Abstract
Cardiovascular disease (CVD) is the greatest cause of death, accounting for nearly one-third of all deaths worldwide. The increase in obesity rates over 3 decades is widespread and threatens the public health in both developed and developing countries. Obesity, the excessive accumulation of visceral fat, causes the clustering of metabolic disorders, such as type 2 diabetes, dyslipidemia, and hypertension, culminating in the development of CVD. Adipose tissue is not only an energy storage organ, but an active endocrine tissue producing various biologically active proteins known as adipokines. Since leptin, a central regulator of food intake and energy expenditure, was demonstrated to be an adipose-specific adipokine, attention has focused on the identification and characterization of unknown adipokines to clarify the mechanisms underlying obesity-related disorders. Numerous adipokines have been identified in the past 2 decades; most adipokines are upregulated in the obese state. Adipokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, and resistin are pro-inflammatory, and exacerbate various metabolic and cardiovascular diseases. However, a small number of adipokines, including adiponectin, are decreased by obesity, and generally exhibit antiinflammatory properties and protective functions against obesity-related diseases. Collectively, an imbalance in the production of pro- and antiinflammatory adipokines in the obese condition results in multiple complications. In this review, we focus on the pathophysiologic roles of adipokines with cardiovascular protective properties.
Cardiovascular disease (CVD) is the greatest cause of death, accounting for nearly one-third of all deaths worldwide.1–3
The increase in obesity rates over 3 decades is widespread and threatens the public health in both developed and developing countries.4
Obesity, the excessive accumulation of visceral fat, causes the clustering of metabolic disorders, such as type 2 diabetes, dyslipidemia, and hypertension, culminating in the development of CVD.5
Recent evidence indicates that obese states (e.g., excess adiposity, adipocyte dysfunction) cause chronic low-grade inflammation, which contributes to the development of obesity-linked disorders, including insulin resistance and atherosclerosis.6,7
Adipose tissue is not only an energy storage organ, but an active endocrine tissue producing various biologically active proteins known as adipokines (also known as adipocytokines).6
The term “adipokine” refers to any bioactive product of adipose tissue origin. Since leptin, a central regulator of food intake and energy expenditure, was demonstrated to be an adipose-specific adipokine, great attention has been given to the identification and characterization of unknown adipokines to clarify the mechanisms underlying obesity-related disorders.8
Numerous adipokines have been identified in the past 2 decades; most adipokines are upregulated in the obese state.9
Adipokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, and resistin are pro-inflammatory, and exacerbate various metabolic and cardiovascular diseases. However, a small number of adipokines, including adiponectin (APN), are decreased by obesity, and generally exhibit antiinflammatory properties and protective functions against obesity-related diseases.6
Collectively, an imbalance in the production of pro- and antiinflammatory adipokines in the obese conditions results in multiple complications. In this review, we focus on the pathophysiologic roles of adipokines with cardiovascular protective properties.
Role of Adipokines in Vascular Diseases
APN
APN is the most abundantly expressed adipokine of adipose origin, identified by systematic analysis of adipose-expressed genes.10
APN regulates cellular function via activation of its specific receptors, APN receptor 1 (AdipoR1) and APN receptor 2 (AdipoR2), each encoded by their respectively named genes. The APN receptors contain 7 transmembrane domains topologically distinct from G protein-coupled receptor proteins. APN binds to the C-terminal extracellular domain of the APN receptor, whereas the N-terminal cytoplasmic domain interacts with an adaptor protein (phosphotyrosine interacting with PH domain and leucine zipper 1, APPL1). Whereas AdipoR1 is abundantly expressed in muscular tissues (including the heart) and endothelial cells, AdipoR2 is predominantly expressed in the liver.11
As such, the relevant importance of AdipoR1 vs. AdipoR2 concerning APN’s biologic effects is largely determined by their expression profile in different tissues.
Abundantly present in human plasma, APN plasma levels negatively correlate with body mass index (BMI).12,13
The plasma APN concentration is significantly lower in coronary artery disease (CAD) patients compared with healthy subjects. Moreover, hypoadiponectinemia (plasma APN <4.0 µg/mL) is significantly associated with CAD prevalence, even after adjustment for well-known CAD risk factors such as diabetes mellitus (DM), dyslipidemia, hypertension, tobacco use, and BMI in male Japanese subjects.14
In prospective studies, high plasma APN levels have predicted lower risk of acute myocardial infarction (MI) in subjects with CAD and type 2 DM.15,16
Hypoadiponectinemia is also associated with complexity of coronary lesions in the patients with stable CAD.17,18
Consistently, higher APN levels are significantly associated with lower risk of CAD in the Framingham Offspring Study.19
Thus, circulating APN levels may be a valuable biomarker of CAD prevalence. In contrast, several reports indicated no association between APN concentration and CAD prevalence.20–22
The APN level is not a predictor of CAD in the American Indian population.21
High APN levels are only associated with reduced risk of non-fatal MI in men, and no association has been observed between APN levels and fatal coronary events in both men and women.20
A meta-analysis indicated that the association of APN level with CAD risk is comparatively weak.22
These inconsistent results might stem from differences in the study populations (e.g., sex, race, comorbidities). Therefore, further studies are warranted to elucidate the significance of APN as a CAD biomarker.
Experimental studies have indicated that APN modulates various vascular disorders. Adenovirus-mediated APN overexpression reduces atherosclerotic lesion formation in atherosclerosis-prone apolipoprotein E (ApoE) knockout (KO) mice, which is accompanied by decreased expression levels of TNF-α and scavenger receptor (SR)-A.23
Furthermore, ApoE/APN double-KO mice exhibit more severe atherosclerosis progression (accompanied by increased T-lymphocyte accumulation in atherosclerotic lesions) compared with ApoE-single KO mice.24
Additionally, APN knockout (APN-KO) mice demonstrate enhanced neointimal formation after vascular wire injury.25
APN-KO mice fed an atherogenic diet manifest reduced endothelium-dependent vasodilation in aortic ring samples.26
APN-deficiency leads to salt-sensitive hypertension, whereas APN administration reduces increased blood pressure in obese mice.27
APN is detected on the endothelial surface of the normal aorta of wild-type mice; it is present on endothelial cells, synthetic smooth muscle cells, and monocytes adherent to endothelial cells.28
By associating with the tethering molecule T-cadherin, APN accumulates in the vascular wall.29
These in vivo data suggest that APN protects against various obesity-inducible vascular diseases such as atherosclerosis, vascular dysfunction, and hypertension by its interaction with vascular component cells.
