chromosome 3p21–2313
was rapidly followed by reports of other variants (Figure 2)14,15
that can either alter protein synthesis16
or processing/trafficking,17
or affect channel gating, kinetics, permeability, and ion selectivity.18,19
In more detail, missense mutations (in BrS these occur in two-thirds of patients) are point mutations in which a single nucleotide change results in a codon that codes for a different amino acid; in smaller amounts, nonsense mutations (protein truncated at the mutation site), and splice-site, frameshift, insertion, and deletion mutations (protein completely altered after the mutation site) have been discovered as well.14
In the past decades, with accumulated findings suggesting that BrS has a heterogeneous genetic basis, a more complex inheritance has been proved. Nowadays, <40% of BrS cases are familial, whereas other cases are sporadic.12,19
Furthermore, unlike most previously reported sodium channelopathies, overlap syndromes displaying recessive inheritance characteristics and not following simple Mendelian rules, can be found.20

In spite of the low yield of genetic screening (<30%,
Table), with 65–70% of BrS patients remaining genetically unresolved, mutations in additional genes,15
including different subunits of the L-type cardiac Ca2+
channel,21
remain to be further investigated.
Table.
Pathogenic Variants Diagnostic of BrS
| Genetics |
| Main genes displaying pathogenic variants |
% of BrS carriers |
| SCN5A |
20–25% cases |
| CACNA1C |
5% cases |
CACNA2D1, KCND3, CACNB2b, GPD1L, HCN4, KCNE3, KCNE5, KCNJ8, RANGRF,
TRPM4, SCN1B, SCN2B, SCN3B, SLMAP |
<1% cases, each |
BrS, Brugada syndrome; CACNA1C, calcium voltage-gated channel subunit α1C; CACNA2D1, calcium voltage-gated channel auxiliary subunit α2δ1; CACNB2b, calcium voltage-gated channel auxiliary subunit β2; GPD1L, glycerol-3-phosphate dehydrogenase 1-like; HCN4, hyperpolarization-activated, cyclic nucleotide-gated K+4; KCND3, potassium voltage-gated channel subfamily D member 3; KCNE3, potassium voltage-gated channel subfamily E regulatory subunit 3; KCNE5, potassium voltage-gated channel subfamily E regulatory subunit 5; KCNJ8, potassium voltage-gated channel subfamily J member 8; RANGRF, RAN guanine nucleotide release factor; SCN1B, sodium voltage-gated channel β subunit 1; SCN2B, sodium voltage-gated channel β subunit 2; SCN3B, sodium voltage-gated channel β subunit 3; SCN5A, sodium voltage-gated channel α subunit 5; SLMAP, sarcolemma associated protein; TRPM4, transient receptor potential cation channel subfamily M member 4.
Although repolarization heterogeneity within the epicardium of the right ventricular outflow tract (RVOT) has been linked to phase 2 reentry VT,22
other authors have delineated conduction disturbances (PR-segment prolongation, different degrees of incomplete RBBB) and late potentials (see later) on the surface ECG, associated with H-V interval prolongation in electrophysiologic studies, as substrates for depolarization disorders.23
Postmortem analyses of unexplained sudden death victims in different series were all concordant in describing peculiar tissue abnormalities at the RVOT,24
characterized by reduced connexin-43 expression, interstitial fibrosis,25
and activation slowing that conditions an absent transmural repolarization gradient, and abnormal conduction restitution.26
To date, both theories are believed to be involved in the etiopathogenesis of BrS as demonstrated by an elegant study of ECG imaging on 25 BrS and 6 RBBB patients; unlike BrS, RBBB showed delayed activation in the entire RV, without ST- segment elevation, fractionation, or repolarization abnormalities.27
