Ischemic Heart Disease
Alamandine Protects the Heart Against Reperfusion Injury via the MrgD Receptor
2018 Volume 82 Issue 10 Pages 2584-2593

Details
2018 Volume 82 Issue 10 Pages 2584-2593
Background: Alamandine differs from angiotensin-(1–7) in a single N-terminal alanine residue. The aim of this study was to investigate whether alamandine protects the heart against reperfusion injury.
Methods and Results: After euthanizing Sprague-Dawley rats, hearts were perfused with Krebs-Henseleit buffer for a 20-min pre-ischemic period with or without alamandine, followed by 20 min global ischemia and 50 min reperfusion. Alamandine (0.1 mg/kg) improved the postischemic left ventricular developed pressure and ±dP/dt, decreased the infarct size, and decreased the lactate dehydrogenase levels in the effluent. Alamandine increased the coronary flow and the amount of atrial natriuretic peptide (ANP) in the coronary effluent, and it decreased the expression of apoptotic proteins and increased the expression of antioxidative proteins. Pretreatment with the MrgD receptor antagonist or PD123319, but not the angiotensin type 1 receptor antagonist, attenuated the cardioprotective effects of alamandine. A similar cardioprotective effect with alamandine was also observed with high plasma ANP levels in an in vivo study. Alamandine directly stimulated ANP secretion from isolated atria, which was completely blocked by pretreatment with the MrgD receptor antagonist and was partially blocked by PD123319.
Conclusions: These results suggest that the cardioprotective effects of alamandine against I/R injury are, in part, related to the activation of antioxidant and antiapoptotic enzymes via the MrgD receptor.
The renin-angiotensin system (RAS) plays an important role in the regulation of blood pressure and is involved in the pathogenesis of various cardiovascular disorders. Alamandine can be formed from angiotensin A (Ang A) by catalysis of the C-terminal residue, phenylalanine, by angiotensin-converting enzyme 2 (ACE2).1 It can also be synthesized from Ang-(1–7) by decarboxylation of aspartate into alanine.2 Alamandine is an endogenous peptide in rat cardiac tissue and human plasma.1 Due to the high degree of homology in the amino acid sequence and structure between alamandine and Ang-(1–7), their biological actions seem to be closely related.3 However, some dissimilarities have been reported. Ang-(1–7) is a vasodilatory, antifibrotic, and antihypertensive agent primarily via the G-protein-coupled Mas receptor.4–7 Like Ang-(1–7), alamandine also has an antifibrotic effect on isoproterenol-treated rats and an antihypertensive effect on spontaneously hypertensive rats via the Mas-related G-protein-coupled receptor D (MrgD).1,8,9 Both alamandine and Ang-(1–7) inhibited Ang A-mediated vasoconstriction in the rabbit aorta, but there was a clear distinction between the two peptides in Ang II-mediated vasoconstriction in atherogenic blood vessels.8 Ang-(1–7), not alamandine, shows an antiproliferative effect1,4,10 and an antiangiogenic effect by decreasing proangiogenic hormones.1,11 In addition, the opposite effect of alamandine on leptin expression and secretion in adipose tissue to Ang-(1–7) was shown.12 Ang-(1–7)/Mas receptor activation stimulates the secretion of atrial natriuretic peptide (ANP)13 and facilitates cardioprotection of ischemic preconditioning and reperfusion arrhythmias in isolated rat hearts.14,15 However, there is no information regarding whether alamandine improves cardiac dysfunction against ischemia-reperfusion (I/R) injury or is involved in ANP secretion. While the beneficial effects of Ang-(1–7) are well known, few studies with alamandine have been performed. Therefore, the aims of this study were to: (1) determine whether alamandine improves cardiac dysfunction against I/R injury; and (2) define whether its cardioprotective effects are related to apoptotic proteins and ANP.
Male Sprague-Dawley rats, weighing 230–250 g (Dae Han Bio Link, Eumsung, Korea), were housed in temperature-controlled rooms with a 12-h light-dark cycle. Animals were given free access to standard laboratory chow (5L79 Purina rat and mouse 18% chow; Charles River Laboratories Inc., Wilmington, MA, USA) and water. All experimental protocols conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH publication number 85-23, revised 1996) and were approved by our institution.