In vitro, treatment with APN protein attenuates lipopolysaccharide (LPS)-induced expression of TNF-α in cultured macrophages via inhibition of nuclear factor (NF)-κB signaling (Figure 1). APN treatment also reduces SR-A expression and prevents transformation of macrophages to foam cells30
(Figure 1). APN increases the tissue inhibitor of metalloproteinase-1 via IL-10 expression in human monocyte-derived macrophages, thereby contributing to stabilization of atherosclerotic plaques.31
Additionally, APN promotes the polarization of macrophages to their antiinflammatory M2 state32,33
(Figure 1). Collectively, APN modulates the phenotype and function of macrophages in its target organs, thereby attenuating the vascular inflammatory response.
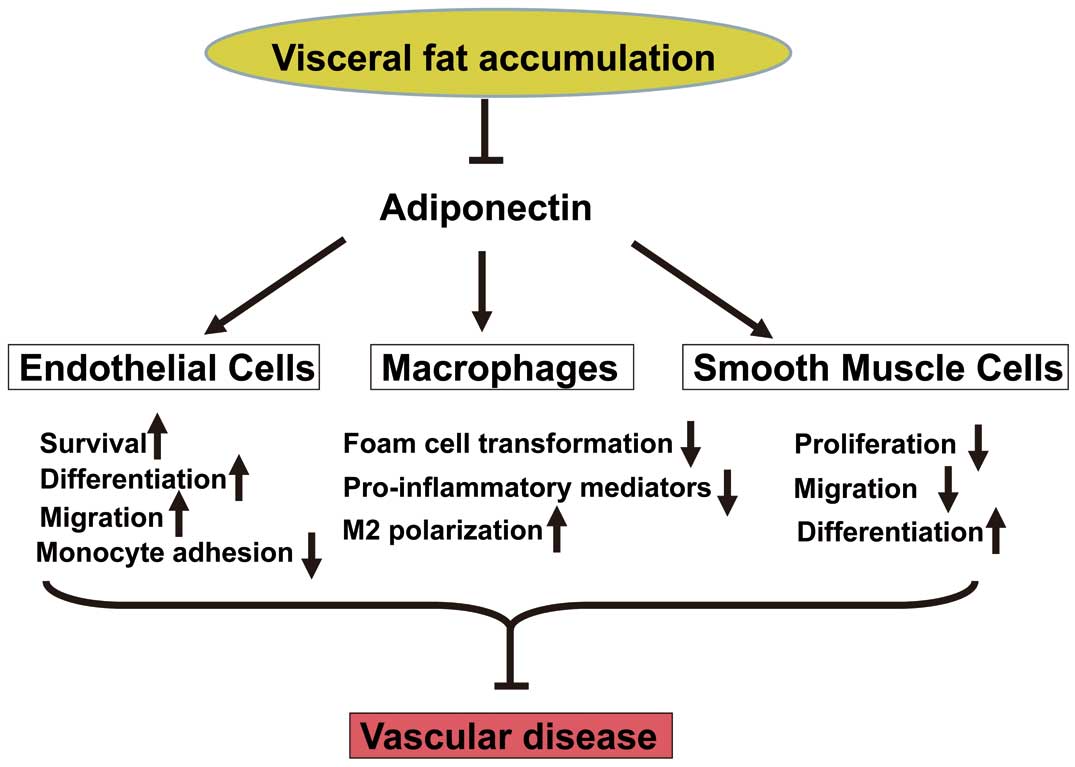
Treatment of endothelial cells with APN promotes differentiation into capillary-like structures and migration, and reduces apoptosis under conditions of serum starvation partly via the AMP-activated protein kinase (AMPK)-eNOS-dependent pathway34,35
(Figure 1). APN also enhances endothelial cell differentiation, migration, and survival through a cyclooxygenase (COX)-2 dependent mechanism.36
APN inhibits monocyte adhesion to TNF-α-stimulated endothelial cells37
(Figure 1). It reduces TNF-α-stimulated expression of vascular cell adhesion molecule (VCAM)-1 in cultured endothelial cells via cyclic AMP (cAMP)/protein kinase A (PKA) signal-mediated inhibition of NF-κB activation.38
APN also inhibits the TNF-α-stimulated inflammatory response in cultured endothelial cells by recruitment and activation of ceramidase in an AdipoR1/caveolin-1 dependent manner.39
Furthermore, APN suppresses reactive oxygen species production stimulated by high glucose in cultured endothelial cells by a cAMP/PKA pathway.40
As outlined above, APN promotes endothelial cell health, attenuates endothelial inflammation, and ultimately inhibits atherogenesis initiation.
Treatment of vascular smooth muscle cells with APN attenuates growth factor induced-proliferation and migration by inhibiting extracellular signal-regulated kinase activation41
(Figure 1). APN reduces cytokine-stimulated expression of heparin-binding EGF-like growth factor (HB-EGF) in endothelial cells.25
Moreover, APN promotes vascular smooth muscle cell differentiation via AMPK-mediated inhibition of mammalian target of rapamycin complex 142
(Figure 1). Collectively, APN prevents atherosclerosis progression by directly affecting the behavior of vascular component cells, including macrophages, endothelial cells, and smooth muscle cells. APN therefore represents a target molecule for the prevention or treatment of atherosclerotic disease.
CTRP Family
In 2004, a highly conserved family of APN paralogs, designated C1q/TNF-related proteins (CTRPs), was reported.43
Each of the 15 known members (CTRP1–CTRP15) consists of 4 distinct domains, comprising an N-terminal signal peptide, a short variable domain, a collagen-like domain, and a C-terminal C1q-like globular domain.43,44
Both CTRPs and APN belong to the C1q/TNF protein superfamily. Investigated for its structural similarity to APN, the CTRP family members exhibit broadly diverse functions.