Fever may induce the appearance of a type 1 BrS ECG pattern and may trigger episodes of pVT/VF in affected patients,28
because of accentuation of the inactivation of the Na+
channel.17
When an increase in body temperature >38℃ occurs, current guidelines recommend close ECG monitoring in combination with antipyretics.3
Hypertestosteronemia appears to be a modulating factor that associates BrS with the male sex (8–10-fold more prevalent than in women);29
the incidence of fatal arrhythmias seems related to ion currents underlying the epicardial action potential notch.2
Diagnosis
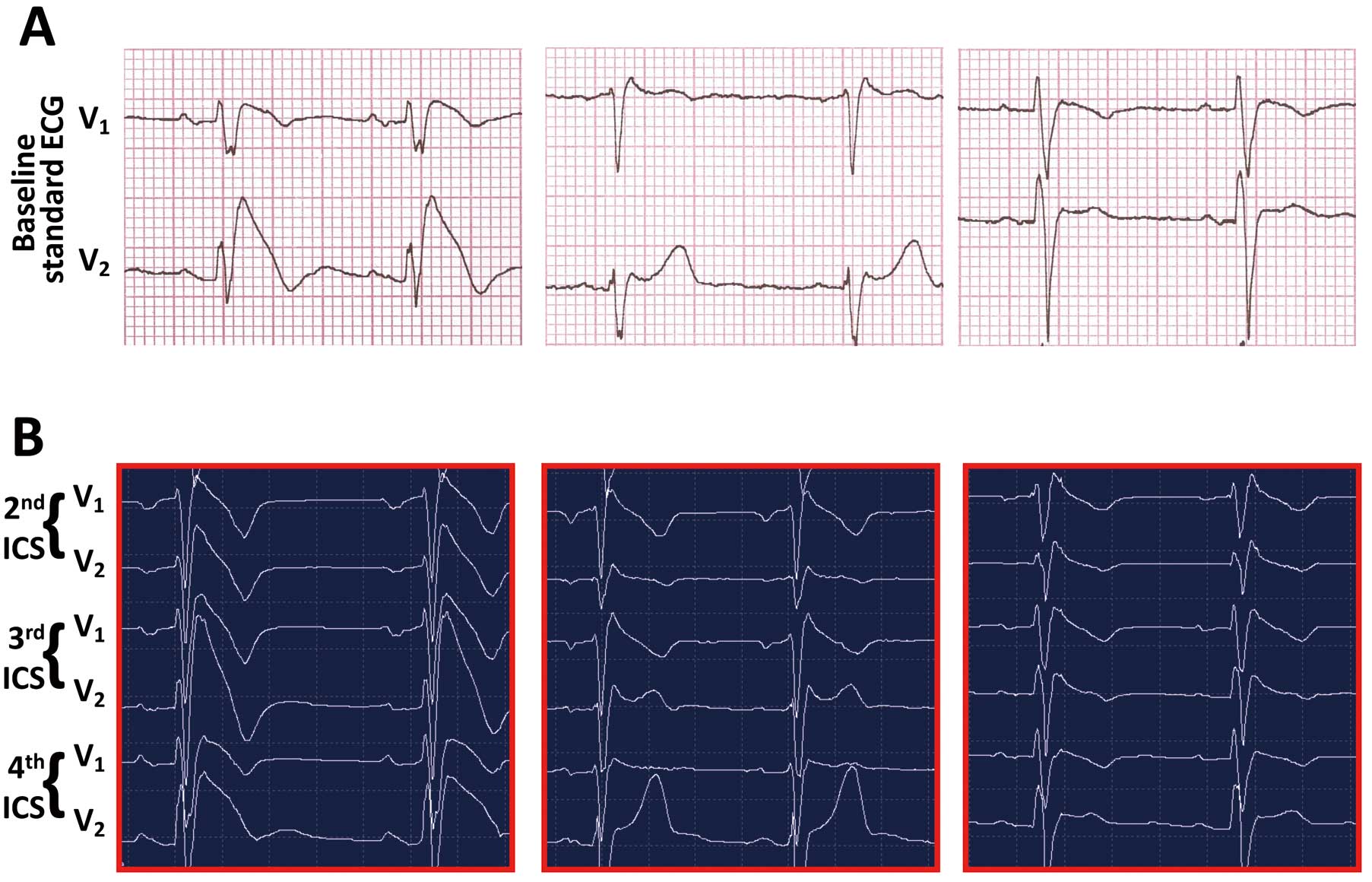
Diagnostic ECG (type 1,
Figure 3A,B) displays a coved ST-segment elevation of at least 2 mm of amplitude recorded in the RPL, either spontaneously or evoked by a 10-min infusion of sodium channel blocking agents (ajmaline 1 mg/kg, flecainide 2 mg/kg, procainamide 10 mg/kg, or pilsicainide 1 mg/kg).1–3,7–10
The saddleback ST-segment elevation ≥2 mm (type 2 BrS ECG,
Figure 3A) or <1 mm (either coved or saddleback, type 3 BrS ECG,
Figure 3A) is suspicious for BrS and requires further investigation.2,3,30–32
The first consensus in 2002 recommended diagnosis when a type 1 ECG pattern was detected in 2 among V1–3
leads with a gradually descending ST segment;7–9
3 years later, the second consensus focused on the relative weight of a few clinical aspects such as (1) documented VF; (2) self-terminating pVT; (3) family history of SCD (<45 years); (4) coved-type ECGs in FMs; (5) inducibility at programmed electrical stimulation (PES); (6) syncope; and (7) nocturnal agonal respiration.10
Following the report on BrS diagnosed in just 1 RPL,33,34
the Heart Rhythm Society (HRS), the European Heart Rhythm Association (EHRA), and the Asia Pacific Heart Rhythm Society (APHRS), released new guidelines according to spontaneous or drug-induced coved ST-segment elevation ≥2 mm in ≥1 lead (either V1
or V2) located in the 4th, 3rd, or 2nd intercostal space (ICS), and type 2 or type 3 ECG that fully converts into a type 1 ECG morphology.2
In this regard, it has been recently demonstrated that the upper displacement of the RPL on either rest ECG or Holter-ECG (Figure 3B) is able to unveil approximately 21% of new BrS cases35
because it reflects the normal projection of the heart (including the RVOT) onto the anterior chest surface.36
Such appraisals prevent the pro-arrhythmic side effects of class IC drugs,30–32,37,38
but also raise concerns about risk stratification (see later) of such individuals, because the diagnostic pattern is found spontaneously. Finally, a recent consensus of the HRS/EHRA/APHRS and the Latin American Society of Cardiac Pacing and Electrophysiology (SOLAECE) proposed the Shanghai Score System3
that calculates points derived by: ECG findings (spontaneous type 1 ECG at nominal or high leads=3.5; fever-induced type 1 ECG at nominal or high leads=3; type 2 or 3 ECG that converts to type 1 after drug infusion=2); clinical history (unexplained CA or documented VF/pVT=3; nocturnal agonal respirations=2; suspected arrhythmic syncope=2; syncope of unclear mechanism/unclear etiology=1; atrial fibrillation or flutter in patients <30 years without alternative etiology=0.5); family history (1st- or 2nd-degree relative with definite BrS=2; suspicious SCD during fever/nocturnal/under medication to be avoided in BrS=1; unexplained SCD <45 years with negative autopsy=0.5); probable pathogenic mutation in BrS susceptibility gene=0.5. According to this score, diagnosis is probable/definite when the cumulative sum is ≥3.5 points, and possible when 2–3 points are obtained; in cases with <2 points diagnosis is not feasible.