ChemicalsAlamandine, D-pro7-Ang-(1–7) (D-pro) and ANP were purchased from Bachem Holding AG (Bubendorf, Switzerland). Losartan, PD123319, NG-nitro-L-arginine methyl ester (L-NAME) hydrochloride, and SB239063 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against Bax, Bcl-2, caspase-3, caspase-9, or GAPDH were purchased from Cell Signaling Technology (Denver, MA, USA), and the primary antibody against catalase was purchased from Calbiochem AG (San Diego, CA, USA). Primary antibodies against Mn-superoxide dismutase (Mn-SOD) or heme oxygenase-1 (HO-1) were purchased from Enzo Life Sciences (Plymouth Meeting, PA, USA).
In Vitro I/R Model PreparationLangendorff experiments were performed as previously described.16,17 The hearts were removed from anesthetized rats, and then the ascending aorta was cannulated and immediately perfused on a Langendorff apparatus with Krebs-Henseleit (K-H) solution (119.0 mmol/L NaCl, 4.7 mmol/L KCl, 1.25 mmol/L CaCl2, 1.24 mmol/L MgSO4, 20.1 mmol/L NaHCO3, 1.24 mmol/L KH2PO4, and 11.2 mmol/L glucose) at a constant pressure of 80 mmHg. The perfusate was bubbled with 95% O2 and 5% CO2 and then kept at 37℃. In order to assess contractile functions, a latex balloon was connected to a pressure transducer (ML-820; ADInstruments, Australia) that was inserted into the left ventricular cavity. The left ventricular end-diastolic pressure (LVEDP) was set at 5–10 mmHg by inflating the balloon with physiological saline. The left ventricular developed pressure (LVDP) and the maximum positive and negative peak of the first derivative of LV pressure (+dP/dtmax, −dP/dtmin) were then continuously recorded. Pacing wires were fixed to the right atrium and left ventricle, and the hearts were paced at 300 beats/min (5 Hz, 2 V, 2 ms). Hearts were perfused for a total of 90 min or 160 min for the measurement of hemodynamic changes and western blots, or myocardial infarct size respectively; 20 min preischemic period, 20 min global ischemia, and 50 or 120 min reperfusion at 37℃ (Figure 1A). Coronary flow was measured by collecting the effluent.

Experimental protocols and improvement of ischemia-reperfusion (I/R)-induced left ventricular dysfunction caused by alamandine. (A) Experimental protocols for the Langendorff experiments. (B) Time course for changes in left ventricular developed pressure (LVDP) (a) and left ventricular end-diastolic pressure (LVEDP) (b), +dP/dt (c), and −dP/dt (d) during reperfusion. For each condition, basal preischemic values are shown as absolute values (left axis) and as a percent (%) of the corresponding values taken over the preischemic value (right axis). I/R+Veh: control I/R group; I/R+Ala: I/R in the alamandine-treated group; I/R+D-pro+Ala, I/R+PD+Ala, and I/R+Los+Ala: I/R with cotreatment of alamandine with D-pro, PD123319, and losartan respectively. Values are presented as means±SEM of 8 rats per group. *vs. the I/R+Veh group, P<0.05, **P<0.01.
The following groups were studied (Figure 1A). In Group 1, hearts were perfused with K-H buffer for a 20-min preischemic period, followed by 20 min global ischemia and 50 min reperfusion (control ischemia group, I/R, n=8). In Group 2, rats were pretreated with alamandine (0.1 mg/kg, i.p.) 2 h before euthanasia and hearts were perfused with K-H buffer for a 20-min preischemic period, followed by 20 min global ischemia and 50 min reperfusion (I/R+alamandine, n=8). In Groups 3, 4, and 5, an MrgD receptor antagonist (D-pro, 0.1 mg/kg) (I/R+alamandine+D-pro, n=8),1 AT2 receptor antagonist (PD123319, 0.1 mg/kg) (I/R+alamandine+PD, n=8), or AT1 receptor antagonist (losartan, 0.1 mg/kg) (I/R+alamandine+Los, n=8) was injected i.p. 30 min before alamandine treatment. Hearts were perfused with K-H buffer according to the I/R protocol. Following the experiments, hearts were frozen in liquid nitrogen and stored at −70℃ for western blotting.