CTRP9
CTRP9 shares the greatest amino acid overlap with APN in its globular C1q domain, sharing 51% amino acid identity with the globular domain of APN.45
Plasma CTRP9 levels are decreased in rodent models of obesity.46
Interestingly, CTRP9 forms heterotrimers with APN, and appears to share the AdipoR1 in both cultured endothelial cells and cardiomyocytes.45–47
CTRP9 stimulates eNOS phosphorylation and nitric oxide production in endothelial cells via AdipoR1-dependent activation of AMPK, and enhances endothelium-dependent vasorelaxation of aortic rings47
(Figure 2). CTRP9 attenuates inflammatory responses to TNF-α in cultured endothelial cells via AMPK activation48
(Figure 2). CTRP9 also reduces high glucose-inducible oxidative damage in cultured endothelial cells through the AdipoR1-dependent pathway49
(Figure 2). CTRP9 therefore serves a protective function in endothelial cells. Furthermore, systemic delivery of CTRP9 via adenoviral vector expression system reduced neointimal formation in a mouse model of vascular injury. CTRP9 reduces the proliferation of cultured vascular smooth muscle cells via the cAMP/PKA-dependent pathway (Figure 2). Of importance, CTRP9 exerts an antiproliferative effect upon vascular smooth muscle cells, which is independent of AdipoR1 or AMPK. CTRP9 reduces the inflammatory response to oxidized low-density lipoprotein in cultured macrophages in an AMPK-dependent mechanism.50
Furthermore, administration of CTRP9 by the lentiviral vector system reduced the macrophage and lipid content of carotid plaques in ApoE-KO mice, indicating CTRP9 may be an antiatherogenic factor.51
Taken together, it is likely CTRP9 possesses vascular protective effects, but future studies using transgenic animal models are necessary to dissect the role endogenous CTRP9 plays in vascular disease.

Omentin-1 was originally identified as a soluble galactofuranose-binding lectin.52
Omentin-1 is abundantly expressed in human visceral fat, and its circulating concentration is decreased in obese subjects.53
Furthermore, plasma omentin-1 levels negatively correlate with carotid intima media thickness, an established marker of early atherosclerosis in healthy men and type 2 DM patients.54,55
Omentin-1 levels are negatively associated with the prevalence and angiographic severity of CAD in patients with metabolic syndrome.56
Furthermore, serum omentin-1 levels are lower in patients with acute coronary syndrome than in subjects with stable angina pectoris.57
These findings support the utility of omentin-1 as a biomarker of CAD.
In experimental studies, omentin-1 exerted protective functions against vascular diseases. Omentin-1 transgenic (OMT-Tg) mice on an ApoE-KO background (ApoE-KO/OMT-Tg mice) exhibited attenuated atherosclerotic areas in the aortic sinus compared with ApoE-single KO mice.58
ApoE-KO/OMT-Tg mice manifested significantly reduced macrophage infiltration and expression of pro-inflammatory cytokines (such as TNF-α, IL-6 and monocyte chemoattractant protein 1) in the aorta compared with ApoE-KO mice. In human monocyte-derived macrophages, omentin-1 protein significantly reduced lipid droplets and cholesteryl ester contents. Omentin-1 also reduced LPS-induced expression of pro-inflammatory genes (such as TNF-α) in macrophages via Akt activation (Figure 3). Consistently, administration of omentin-1 in ApoE-KO mice decreased atherosclerosis.59
Omentin-1 treatment also enhanced vasodilation in isolated aortic rings of mice in an eNOS-dependent pathway60
(Figure 3). Omentin-1 enhances endothelial cell differentiation into capillary-like structures, and decreases endothelial cell apoptosis in an AMPK/eNOS-dependent fashion (Figure 3). Systemic delivery of omentin-1 promoted blood flow recovery and capillary formation in a mouse model of vascular insufficiency via eNOS signaling.61
Omentin-1 has also inhibited TNF-α-induced expression of intracellular adhesion molecule-1 and VCAM-1 in cultured endothelial cells.62
Thus, omentin-1 exerts salutary effects on endothelial cell function and activation. Furthermore, systemic delivery of omentin-1 attenuated neointimal thickening and vascular cell proliferation in the injured arteries of mice.63
Omentin-1 reduces the growth of vascular smooth muscle cells in an AMPK-dependent mechanism (Figure 3). Transgenic overexpression of omentin-1 reduces neointimal formation in response to injury, accompanied by attenuated inflammatory response in injured vessels. Taken together, the evidence shows that omentin-1 functions as an adipokine with antiatherogenic and antiinflammatory properties, similar to both APN and CTRP9.

Role of Adipokines in Cardiac Diseases
Adiponectin
Clinical studies have demonstrated significantly reduced APN levels in type 2 diabetic patients.64
Furthermore, total plasma APN concentrations are inversely correlated with MI risk.14,15
In 2002, the metabolic effects of an APN-deficient state in mice were reported (delayed clearance of free fatty acid in plasma, decreased muscle fatty acid-transport protein-1 expression, and increased TNF-α concentration).65
In 2005, exacerbated myocardial ischemic injury was reported in APN-KO animals, the first direct evidence that APN was an intrinsic protein protecting the heart against ischemia/reperfusion (I/R) injury.66
Since then, numerous animal experiments have demonstrated that exogenous recombinant APN significantly protects the heart against I/R injury.67,68
Mechanistic studies have been performed to determine the manner by which APN is cardioprotective against I/R injury. APN exerts its metabolic-regulating, antiinflammatory, and vasculoprotective actions largely through AMP-dependent protein kinase (AMPK),69
a serine-threonine kinase that acts as an energy sensor that can be activated by hypoxic stress.70
APN exerts its cardioprotective effect via several important signaling pathways. APN increases nitric oxide (NO) production by activation of the AMPK-Akt-eNOS phosphorylation pathway.35,71
APN is also cardioprotective by inhibiting both oxidative stress (by decreasing gp91phox
overexpression in I/R cardiac tissue) and nitrative stress (by preventing NO overproduction by inhibiting iNOS expression during pathologic states).67
Furthermore, this antioxidative/antinitrative cardioprotective effect of APN is largely AMPK independent,72,73
and largely mediated via PKA-dependent NFκB inhibition.74
Despite being traditionally viewed as an adipocyte-specific endocrine molecule with cardioprotective effects, APN is also expressed locally in adult cardiomyocytes, its production increased by activation of peroxisome proliferator-activated receptor-γ (PPARγ).75–77
Moreover, cardiomyocyte-derived APN is biologically active in protecting against myocardial I/R injury, achieving the protective effect primarily via paracrine/autocrine activation of APN receptors.78
Additionally, in a related fashion, the cardioprotective effects of PPAR-γ agonists are critically dependent on intact APN expression. PPARγ agonists improve insulin sensitivity by upregulating APN expression.79
During pathologic conditions in which plasma APN levels are markedly reduced and/or the APN stimulatory action of PPAR-γ agonists is impaired (e.g., advanced DM or elderly DM patients), PPAR-γ agonists may upregulate superoxide production, culminating in unfavorable cardiovascular outcomes.80
In a recent study investigating the direct mechanisms underlying both APN signaling and APN resistance, GRK2 phosphorylated APN receptor 1 post-MI impaired the cardioprotective functions of APN.81
Inhibition of GRK2-mediated AdipoR1 phosphorylation represents a completely novel manner of restoring APN cardioprotective signaling in the post-MI injury period.