Risk Stratification
Symptoms
Asymptomatic patients are usually discovered during (1) routine check-ups (pre-operation, sports, insurance), (2) family screening; (3) antiarrhythmic treatment for palpitations or (4) fever.
The complete syndrome is characterized by episodes of rapid pVT in patients with an ECG pattern of RBBB and coved ST-segment elevation in the RPL. When the episodes terminate spontaneously, the patient develops syncopal attacks. When the episodes are sustained, full blown CA and eventually SCD occur. Such a distinction has been questioned recently, because syncope with prodrome, especially blurred vision, suggests a benign etiology of syncope itself in BrS patients;39
in contrast, many patients with suspected autonomic dysfunction who have undergone a tilt-table test, which was positive, and who have been treated accordingly have subsequently died suddenly.40
Family History of Sudden Death
Although a family history of BrS was initially considered to play an important role in the diagnostic process,8
different ethnic groups, population selection in each study, inclusion criteria of ECG with different ICS, and the number/timing of the ECG recordings did not confirm such an association.2,3
Sex Difference
Although the original descriptions noted that men present with a greater risk clinical profile than women and have a worse prognosis,41
a recent report provided the first event rate in the female sex (0.7%/year), and focused on SCD or previous sinus node dysfunction as a high-risk factor in future arrhythmic events in women as well.42
Risk and Prognosis in Life Stages
After the first case of CA in a Polish boy during a febrile illness,5
research groups confirmed the higher prevalence in adults,1–11,28–42
not excluding involvement in children when particularly aggressive phenotypes were manifested.43
In fact, rapid VT and conduction abnormalities,44
as well as sinus node dysfunction and atrial tachyarrhythmias45
in the absence of structural or metabolic abnormalities, have been observed in infancy.46
On the other hand, the elderly have been recently assessed as an epoch of the life with low BrS occurrence,47
although management with ICDs is associated with more inappropriate shocks and lead failure in old patients,48
who perhaps might benefit more from pacemaker therapy.49
Programmed Electrical Stimulation
For identification of individuals at major risk who would benefit from ICD therapy, pVT/VF inducibility at PES, became widely accepted.1,9,10,50
Brugada et al1,5,7,33,34
showed that PES is highly sensitive in predicting high risk, whereas the cohorts analyzed by Priori et al,51
Eckardt et al,52
and the FINGER study53
did not provide evidence that PES is a good indicator. Accordingly, PES in BrS has received a Class IIb recommendation.2
The last pooled analysis suggested that induced pVT/VF during PES up to double extrastimuli is associated with a higher risk of CA,54
although non-inducibility is not synonymous with low risk. To consolidate the usefulness of PES, multicenter prospective studies that include large numbers of patients are required.55
Noninvasive Instrumental Features
Nowadays there is an increasing interest in other noninvasive assessments, such as late potentials analysis and T-wave amplitude variability. These parameters reflect disorders in ventricular depolarization and repolarization, respectively.56
In agreement with these findings, a multicenter prospective registry identified fragmentation of the QRS (defined as ≥2 spikes within the QRS complex in leads V1
–V3) as a predictor of arrhythmic events in BrS patients without a history of pVT/VF.57
However, so far none of the above has proven effective in risk prediction. Considerable evidence supports the concept of the spontaneous type 1 pattern on either ECG or 12-lead Holter-ECG,51
recorded in whichever ICS,58
being associated with worse prognosis.50,51,59
In agreement with such findings, in a cohort of 300 suspected BrS patients, 64 cases that were diagnosed according to the new guidelines displayed 0.11% annual incidence of CA over a mean observation time of 41 years.35
Along the same line, others showed that in highly symptomatic (55% events) patients, type 1 ECG in the 4th and 3rd ICS did not influence prognosis, although the occurrence of diagnosis in only High-ICS and with drug test were very low (19.3 and 11.2%, respectively);58
therefore, both of these clinical observations reinforce the concept that a spontaneous BrS ECG pattern retains its prognostic value even when present in high precordial leads.35,58
On account of the delineated clinical features, a new category of risk, namely intermediate-low, can be hypothesized and included in the pyramid of risk (Figure 4).