In Vivo I/R Model PreparationRats were anesthetized with i.p. ketamine (100 mg/kg) and xylazine (10 mg/kg) and intubated endotracheally and connected to a respirator (Biological Research Apparatus, Varese, Italy; 60–80 strokes/min) for mechanical ventilation at a rate adjusted to maintain normal blood gases.16 A left thoracotomy was performed and the left anterior descending (LAD) coronary artery was ligated using a 6-0 black silk suture. The sham group only underwent a left thoracotomy. Rats were subjected to 45 min of LAD coronary artery ischemia followed by reperfusion. Rats received vehicle or alamandine (1 mg/kg, i.p.) 2 h before surgery. After reperfusion for 1 day, rats were sacrificed by decapitation and blood was collected into prechilled tubes containing heparin.
Preparation of Perfused Beating AtriaIsolated perfused beating atria were prepared as previously described.18,19 In brief, hearts were rapidly excised from rats and a cannula was fixed into the left atria. The cannulated atria were fixed in an organ chamber, perfused with oxygenated 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffered saline (10 mmol/L HEPES, 118 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 25 mnol/L NaHCO3, 10 mmol/L glucose, and 0.1% bovine serum albumin) at 37℃, and then paced at 1.2 Hz (30 V, 0.4 ms). The intra-atrial pressure was recorded using a PowerLab (ADInstruments); pulse pressure (PP) is defined as the difference between the systolic and the diastolic pressure. The pericardial buffer solution containing [3H]-inulin (Amersham Biosciences, Sweden) that was used for measuring the translocation of extracellular fluid (ECF) was also oxygenated through silicone tube coils inside the organ chamber. After stabilization of the ANP secretion and ECF translocation for 80 min, the atrial perfusate was collected at 2-min intervals for 50 min at 4℃.
The following groups were studied. Group 1 comprised the control atria (n=10). After a 10-min control period, atria were high-stretched by increasing the height of the outflow catheter from 5 to 7.5 cmH2O by connecting a 2.5-cm long catheter, and the atrial perfusate was continuously collected. The loaded volume to the atria during diastole was 736 μL.18,19 Group 2 comprised the atria perfused with alamandine. After a 10-min control period, the atria were perfused with various doses of alamandine (1, 3, 10 μmol/L; n=10 for each group) and the height of the outflow catheter was increased from 5 to 7.5 cmH2O. Group 3 comprised the atria perfused with alamandine (3 μmol/L) in the presence of the antagonist for the AT1 receptor (losartan, 10 μmol/L), AT2 receptor (PD123319, 10 μmol/L), or MrgD receptor (D-pro, 10 μmol/L). Atria were perfused with losartan, PD123319, or D-pro 20 min before sample collection, and alamandine (3 μmol/L, n=10 for each group) was simultaneously perfused into the high-stretched atria after a 10-min control period. Group 4 comprised the atria perfused with alamandine (3 μmol/L) in the presence of an inhibitor for nitric oxide synthase (NOS) (L-NAME, 10 μmol/L), phospholipase C (PLC; U73122, 10 μmol/L), or p38 mitogen-activated protein kinase (p38 MAP kinase; SB239063, 10 μmol/L). Atria were perfused with L-NAME, U73122, or SB239063 20 min before sample collection, and alamandine (3 μmol/L, n=10 for each group) was simultaneously perfused into the high-stretched atria after a 10-min control period.