Adiponectin, Heart Disease, and DM
Ischemic heart disease (IHD) is the leading cause of death in patients with DM. Hyperglycemia and hyperlipidemia directly adversely effect ischemic cardiomyocytes, initiating larger infarct size and more severe heart failure after myocardial ischemia.82
Significant research has been done to find the molecular basis linking DM and IHD to identify modalities that may decrease cardiovascular mortality in diabetic patients. Hypoadiponectinemia in type-2 diabetic patients contributes to increased cardiomyocyte death and the patient’s death after MI.83
Significant work elucidating the transmembrane signaling mechanisms responsible for APN’s cardiomyocyte-protective effect has yielded important information. A signaling complex has been demonstrated to be essential for the anti-ischemic/cardioprotective effects of APN (Figure 4).39
It is composed of the structural protein caveolin (Cav), AdipoR1, APPL1 (the most important molecule in APN’s AMPK-dependent signaling), and adenylate cyclase and neutral ceramidase (2 important molecules in APN’s AMPK-independent signaling).84
The interaction between AdipoR1 and caveolin is required for proper APN-mediated signaling. Treatment with high glucose/high lipid disrupts the AdipoR1/Cav signalosome, leading to APN resistance.85

The diabetic condition itself may cause cardiac APN resistance, diminishing the cardioprotective effects of APN. High glucose/high lipid treatments impair APN activation of AMPK-dependent ACC (Acetyl-CoA Carboxylase) signaling, as well as AMPK-independent antioxidative/antinitrative protection, severely inhibiting the cardioprotective effect of APN.84
As mentioned before, the efficacy of recombinant exogenous APN in ameliorating myocardial I/R disease has been demonstrated in multiple animal studies. The practical clinical application of exogenous recombinant APN is limited for multiple reasons such as APN’s complex quaternary structure, rapid turnover, and the high cost of production to generate the amounts required for clinical care. The recently discovered APN receptor agonist, AdipoRon, is a synthetic molecule that is orally active, binds both APN receptors 1 and 2, ameliorates insulin resistance and type 2 DM, and prolongs the shortened lifespan of genetic diabetic mice.86
AdipoRon attenuates post-ischemic cardiac injury and possesses potent antinitrative and antioxidative effects, and may represent a novel therapeutic approach to treating cardiovascular complications from obesity-related disorders such as DM.87
In a recent work by Wang and colleagues, AdipoRon restored deranged autophagic flux in the diabetic state. Hypoadiponectinemia impairs autophagic flux, which contributes to enhanced myocardial I/R injury in the diabetic state. APN receptor activation by AdipoRon restored PRKAA-mediated autophagosome formation and antioxidant-mediated autophagosome clearance, a novel intervention effective against myocardial I/R injury in the diabetic condition (accepted, but yet unpublished data at the time of writing).
CTRP3
CTRP3 (also known as cartducin) is a growth plate secretory protein of primarily cartilaginous origin, but monocytic and osteosarcoma cells also secrete it.88
Initially identified during a search for genes responsible for chondrocyte differentiation induction, CTRP3 regulates APN secretion from adipocytes.89,90
CTRP3 additionally regulates glucose homeostasis,91
and stimulates in vitro endothelial cell proliferation and migration.92
MI decreases the circulating levels of CTRP3. CTRP3 replenishment improves survival and restores cardiac function after MI and attenuates post-ischemic pathologic remodeling (evidenced by echocardiographic and histologic parameters).93
Preventing post-MI CTRP3 inhibition or CTRP3 supplementation may aid in the restoration of cardiac function and mitigate heart failure sequelae (Figure 5).
CTRP6
Expressed in adipose tissue, brain, heart, and placenta, the adipokine CTRP6 regulates metabolism and inflammation.94,95
Additionally, CTRP6 improves cardiac function and ameliorates ventricular remodeling post-MI, protecting against cardiac fibrosis by inhibiting myofibroblast differentiation, extracellular matrix production, and cardiac fibroblast migration via Smad-independent RhoA/MRTF-A (myocardin-related transcription factor-A) signaling (Figure 5).96
CTRP9
As mentioned earlier in this review, of the entire known CTRP family, CTRP9 shares the most homology and amino acid overlap (51%) with APN.45
In comparison with its family members, CTRP9 has been the subject of significantly more published research concerning its CVD implications. Similar to APN, total plasma CTRP9 levels are significantly reduced in diabetic animals.97
However, in dissimilar fashion to APN-deficient mice (which lack phenotypic changes under physiologic conditions), CTRP9-deficient mice are obese, insulin resistant, and exhibit hepatic steatosis with reduced skeletal muscle AMPK activation and mitochondrial content.84
Such results are genetic evidence for a physiologic role of CTRP9, suggesting that CTRP9 may play a significant role in cardiovascular homeostasis. Additionally, CTRP9 primarily circulates in the plasma as the gCTRP9 isoform. This is different from APN, which circulates in the plasma as multimers containing full-length APN.98
The cellular secretome’s important influence on the local microenvironment continues to gain recognition.99
The heart itself produces secretomes (known as “cardiokines”), essential regulators of cardiac physiology and pathology.100
CTRP9, unlike APN, is not a typical adipokine and functions as a cardiokine. The initial CTRP tissue distribution study (using semiquantitative PCR) demonstrated that the heart is the third most abundant CTRP9-expressing organ.45
CTRP9 is the most abundantly expressed adipokine in the heart, exceeding local APN expression more than 100-fold, with local cardiac CTRP9 levels exceeding plasma CTRP9 levels more than 2-fold.97
CTRP9 is a potent cardioprotective molecule capable of attenuating acute I/R injury.46,97,98
Additionally, CTRP9 mitigates pathologic ventricular remodeling post-MI and ischemic heart failure in nondiabetic and diabetic animals, largely via a PKA-dependent pathway.101
In the diabetic condition, TNF-α-initiated oxidative PPARγ suppression downregulates CTRP9, which contributes to exacerbated cardiac injury (Figure 5).97
Perspectives
The concept that adipose tissue is a biologically active organ secreting proteins of significant metabolic consequence is a dramatic paradigm shift in the past 2 decades. In this review, we have discussed the pathophysiologic roles of adipokines in both the vascular and cardiac systems. As our understanding of adipokines continues to grow, we continue to discover novel therapeutic modalities mitigating both vascular and cardiac injury exacerbated by metabolic disorders such as type 2 DM, dyslipidemia, and hypertension.