Events Rate in BrS
Overall, the risk of lethal or near-lethal arrhythmic episodes among previously asymptomatic patients with BrS varies according to the series: 8% event rate at 33±39 months of follow-up reported by Brugada et al;33
6% event rate at 34±44 months by Priori et al;57
1% event rate after 40±50 months and 30±21 months of follow-up, respectively by Eckardt et al60
and Giustetto et al61
and finally, Probst et al53
reported a 1.5% event rate at 31 months of follow-up.
Implantable Devices
In the absence of effective pharmacologic therapies that prevent from SCD, the ICD is the only therapeutic option for BrS patients (Figure 5A). It is important to remark that ICDs are not free from several disadvantages, especially in young patients who will undergo plural device replacements for battery exhaustion.62
To this regard, at 10-years post-implantation, the rates of inappropriate shocks and/or device malfunction caused by lead failure are 37% and 29%, respectively.63
Endocardial lead dysfunction requires extraction and replacement, with further complications. Recently, the possibility of an entirely subcutaneous device (S-ICD) that avoids the side effects associated with transvenous electro-catheters has added options to the available armamentarium.64
Such devices have recently overcome the initial concerns of a potential risk of undersensing tachyarrhythmias (Figure 5B). To date, the role of the anti-tachypacing modality offered by traditional transvenous ICD in preventing degeneration of monomorphic VT is not considered essential in this category of patients, because low amplitude wave VF and polymorphic VT are the most common arrhythmias found in BrS patients; intriguingly, a recent multicenter retrospective study observed that monomorphic VTs account for only 4.2% of BrS patients implanted with an ICD and that there is a strong likelihood of arrhythmia disappearance after endocardial and/or epicardial ablation;65
this technical aspect corroborates the therapeutic role of S-ICD, and its association with fewer complications over a lifetime qualifies the subcutaneous defibrillator as the present and future indication for this complex arrhythmogenic disease.

Radiofrequency Catheter Ablation
The early attempts at catheter ablation to treat BrS patients were limited to a few reported cases of patients with electrical storms.
The former approach was designed to target the initiating PVCs that trigger VF with radiofrequency ablation (RFA) at the endocardial site of the RVOT.66
However, this approach did not prove successful in all cases, because BrS patients rarely have PVCs frequently enough to allow mapping and to provide a precise target for ablation.
The first epicardial RFA of the RVOT was followed by pVT/VF inducibility in 22% cases, while BrS pattern disappeared after procedure in 3 above 9 patients at follow-up.67
A recent report evaluated 14 BrS patients with ICD for targeting fragmented and delayed potentials and low voltage areas in the basal condition and after flecainide test. In a short time, data reported 100% pattern disappearance under basal conditions and after drug challenge, no pVT/VF inducibility, and no more episodes.68
Although larger studies with longer follow-up are required, these results provide new insights into the electrophysiological mechanisms of BrS; however, areas of uncertainty regarding the clinical outcome after embellishment of the ventricular repolarization have been already observed in the history of cardiac electrophysiology, when digoxin administration, which shortens the QT segment,69
was not associated with better outcome in long QT (LQT) syndrome patients.70
Moreover, whether the elimination of the phenotypic manifestations of the BrS will result in less arrhythmic events during follow-up remains to be established.
Finally, it should be taken into account that in patients in whom BrS is associated with the early repolarization pattern/syndrome, selective ablation of the anterior RV epicardium (including the RVOT) is not ameliorative.3
Experimental Therapeutic Options and Future Perspectives
Because translation to humans of the experimental results in homozygous Scn5a−/−
mice with conduction block and reentrant VT71
has been disappointing, mainly because of the lack of testosterone-mediated
Ito
currents or concordance between the RVOT and ECG in non-human settings, selective pharmacologic strategies have been developed.
Isoproterenol is effective in reducing the arrhythmic burden and electrical storm during energy delivery by the ICD;72,73
its pharmacodynamics seem related to increased Ca2+
currents through L-type Ca2+
channels. Dimethyl lithospermate B slows inactivation of
INa, thus increasing
INa
during the early phases of AP and suppressing arrhythmogenesis.2
Bepridil suppressed VT/VF in several studies of patients with BrS, mainly through
Ito
inhibition and
INa
increment (peak and late currents) via upregulation of the sodium channels, while prolonging the QT interval at slow rates.74
Hydroquinidine, a class IA antiarrhythmic drug, inhibits
Ito
to a greater extent in the epicardium than in the endocardium, and has proven efficacy and safety in a long-term follow-up study.75
Cilostazol, a phosphodiesterase III inhibitor, normalizes the ST-segment, most likely by augmenting
ICa
as well as by reducing
Ito
secondary to an increase in cAMP and heart rate.76
Finally, combined use of a Chinese herb extract that inhibits
Ito
and hydroquinidine has been hypothesized.77
Cellular reprogramming through the technology of human-induced pluripotent stem cells (hIPSCs) has been described for different primary electrical diseases, including LQT1, LQT2, LQT3, LQT8/Timothy syndrome and catecholaminergic pVT, as well as for BrS.78,79
If optimized, a hypothetical hiPSC-based approach could be successful in cases of frequent ICD shocks attributable to VT in spite of aggressive antiarrhythmic treatment, thus realizing “patient-tailored therapy”.