APN is an adipocyte-derived protein with diverse biologic function in multiple organ systems, including vascular tissue and the heart. Moreover, similar to insulin resistance, APN resistance develops in obesity, DM, and heart failure. However, the molecular mechanisms underlying APN resistance development in different cell types under different pathologic conditions require clarification. The underlying molecular mechanisms responsible for APN receptor downregulation (demonstrated in basic and clinical observations) remain unknown. Successful restoration of APN receptor expression may ameliorate the clinical cardiac and vascular complications of I/R injury. Additionally, AdipoR1 phosphorylation inhibits APN signaling, albeit via unknown molecular mechanisms.81
It is important to determine whether pharmacologic or genetic interventions capable of blocking AdipoR1 phosphorylation will re-establish the cardiometabolic regulatory, antiinflammatory, and cardioprotective effects of APN.
The practical clinical application of exogenous recombinant APN is limited by multiple factors discussed in this review. The promising studies discussed in this review concerning AdipoRon, the first orally active APN receptor agonist, may circumvent the limitations of APN for clinical applicability.
The relatively newly discovered CTRP family consists of adipokines with exciting therapeutic potential. Several CTRPs (including CTRP1, CTRP4, CTRP7, and CTRP9) are expressed in the heart at levels significantly greater than APN. CTRP9 remains the most abundantly expressed adipokine in the adult mouse heart. Cardiac-produced CTRP9 levels significantly exceed those in plasma circulation, suggesting that alteration of cardiac-produced CTRP9 may significantly contribute to cardiac pathophysiology.97
The closest APN paralog, CTRP9, shares functions (such as aging and sex regulation) with APN.45
Evidence supports important metabolic,97
vasoprotective,47
and cardioprotective roles for CTRP9 in diabetic97
and nondiabetic46
mice. Preservation of CTRP9 expression or augmentation of CTRP9-initiated signaling mechanisms may be potential avenues of ameliorating ischemic cardiovascular injury in metabolic disorders.
In the past decade, the comprehension of adipokine biology and signaling in the cardiovascular system has seen great advances. Knowledge gaps remain, and represent opportunities for novel work seeking improved therapeutics against the cardiovascular complications of metabolic diseases.
Acknowledgments
This work was supported by following grants: Grant-in-Aid for Scientific Research (#26293185) and grants from Takeda Science Foundation and the Uehara Memorial Foundation (N.O.), American Diabetes Association 1-14-BS-218 (Y.W.), National Institutes of Health (HL-96686 and HL-123404), and the American Diabetes Association 1-15-BS-122 (X.-L.M.).
Conflict of Interest
Research grants were received from Astellas Pharma Inc., Daiichi Sankyo Co. Ltd. and Takeda Pharma Co. Ltd. (to N.O.). However, the research topics of these grants were not restricted. Lecture fees were received from Kowa Pharmaceutical Co. Ltd. (to N.O.). Molecular Cardiovascular Medicine was endowed by Kowa Pharmaceutical Co. Ltd. (to N.O. and K.O.).
References
- 1.
Yatsuya H, Li Y, Hilawe EH, Ota A, Wang C, Chiang C, et al. Global trend in overweight and obesity and its association with cardiovascular disease incidence. Circ J 2014; 78: 2807–2818.
- 2.
Tsushima H, Yamamoto H, Kitagawa T, Urabe Y, Tatsugami F, Awai K, et al. Association of epicardial and abdominal visceral adipose tissue with coronary atherosclerosis in patients with a coronary artery calcium score of zero. Circ J 2015; 79: 1084–1091.
- 3.
Wong ND. Epidemiological studies of CHD and the evolution of preventive cardiology. Nat Rev Cardiol 2014; 11: 276–289.
- 4.
Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014; 384: 766–781.
- 5.
Matsuzawa Y, Funahashi T, Nakamura T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J Atheroscler Thromb 2011; 18: 629–639.
- 6.
Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011; 11: 85–97.
- 7.
Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011; 145: 341–355.
- 8.
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425–432.
- 9.
Ohashi K, Shibata R, Murohara T, Ouchi N. Role of anti-inflammatory adipokines in obesity-related diseases. Trends Endocrinol Metab 2014; 25: 348–355.
- 10.
Maeda K, Okubo K, Shimomura I, Funahashi T, Matsuzawa Y, Matsubara K. cDNA cloning and expression of a novel adipose specific collagen-like factor, apM1 (AdiPose Most abundant Gene transcript 1). Biochem Biophys Res Commun 1996; 221: 286–289.
- 11.
Yan J, Yang H, Gan L, Sun C. Adiponectin-impaired adipocyte differentiation negatively regulates fat deposition in chicken. J Anim Physiol Anim Nutr (Berl) 2014; 98: 530–537.
- 12.
Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999; 257: 79–83.
- 13.
Ryo M, Nakamura T, Kihara S, Kumada M, Shibazaki S, Takahashi M, et al. Adiponectin as a biomarker of the metabolic syndrome. Circ J 2004; 68: 975–981.
- 14.
Kumada M, Kihara S, Sumitsuji S, Kawamoto T, Matsumoto S, Ouchi N, et al. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol 2003; 23: 85–89.
- 15.
Pischon T, Girman CJ, Hotamisligil GS, Rifai N, Hu FB, Rimm EB. Plasma adiponectin levels and risk of myocardial infarction in men. JAMA 2004; 291: 1730–1737.
- 16.
Schulze MB, Shai I, Rimm EB, Li T, Rifai N, Hu FB. Adiponectin and future coronary heart disease events among men with type 2 diabetes. Diabetes 2005; 54: 534–539.
- 17.
Wang XY, Guo YH, Guo LJ. Association between plasma adiponectin levels and coronary lesion complexity. Beijing Da Xue Xue Bao 2007; 39: 599–602 (in Chinese).
- 18.
Otsuka F, Sugiyama S, Kojima S, Maruyoshi H, Funahashi T, Matsui K, et al. Plasma adiponectin levels are associated with coronary lesion complexity in men with coronary artery disease. J Am Coll Cardiol 2006; 48: 1155–1162.
- 19.
Ai M, Otokozawa S, Asztalos BF, White CC, Cupples LA, Nakajima K, et al. Adiponectin: An independent risk factor for coronary heart disease in men in the Framingham offspring Study. Atherosclerosis 2011; 217: 543–548.
- 20.
Laughlin GA, Barrett-Connor E, May S, Langenberg C. Association of adiponectin with coronary heart disease and mortality: The Rancho Bernardo study. Am J Epidemiol 2007; 165: 164–174.
- 21.
Lindsay RS, Resnick HE, Zhu J, Tun ML, Howard BV, Zhang Y, et al. Adiponectin and coronary heart disease: The Strong Heart Study. Arterioscler Thromb Vasc Biol 2005; 25: e15–e16.
- 22.
Sattar N, Wannamethee G, Sarwar N, Tchernova J, Cherry L, Wallace AM, et al. Adiponectin and coronary heart disease: A prospective study and meta-analysis. Circulation 2006; 114: 623–629.
- 23.
Okamoto Y, Kihara S, Ouchi N, Nishida M, Arita Y, Kumada M, et al. Adiponectin reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 2002; 106: 2767–2770.
- 24.
Okamoto Y, Folco EJ, Minami M, Wara AK, Feinberg MW, Sukhova GK, et al. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ Res 2008; 102: 218–225.
- 25.
Matsuda M, Shimomura I, Sata M, Arita Y, Nishida M, Maeda N, et al. Role of adiponectin in preventing vascular stenosis: The missing link of adipo-vascular axis. J Biol Chem 2002; 277: 37487–37491.
- 26.
Ouchi N, Ohishi M, Kihara S, Funahashi T, Nakamura T, Nagaretani H, et al. Association of hypoadiponectinemia with impaired vasoreactivity. Hypertension 2003; 42: 231–234.
- 27.
Ohashi K, Kihara S, Ouchi N, Kumada M, Fujita K, Hiuge A, et al. Adiponectin replenishment ameliorates obesity-related hypertension. Hypertension 2006; 47: 1108–1116.
- 28.
Mori T, Koyama Y, Maeda N, Nakamura Y, Fujishima Y, Matsuda K, et al. Ultrastructural localization of adiponectin protein in vasculature of normal and atherosclerotic mice. Sci Rep 2014; 4: 4895.
- 29.
Fujishima Y, Maeda N, Matsuda K, Masuda S, Mori T, Fukuda S, et al. Adiponectin association with T-cadherin protects against neointima proliferation and atherosclerosis. FASEB J 2017; 31: 1571–1583.
- 30.
Ouchi N, Kihara S, Arita Y, Nishida M, Matsuyama A, Okamoto Y, et al. Adipocyte-derived plasma protein, adiponectin, suppresses lipid accumulation and class A scavenger receptor expression in human monocyte-derived macrophages. Circulation 2001; 103: 1057–1063.
- 31.
Kumada M, Kihara S, Ouchi N, Kobayashi H, Okamoto Y, Ohashi K, et al. Adiponectin specifically increased tissue inhibitor of metalloproteinase-1 through interleukin-10 expression in human macrophages. Circulation 2004; 109: 2046–2049.
- 32.
Ohashi K, Parker JL, Ouchi N, Higuchi A, Vita JA, Gokce N, et al. Adiponectin promotes macrophage polarization toward an anti-inflammatory phenotype. J Biol Chem 2010; 285: 6153–6160.
- 33.
Lovren F, Pan Y, Quan A, Szmitko PE, Singh KK, Shukla PC, et al. Adiponectin primes human monocytes into alternative anti-inflammatory M2 macrophages. Am J Physiol Heart Circ Physiol 2010; 299: H656–H663.
- 34.
Kobayashi H, Ouchi N, Kihara S, Walsh K, Kumada M, Abe Y, et al. Selective suppression of endothelial cell apoptosis by the high molecular weight form of adiponectin. Circ Res 2004; 94: e27–e31.
- 35.
Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, et al. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem 2004; 279: 1304–1309.
- 36.
Ohashi K, Ouchi N, Sato K, Higuchi A, Ishikawa TO, Herschman HR, et al. Adiponectin promotes revascularization of ischemic muscle through a cyclooxygenase 2-dependent mechanism. Mol Cell Biol 2009; 29: 3487–3499.
- 37.
Ouchi N, Kihara S, Arita Y, Maeda K, Kuriyama H, Okamoto Y, et al. Novel modulator for endothelial adhesion molecules: Adipocyte-derived plasma protein adiponectin. Circulation 1999; 100: 2473–2476.
- 38.
Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation 2000; 102: 1296–1301.
- 39.
Wang Y, Wang X, Lau WB, Yuan Y, Booth D, Li JJ, et al. Adiponectin inhibits tumor necrosis factor-alpha-induced vascular inflammatory response via caveolin-mediated ceramidase recruitment and activation. Circ Res 2014; 114: 792–805.
- 40.
Ouedraogo R, Wu X, Xu SQ, Fuchsel L, Motoshima H, Mahadev K, et al. Adiponectin suppression of high-glucose-induced reactive oxygen species in vascular endothelial cells: Evidence for involvement of a cAMP signaling pathway. Diabetes 2006; 55: 1840–1846.
- 41.
Arita Y, Kihara S, Ouchi N, Maeda K, Kuriyama H, Okamoto Y, et al. Adipocyte-derived plasma protein adiponectin acts as a platelet-derived growth factor-BB-binding protein and regulates growth factor-induced common postreceptor signal in vascular smooth muscle cell. Circulation 2002; 105: 2893–2898.
- 42.
Ding M, Xie Y, Wagner RJ, Jin Y, Carrao AC, Liu LS, et al. Adiponectin induces vascular smooth muscle cell differentiation via repression of mammalian target of rapamycin complex 1 and FoxO4. Arterioscler Thromb Vasc Biol 2011; 31: 1403–1410.
- 43.
Wong GW, Wang J, Hug C, Tsao TS, Lodish HF. A family of Acrp30/adiponectin structural and functional paralogs. Proc Natl Acad Sci USA 2004; 101: 10302–10307.
- 44.
Kishore U, Gaboriaud C, Waters P, Shrive AK, Greenhough TJ, Reid KB, et al. C1q and tumor necrosis factor superfamily: Modularity and versatility. Trends Immunol 2004; 25: 551–561.
- 45.
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Ge G, Spooner E, Hug C, et al. Identification and characterization of CTRP9, a novel secreted glycoprotein, from adipose tissue that reduces serum glucose in mice and forms heterotrimers with adiponectin. FASEB J 2009; 23: 241–258.
- 46.
Kambara T, Ohashi K, Shibata R, Ogura Y, Maruyama S, Enomoto T, et al. CTRP9 protein protects against myocardial injury following ischemia-reperfusion through AMP-activated protein kinase (AMPK)-dependent mechanism. J Biol Chem 2012; 287: 18965–18973.
- 47.
Zheng Q, Yuan Y, Yi W, Lau WB, Wang Y, Wang X, et al. C1q/TNF-related proteins, a family of novel adipokines, induce vascular relaxation through the adiponectin receptor-1/AMPK/eNOS/nitric oxide signaling pathway. Arterioscler Thromb Vasc Biol 2011; 31: 2616–2623.