Conclusions
BrS is a primary electrical disorder, characterized by typical ECG signs, and it predisposes to death secondary to VT in the absence of cardiac dysfunction. Recommendations released by scientific societies have been updated several times, mainly for ascertaining diagnosis. Therefore, patients should be evaluated in a dedicated clinic with appropriately trained staff. In the meantime, the increased number of subjects with new diagnosis will require a modified clinical approach, such as hospitalization during fever, as well as extending the already wide list of drugs to be avoided (www.brugadadrugs.org) in all patients affected by BrS.
Acknowledgments
This work was partially supported by a grant of the Italian Ministry of Education, University and Research (MIUR):PON03PE_00009_4 OPTIMA Cardiopaths.
References
- 1.
Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: A marker for sudden death in patients without demonstrable structural heart disease. Circulation 1998; 97: 457–460.
- 2.
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013; 15: 1389–1406.
- 3.
Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-wave syndromes expert consensus conference report: Emerging concepts and gaps in knowledge. Heart Rhythm 2016; 13: e295–e324.
- 4.
Martini B, Nava A, Thiene G, Buja GF, Canciani B, Scognamiglio R, et al. Ventricular fibrillation without apparent heart disease: Description of six cases. Am Heart J 1989; 118: 1203–1209.
- 5.
Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: A distinct clinical and electrocardiographic syndrome: A multicenter report. J Am Coll Cardiol 1992; 20: 1391–1396.
- 6.
Miyazaki T, Mitamura H, Miyoshi S, Soejima K, Aizawa Y, Ogawa S. Autonomic and antiarrhythmic drug modulation of ST segment elevation in patients with Brugada syndrome. J Am Coll Cardiol 1996; 27: 1061–1070.
- 7.
Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, persistent ST-segment elevation, and sudden cardiac death. J Cardiovasc Electrophysiol 1997; 8: 325–331.
- 8.
Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Brignole M, et al. Clinical and genetic heterogeneity of right bundle branch block and ST-segment elevation syndrome: A prospective evaluation of 52 families. Circulation 2000; 102: 2509–2515.
- 9.
Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, et al. Proposed diagnostic criteria for the Brugada syndrome: Consensus report. Circulation 2002; 106: 2514–2519.
- 10.
Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: Report of the Second Consensus Conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111: 659–670.
- 11.
Ikeda T, Abe A, Yusu S, Nakamura K, Ishiguro H, Mera H, et al. The full stomach test as a novel diagnostic technique for identifying patients at risk of Brugada syndrome. J Cardiovasc Electrophysiol 2006; 17: 602–607.
- 12.
Juang JJ, Horie M. Genetics of Brugada syndrome. J Arrhythm 2016; 32: 418–425.
- 13.
Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998; 392: 293–296.
- 14.
Van Driest SL, Wells QS, Stallings S, Bush WS, Gordon A, Nickerson DA, et al. Association of arrhythmia-related genetic variants with phenotypes documented in electronic medical records. JAMA 2016; 315: 47–57.
- 15.
Crotti L, Marcou CA, Tester DJ, Castelletti S, Giudicessi JR, Torchio M, et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: Implications for genetic testing. J Am Coll Cardiol 2012; 60: 1410–1418.
- 16.
Bezzina C, Veldkamp MW, van den Berg MP, Postma AV, Rook MB, Viersma JW, et al. A single Na+ channel mutation causing both long-QT and Brugada syndromes. Circ Res 1999; 85: 1206–1213.
- 17.
Dumaine R, Towbin JA, Brugada P, Vatta M, Nesterenko DV, Nesterenko VV, et al. Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ Res 1999; 85: 803–809.
- 18.
Ackerman MJ, Splawski I, Makielski JC, Tester DJ, Will ML, Timothy KW, et al. Spectrum and prevalence of cardiac sodium channel variants among black, white, Asian, and Hispanic individuals: Implications for arrhythmogenic susceptibility and Brugada/long QT syndrome genetic testing. Heart Rhythm 2004; 1: 600–607.
- 19.
Schulze-Bahr E, Eckardt L, Breithardt G, Seidl K, Wichter T, Wolpert C, et al. Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: Different incidences in familial and sporadic disease. Hum Mutat 2003; 21: 651–652.
- 20.
Neu A, Eiselt M, Paul M, Sauter K, Stallmeyer B, Isbrandt D, et al. A homozygous SCN5A mutation in a severe, recessive type of cardiac conduction disease. Hum Mutat 2010; 31: E1609–E1621.
- 21.
Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 2007; 115: 442–449.
- 22.
Antzelevitch C. In vivo human demonstration of phase 2 reentry. Heart Rhythm 2005; 2: 804–806.
- 23.
Postema PG, van Dessel PF, Kors JA, Linnenbank AC, van Herpen G, Ritsema van Eck HJ, et al. Local depolarization abnormalities are the dominant pathophysiologic mechanism for type 1 electrocardiogram in brugada syndrome a study of electrocardiograms, vectorcardiograms, and body surface potential maps during ajmaline provocation. J Am Coll Cardiol 2010; 55: 789–797.
- 24.
Corrado D, Basso C, Buja G, Nava A, Rossi L, Thiene G. Right bundle branch block, right precordial ST-segment elevation, and sudden death in young people. Circulation 2001; 103: 710–717.
- 25.
Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJ, Verkerk AO, de Groot JR, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005; 112: 2769–2777.
- 26.
Nademanee K, Raju H, de Noronha SV, Papadakis M, Robinson L, Rothery S, et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol 2015; 66: 1976–1986.
- 27.
Zhang J, Sacher F, Hoffmayer K, O’Hara T, Strom M, Cuculich P, et al. Cardiac electrophysiologic substrate underlying the ECG phenotype and electrogram abnormalities in Brugada syndrome patients. Circulation 2015; 131: 1950–1959.
- 28.
Mizusawa Y, Morita H, Adler A, Havakuk O, Thollet A, Maury P, et al. Prognostic significance of fever-induced Brugada syndrome. Heart Rhythm 2016; 13: 1515–1520.
- 29.
Shimizu W, Matsuo K, Kokubo Y, Satomi K, Kurita T, Noda T, et al. Sex hormone and gender difference: Role of testosterone on male predominance in Brugada syndrome. J Cardiovasc Electrophysiol 2007; 18: 415–421.
- 30.
Shimizu W, Antzelevitch C, Suyama K, Kurita T, Taguchi A, Aihara N, et al. Effect of sodium channel blockers on ST segment, QRS duration, and corrected QT interval in patients with Brugada syndrome. J Cardiovasc Electrophysiol 2000; 11: 1320–1329.
- 31.
Batchvarov VN, Govindan M, Camm AJ, Behr ER. Significance of QRS prolongation during diagnostic ajmaline test in patients with suspected Brugada syndrome. Heart Rhythm 2009; 6: 625–631.
- 32.
Evain S, Briec F, Kyndt F, Schott JJ, Lande G, Albuisson J, et al. Sodium channel blocker tests allow a clear distinction of electrophysiological characteristics and prognosis in patients with a type 2 or 3 Brugada electrocardiogram pattern. Heart Rhythm 2008; 5: 1561–1564.
- 33.
Brugada J, Brugada R, Antzelevitch C, Towbin J, Nademanee K, Brugada P. Long-term follow-up of individuals with the electrocardiographic pattern of right bundle-branch block and ST-segment elevation in precordial leads V1 to V3. Circulation 2002; 105: 73–78.
- 34.
Richter S, Sarkozy A, Paparella G, Henkens S, Boussy T, Chierchia GB, et al. Number of electrocardiogram leads displaying the diagnostic coved-type pattern in Brugada syndrome: A diagnostic consensus criterion to be revised. Eur Heart J 2010; 31: 1357–1364.
- 35.
Curcio A, Mazzanti A, Bloise R, Monteforte N, Indolfi C, Priori SG, et al. Clinical presentation and outcome of Brugada syndrome diagnosed with the new 2013 criteria. J Cardiovasc Electrophysiol 2016; 27: 937–943.
- 36.
Veltmann C, Papavassiliu T, Konrad T, Doesch C, Kuschyk J, Streitner F, et al. Insights into the location of type I ECG in patients with Brugada syndrome: Correlation of ECG and cardiovascular magnetic resonance imaging. Heart Rhythm 2012; 9: 414–421.
- 37.
Veltmann C, Wolpert C, Sacher F, Mabo P, Schimpf R, Streitner F, et al. Response to intravenous ajmaline: A retrospective analysis of 677 ajmaline challenges. Europace 2009; 11: 1345–1352.
- 38.
Conte G, Sieira J, Sarkozy A, de Asmundis C, Di Giovanni G, Chierchia GB, et al. Life-threatening ventricular arrhythmias during ajmaline challenge in patients with Brugada syndrome: Incidence, clinical features, and prognosis. Heart Rhythm 2013; 10: 1869–1874.
- 39.
Take Y, Morita H, Toh N, Nishii N, Nagase S, Nakamura K, et al. Identification of high-risk syncope related to ventricular fibrillation in patients with Brugada syndrome. Heart Rhythm 2012; 9: 752–759.
- 40.
Sacher F, Arsac F, Wilton SB, Derval N, Denis A, de Guillebon M, et al. Syncope in Brugada syndrome patients: Prevalence, characteristics, and outcome. Heart Rhythm 2012; 9: 1272–1279.
- 41.
Benito B, Sarkozy A, Mont L, Henkens S, Berruezo A, Tamborero D, et al. Gender differences in clinical manifestations of Brugada syndrome. J Am Coll Cardiol 2008; 52: 1567–1573.