- 48.
Jung CH, Lee MJ, Kang YM, Lee YL, Seol SM, Yoon HK, et al. C1q/TNF-related protein-9 inhibits cytokine-induced vascular inflammation and leukocyte adhesiveness via AMP-activated protein kinase activation in endothelial cells. Mol Cell Endocrinol 2016; 419: 235–243.
- 49.
Cheng L, Li B, Chen X, Su J, Wang H, Yu S, et al. CTRP9 induces mitochondrial biogenesis and protects high glucose-induced endothelial oxidative damage via AdipoR1-SIRT1-PGC-1alpha activation. Biochem Biophys Res Commun 2016; 477: 685–691.
- 50.
Zhang P, Huang C, Li J, Li T, Guo H, Liu T, et al. Globular CTRP9 inhibits oxLDL-induced inflammatory response in RAW 264.7 macrophages via AMPK activation. Mol Cell Biochem 2016; 417: 67–74.
- 51.
Li J, Zhang P, Li T, Liu Y, Zhu Q, Chen T, et al. CTRP9 enhances carotid plaque stability by reducing pro-inflammatory cytokines in macrophages. Biochem Biophys Res Commun 2015; 458: 890–895.
- 52.
Tsuji S, Uehori J, Matsumoto M, Suzuki Y, Matsuhisa A, Toyoshima K, et al. Human intelectin is a novel soluble lectin that recognizes galactofuranose in carbohydrate chains of bacterial cell wall. J Biol Chem 2001; 276: 23456–23463.
- 53.
de Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes 2007; 56: 1655–1661.
- 54.
Shibata R, Takahashi R, Kataoka Y, Ohashi K, Ikeda N, Kihara S, et al. Association of a fat-derived plasma protein omentin with carotid artery intima-media thickness in apparently healthy men. Hypertens Res 2011; 34: 1309–1312.
- 55.
Yoo HJ, Hwang SY, Hong HC, Choi HY, Yang SJ, Seo JA, et al. Association of circulating omentin-1 level with arterial stiffness and carotid plaque in type 2 diabetes. Cardiovasc Diabetol 2011; 10: 103.
- 56.
Shang FJ, Wang JP, Liu XT, Zheng QS, Xue YS, Wang B, et al. Serum omentin-1 levels are inversely associated with the presence and severity of coronary artery disease in patients with metabolic syndrome. Biomarkers 2011; 16: 657–662.
- 57.
Zhong X, Zhang HY, Tan H, Zhou Y, Liu FL, Chen FQ, et al. Association of serum omentin-1 levels with coronary artery disease. Acta Pharmacol Sin 2011; 32: 873–878.
- 58.
Hiramatsu-Ito M, Shibata R, Ohashi K, Uemura Y, Kanemura N, Kambara T, et al. Omentin attenuates atherosclerotic lesion formation in apolipoprotein E-deficient mice. Cardiovasc Res 2016; 110: 107–117.
- 59.
Watanabe K, Watanabe R, Konii H, Shirai R, Sato K, Matsuyama TA, et al. Counteractive effects of omentin-1 against atherogenesisdagger. Cardiovasc Res 2016; 110: 118–128.
- 60.
Yamawaki H, Tsubaki N, Mukohda M, Okada M, Hara Y. Omentin, a novel adipokine, induces vasodilation in rat isolated blood vessels. Biochem Biophys Res Commun 2010; 393: 668–672.
- 61.
Maruyama S, Shibata R, Kikuchi R, Izumiya Y, Rokutanda T, Araki S, et al. Fat-derived factor omentin stimulates endothelial cell function and ischemia-induced revascularization via endothelial nitric oxide synthase-dependent mechanism. J Biol Chem 2012; 287: 408–417.
- 62.
Zhong X, Li X, Liu F, Tan H, Shang D. Omentin inhibits TNF-alpha-induced expression of adhesion molecules in endothelial cells via ERK/NF-kappaB pathway. Biochem Biophys Res Commun 2012; 425: 401–406.
- 63.
Uemura Y, Shibata R, Kanemura N, Ohashi K, Kambara T, Hiramatsu-Ito M, et al. Adipose-derived protein omentin prevents neointimal formation after arterial injury. FASEB J 2015; 29: 141–151.
- 64.
Hotta K, Funahashi T, Arita Y, Takahashi M, Matsuda M, Okamoto Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000; 20: 1595–1599.
- 65.
Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 2002; 8: 731–737.
- 66.
Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med 2005; 11: 1096–1103.
- 67.
Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, et al. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation 2007; 115: 1408–1416.
- 68.
Goldstein BJ, Scalia RG, Ma XL. Protective vascular and myocardial effects of adiponectin. Nat Clin Pract Cardiovasc Med 2009; 6: 27–35.
- 69.
Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 2005; 26: 439–451.
- 70.
Tian R, Balschi JA. Interaction of insulin and AMPK in the ischemic heart: Another chapter in the book of metabolic therapy? Circ Res 2006; 99: 3–5.
- 71.
Chen H, Montagnani M, Funahashi T, Shimomura I, Quon MJ. Adiponectin stimulates production of nitric oxide in vascular endothelial cells. J Biol Chem 2003; 278: 45021–45026.
- 72.
Wang Y, Gao E, Tao L, Lau WB, Yuan Y, Goldstein BJ, et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation 2009; 119: 835–844.
- 73.
Wang Y, Tao L, Yuan Y, Lau WB, Li R, Lopez BL, et al. Cardioprotective effect of adiponectin is partially mediated by its AMPK-independent antinitrative action. Am J Physiol Endocrinol Metab 2009; 297: E384–E391.
- 74.
Zhang Y, Wang XL, Zhao J, Wang YJ, Lau WB, Yuan YX, et al. Adiponectin inhibits oxidative/nitrative stress during myocardial ischemia and reperfusion via PKA signaling. Am J Physiol Endocrinol Metab 2013; 305: E1436–E1443.
- 75.
Ding G, Qin Q, He N, Francis-David SC, Hou J, Liu J, et al. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J Mol Cell Cardiol 2007; 43: 73–84.
- 76.
Guo Z, Xia Z, Yuen VG, McNeill JH. Cardiac expression of adiponectin and its receptors in streptozotocin-induced diabetic rats. Metabolism 2007; 56: 1363–1371.
- 77.
Pineiro R, Iglesias MJ, Gallego R, Raghay K, Eiras S, Rubio J, et al. Adiponectin is synthesized and secreted by human and murine cardiomyocytes. FEBS Lett 2005; 579: 5163–5169.