- 42.
Sieira J, Conte G, Ciconte G, de Asmundis C, Chierchia GB, Baltogiannis G, et al. Clinical characterisation and long-term prognosis of women with Brugada syndrome. Heart 2016; 102: 452–458.
- 43.
Probst V, Denjoy I, Meregalli PG, Amirault JC, Sacher F, Mansourati J, et al. Clinical aspects and prognosis of Brugada syndrome in children. Circulation 2007; 115: 2042–2048.
- 44.
Kanter RJ, Pfeiffer R, Hu D, Barajas-Martinez H, Carboni MP, Antzelevitch C. Brugada-like syndrome in infancy presenting with rapid ventricular tachycardia and intraventricular conduction delay. Circulation 2012; 125: 14–22.
- 45.
Gonzalez Corcia MC, Sieira J, Sarkozy A, de Asmundis C, Chierchia GB, Hernandez Ojeda J, et al. Brugada syndrome in the young: An assessment of risk factors predicting future events. Europace, doi:10.1093/europace/euw206.
- 46.
Conte G, Dewals W, Sieira J, de Asmundis C, Ciconte G, Chierchia GB, et al. Drug-induced brugada syndrome in children: Clinical features, device-based management, and long-term follow-up. J Am Coll Cardiol 2014; 63: 2272–2279.
- 47.
Kamakura T, Wada M, Nakajima I, Ishibashi K, Miyamoto K, Okamura H, et al. Evaluation of the necessity for cardioverter-defibrillator implantation in elderly patients with Brugada syndrome. Circ Arrhythm Electrophysiol 2015; 8: 785–791.
- 48.
Conte G, DE Asmundis C, Sieira J, Levinstein M, Chierchia GB, DI Giovanni G, et al. Clinical characteristics, management, and prognosis of elderly patients with Brugada syndrome. J Cardiovasc Electrophysiol 2014; 25: 514–519.
- 49.
Postema PG, Tan HL, Wilde AA. Ageing and Brugada syndrome: Considerations and recommendations. J Geriatr Cardiol 2013; 10: 75–81.
- 50.
Adler A, Rosso R, Chorin E, Havakuk O, Antzelevitch C, Viskin S. Risk stratification in Brugada syndrome: Clinical characteristics, electrocardiographic parameters, and auxiliary testing. Heart Rhythm 2016; 13: 299–310.
- 51.
Priori SG, Napolitano C, Gasparini M, Pappone C, Della Bella P, Giordano U, et al. Natural history of Brugada syndrome: Insights for risk stratification and management. Circulation 2002; 105: 1342–1347.
- 52.
Eckardt L, Kirchhof P, Schulze-Bahr E, Rolf S, Ribbing M, Loh P, et al. Electrophysiologic investigation in Brugada syndrome; yield of programmed ventricular stimulation at two ventricular sites with up to three premature beats. Eur Heart J 2002; 23: 1394–1401.
- 53.
Probst V, Veltmann C, Eckardt L, Meregalli PG, Gaita F, Tan HL, et al. Long-term prognosis of patients diagnosed with Brugada syndrome: Results from the FINGER Brugada Syndrome Registry. Circulation 2010; 121: 635–643.
- 54.
Sroubek J, Probst V, Mazzanti A, Delise P, Hevia JC, Ohkubo K, et al. Programmed ventricular stimulation for risk stratification in the Brugada syndrome: A pooled analysis. Circulation 2016; 133: 622–630.
- 55.
Priori SG, Napolitano C, Brugada P, Brugada R, Brugada J. Should patients with an asymptomatic Brugada electrocardiogram undergo pharmacological and electrophysiological testing? Circulation 2005; 112: 279–292.
- 56.
Yoshioka K, Amino M, Zareba W, Shima M, Matsuzaki A, Fujii T, et al. Identification of high-risk Brugada syndrome patients by combined analysis of late potential and T-wave amplitude variability on ambulatory electrocardiograms. Circ J 2013; 77: 610–618.
- 57.
Priori SG, Gasparini M, Napolitano C, Della Bella P, Ottonelli AG, Sassone B, et al. Risk stratification in Brugada syndrome: Results of the PRELUDE (PRogrammed ELectrical stimUlation preDictive valuE) registry. J Am Coll Cardiol 2012; 59: 37–45.
- 58.
Miyamoto K, Yokokawa M, Tanaka K, Nagai T, Okamura H, Noda T, et al. Diagnostic and prognostic value of a type 1 Brugada electrocardiogram at higher (third or second) V1 to V2 recording in men with Brugada syndrome. Am J Cardiol 2007; 99: 53–57.
- 59.
Kamakura S, Ohe T, Nakazawa K, Aizawa Y, Shimizu A, Horie M, et al. Long-term prognosis of probands with Brugada-pattern ST-elevation in leads V1–V3. Circ Arrhythm Electrophysiol 2009; 2: 495–503.
- 60.