- 78.
Wang Y, Lau WB, Gao E, Tao L, Yuan Y, Li R, et al. Cardiomyocyte-derived adiponectin is biologically active in protecting against myocardial ischemia-reperfusion injury. Am J Physiol Endocrinol Metab 2010; 298: E663–E670.
- 79.
Gable DR, Hurel SJ, Humphries SE. Adiponectin and its gene variants as risk factors for insulin resistance, the metabolic syndrome and cardiovascular disease. Atherosclerosis 2006; 188: 231–244.
- 80.
Tao L, Wang Y, Gao E, Zhang H, Yuan Y, Lau WB, et al. Adiponectin: An indispensable molecule in rosiglitazone cardioprotection following myocardial infarction. Circ Res 2010; 106: 409–417.
- 81.
Wang Y, Gao E, Lau WB, Wang Y, Liu G, Li JJ, et al. G-protein-coupled receptor kinase 2-mediated desensitization of adiponectin receptor 1 in failing heart. Circulation 2015; 131: 1392–1404.
- 82.
Norhammar A, Lindback J, Ryden L, Wallentin L, Stenestrand U; Register of Information and Knowledge about Swedish Heart Intensive Care Admission (RIKS-HIA). Improved but still high short- and long-term mortality rates after myocardial infarction in patients with diabetes mellitus: A time-trend report from the Swedish Register of Information and Knowledge about Swedish Heart Intensive Care Admission. Heart 2007; 93: 1577–1583.
- 83.
Basu R, Pajvani UB, Rizza RA, Scherer PE. Selective downregulation of the high molecular weight form of adiponectin in hyperinsulinemia and in type 2 diabetes: Differential regulation from nondiabetic subjects. Diabetes 2007; 56: 2174–2177.
- 84.
Yi W, Sun Y, Gao E, Wei X, Lau WB, Zheng Q, et al. Reduced cardioprotective action of adiponectin in high-fat diet-induced type II diabetic mice and its underlying mechanisms. Antioxid Redox Signal 2011; 15: 1779–1788.
- 85.
Liu GZ, Liang B, Lau WB, Wang Y, Zhao J, Li R, et al. High glucose/High Lipids impair vascular adiponectin function via inhibition of caveolin-1/AdipoR1 signalsome formation. Free Radic Biol Med 2015; 89: 473–485.
- 86.
Fujita H, Asou N, Iwanaga M, Hyo R, Nomura S, Kiyoi H, et al. Role of hematopoietic stem cell transplantation for relapsed acute promyelocytic leukemia: A retrospective analysis of JALSG-APL97. Cancer Sci 2013; 104: 1339–1345.
- 87.
Zhang Y, Zhao J, Li R, Lau WB, Yuan YX, Liang B, et al. AdipoRon, the first orally active adiponectin receptor activator, attenuates postischemic myocardial apoptosis through both AMPK-mediated and AMPK-independent signalings. Am J Physiol Endocrinol Metab 2015; 309: E275–E282.
- 88.
Weigert J, Neumeier M, Schaffler A, Fleck M, Scholmerich J, Schutz C, et al. The adiponectin paralog CORS-26 has anti-inflammatory properties and is produced by human monocytic cells. FEBS Lett 2005; 579: 5565–5570.
- 89.
Akiyama H, Furukawa S, Wakisaka S, Maeda T. CTRP3/cartducin promotes proliferation and migration of endothelial cells. Mol Cell Biochem 2007; 304: 243–248.
- 90.
Wolfing B, Buechler C, Weigert J, Neumeier M, Aslanidis C, Schoelmerich J, et al. Effects of the new C1q/TNF-related protein (CTRP-3) “cartonectin” on the adipocytic secretion of adipokines. Obesity (Silver Spring) 2008; 16: 1481–1486.
- 91.
Peterson JM, Wei Z, Wong GW. C1q/TNF-related protein-3 (CTRP3), a novel adipokine that regulates hepatic glucose output. J Biol Chem 2010; 285: 39691–39701.
- 92.
Kopp A, Bala M, Buechler C, Falk W, Gross P, Neumeier M, et al. C1q/TNF-related protein-3 represents a novel and endogenous lipopolysaccharide antagonist of the adipose tissue. Endocrinology 2010; 151: 5267–5278.
- 93.
Yi W, Sun Y, Yuan Y, Lau WB, Zheng Q, Wang X, et al. C1q/tumor necrosis factor-related protein-3, a newly identified adipokine, is a novel antiapoptotic, proangiogenic, and cardioprotective molecule in the ischemic mouse heart. Circulation 2012; 125: 3159–3169.
- 94.
Wong GW, Krawczyk SA, Kitidis-Mitrokostas C, Revett T, Gimeno R, Lodish HF. Molecular, biochemical and functional characterizations of C1q/TNF family members: Adipose-tissue-selective expression patterns, regulation by PPAR-gamma agonist, cysteine-mediated oligomerizations, combinatorial associations and metabolic functions. Biochem J 2008; 416: 161–177.
- 95.
Xu W, Chen J, Lin J, Liu D, Mo L, Pan W, et al. Exogenous H2S protects H9c2 cardiac cells against high glucose-induced injury and inflammation by inhibiting the activation of the NF-kappaB and IL-1beta pathways. Int J Mol Med 2015; 35: 177–186.
- 96.
Wu D, Lei H, Wang JY, Zhang CL, Feng H, Fu FY, et al. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J Mol Med (Berl) 2015; 93: 1311–1325.
- 97.
Su H, Yuan Y, Wang XM, Lau WB, Wang Y, Wang X, et al. Inhibition of CTRP9, a novel and cardiac-abundantly expressed cell survival molecule, by TNFalpha-initiated oxidative signaling contributes to exacerbated cardiac injury in diabetic mice. Basic Res Cardiol 2013; 108: 315.
- 98.
Yuan Y, Lau WB, Su H, Sun Y, Yi W, Du Y, et al. C1q-TNF-related protein-9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am J Physiol Endocrinol Metab 2015; 308: E891–E898.
- 99.
Ranganath SH, Levy O, Inamdar MS, Karp JM. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell 2012; 10: 244–258.
- 100.
Shimano M, Ouchi N, Walsh K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012; 126: e327–e332.
- 101.
Sun Y, Yi W, Yuan Y, Lau WB, Yi D, Wang X, et al. C1q/tumor necrosis factor-related protein-9, a novel adipocyte-derived cytokine, attenuates adverse remodeling in the ischemic mouse heart via protein kinase A activation. Circulation 2013; 128: S113–S120.