Eckardt L, Probst V, Smits JP, Bahr ES, Wolpert C, Schimpf R, et al. Long-term prognosis of individuals with right precordial ST-segment-elevation Brugada syndrome. Circulation 2005; 111: 257–263.
- 61.
Giustetto C, Drago S, Demarchi PG, Dalmasso P, Bianchi F, Masi AS, et al. Risk stratification of the patients with Brugada type electrocardiogram: A community-based prospective study. Europace 2009; 11: 507–513.
- 62.
Casado-Arroyo R, Berne P, Rao JY, Rodriguez-Mañero M, Levinstein M, Conte G, et al. Long-term trends in newly diagnosed Brugada syndrome: Implications for risk stratification. J Am Coll Cardiol 2016; 68: 614–623.
- 63.
Sacher F, Probst V, Maury P, Babuty D, Mansourati J, Komatsu Y, et al. Outcome after implantation of cardioverter-defibrillator in patients with Brugada syndrome: A multicenter study–part 2. Circulation 2013; 128: 1739–1747.
- 64.
Olde Nordkamp LR, Conte G, Rosenmöller BR, Warnaars JL, Tan HL, Caputo ML, et al. Brugada syndrome and the subcutaneous implantable cardioverter-defibrillator. J Am Coll Cardiol 2016; 68: 665–666.
- 65.
Rodríguez-Mañero M, Sacher F, de Asmundis C, Maury P, Lambiase PD, Sarkozy A, et al. Monomorphic ventricular tachycardia in patients with Brugada syndrome: A multicenter retrospective study. Heart Rhythm 2016; 13: 669–682.
- 66.
Shah AJ, Hocini M, Lamaison D, Sacher F, Derval N, Haissaguerre M. Regional substrate ablation abolishes Brugada syndrome. J Cardiovasc Electrophysiol 2011; 22: 1290–1291.
- 67.
Nademanee K, Veerakul G, Chandanamattha P, Chaothawee L, Ariyachaipanich A, Jirasirirojanakorn K, et al. Prevention of ventricular fibrillation episodes in Brugada syndrome by catheter ablation over the anterior right ventricular outflow tract epicardium. Circulation 2011; 123: 1270–1279.
- 68.
Brugada J, Pappone C, Berruezo A, Vicedomini G, Manguso F, Ciconte G, et al. Brugada syndrome phenotype elimination by epicardial substrate ablation. Circ Arrhythm Electrophysiol 2015; 8: 1373–1381.
- 69.
Goldberger AL. Clinical electrocardiography: A simplified approach. In: Braunwald’s heart disease: A textbook of cardiovascular medicine, 7th edn. Philadelphia: Saunders, 2005; 141–142.
- 70.
Jervell A. The surdo-cardiac syndrome. Eur Heart J 1985; 6(Suppl D): 97–102.
- 71.
Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, et al. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci USA 2002; 99: 6210–6215.
- 72.
Watanabe A, Fukushima Kusano K, Morita H, Miura D, Sumida W, Hiramatsu S, et al. Low-dose isoproterenol for repetitive ventricular arrhythmia in patients with Brugada syndrome. Eur Heart J 2006; 27: 1579–1583.
- 73.
Ohgo T, Okamura H, Noda T, Satomi K, Suyama K, Kurita T, et al. Acute and chronic management in patients with Brugada syndrome associated with electrical storm of ventricular fibrillation. Heart Rhythm 2007; 4: 695–700.
- 74.
Murakami M, Nakamura K, Kusano KF, Morita H, Nakagawa K, Tanaka M, et al. Efficacy of low-dose bepridil for prevention of ventricular fibrillation in patients with Brugada syndrome with and without SCN5A mutation. J Cardiovasc Pharmacol 2010; 56: 389–395.
- 75.
Bouzeman A, Traulle S, Messali A, Extramiana F, Denjoy I, Narayanan K, et al. Long-term follow-up of asymptomatic Brugada patients with inducible ventricular fibrillation under hydroquinidine. Europace 2014; 16: 572–577.
- 76.
Szél T, Koncz I, Antzelevitch C. Cellular mechanisms underlying the effects of milrinone and cilostazol to suppress arrhythmogenesis associated with Brugada syndrome. Heart Rhythm 2013; 10: 1720–1727.
- 77.
Minoura Y, Panama BK, Nesterenko VV, Betzenhauser M, Barajas-Martínez H, Hu D, et al. Effect of Wenxin Keli and quinidine to suppress arrhythmogenesis in an experimental model of Brugada syndrome. Heart Rhythm 2013; 10: 1054–1062.
- 78.
Veerman CC, Mengarelli I, Guan K, Stauske M, Barc J, Tan HL, et al. hiPSC-derived cardiomyocytes from Brugada Syndrome patients without identified mutations do not exhibit clear cellular electrophysiological abnormalities. Sci Rep 2016; 6: 30967.
- 79.
Okata S, Yuasa S, Suzuki T, Ito S, Makita N, Yoshida T, et al. Embryonic type Na+ channel β-subunit, SCN3B masks the disease phenotype of Brugada syndrome. Sci Rep 2016; 6: 34198.