Measurement of ANP Concentration and ECF TranslocationThe ANP concentration in the perfusate was measured using a specific radioimmunoassay (RIA) method as previously described.7 The ANP in the plasma and coronary effluent was extracted using Sep-Pak C18 cartridges, dried, and measured by RIA.16 The intra- and interassay coefficients of variation were 6.3% (n=9) and 7.8% (n=11) respectively.20 The amount of secreted ANP is presented as ng of ANP/g atrial wet weight/min. The equation for calculating the molar concentration of ANP released is as follows:

where 3,063 is the molecular mass of ANP.20
The radioactivity of [3H]-inulin in the perfusates and pericardial buffer solution was measured with a 2 mL Ultima Gold cocktail solution (PerkinElmer, Massachusetts, MA, USA) using a liquid scintillation system (Tri-Card 300 C; Packard, Downers Grove, IL, USA). The ECF translocation was calculated as follows:21,22

In order to determine the area at risk (AAR), coronary arteries are reoccluded at the end of reperfusion and the hearts were perfused with 5 mL of 0.1% Evans Blue dye dissolved in K-H solution. Hearts were then sectioned (2–3 mm) and incubated in phosphate buffer (pH 7.4), which contained 0.75% triphenyltetrazolium chloride (TTC), for 6 min at 37℃ and fixed in 10% formalin. Infarcted areas were determined by planimetry (Analysis Pro ver. 3.2; Soft Imaging System GmbH, Germany). The infarct size was calculated as the percentage of the area at risk (% IS/AAR).17,23 Activities of CK and LDH were measured using a CK activity colorimetric assay kit (S. Milpitas Blvd.; Milpitas, CA, USA) and LDH ELISA kit (Takara Bio, Inc., Otsu, Japan) respectively.
Western Blot AnalysisTotal proteins were extracted from the left ventricle of the heart.16,23 Briefly, the samples were placed in a lysis buffer containing protease inhibitors, homogenized, incubated on ice for 30 min, and then centrifuged at 16,000 g for 15 min. Protein concentration was determined using a modified Bradford assay. And 30 μg of total protein was boiled in a loading buffer for 5 min, loaded onto gradient SDS-PAGE gels, and then transferred to a PVDF membrane. The membrane was incubated with primary antibodies against Bax, Bcl-2, caspase-3, caspase-9, catalase, Mn-SOD, or HO-1. The immunoreactivity of the secondary antibody was detected by chemiluminescence.16,23
Statistical AnalysisAll values are expressed as means±SEM. Differences in hemodynamic variables over time between the control and the treatment groups were assessed using ANOVA followed by Bonferroni’s multiple comparison test (GraphPAD Software, San Diego, CA, USA). The significance level was set at P<0.05.
During the preischemic period, all the measured parameters, such as LVDP, LVEDP, ±dP/dt, and coronary flow, were comparable among groups. There were some individual variations in the basal value, but without significance. Figure 1B shows the time course of several parameters expressed in relative percent change calculated using preischemic control values and experimental values during reperfusion. Postischemic LVDP appeared to recover slowly and reached 42.0±3.6% of the control value 50 min after the start of reperfusion. Alamandine improved LVDP (51.9±2.5%, P<0.01), which was completely blocked by the pretreatment with D-pro (36.4±3.0%) and PD123319 (35.3±1.7%), but not by pretreatment with losartan (52.3±1.8%) (Figure 1B-a). LVEDP abruptly increased after reperfusion and then slowly decreased down to 50.4±3.1% of the control value 50 min after the start of reperfusion. Pretreatment with alamandine improved the postischemic increase in LVEDP significantly (39.1±2.1% of the control value at 50 min) compared to the untreated control hearts. This effect was completely ameliorated by pretreatment with D-pro (49.5±4.5%), but not with PD123319 (41.3±3.6%) or losartan (33.8±5.7%) (Figure 1B-b). Positive and negative dP/dt abruptly decreased after reperfusion and then slowly recovered to 40.2±3.9% and −36.0±3.4% of the control values at 50 min after the start of reperfusion respectively. Alamandine treatment also improved postischemic changes in ±dP/dt to 49.6±1.9% and −43.0±2.1% of the control value respectively. This effect was completely blocked by pretreatment with D-pro or PD123319, but not by losartan (Figure 1B-c,d).
Changes in Coronary Flow, LDH Activity and ANP Concentration in Effluents, and Infarct Size During Perfusion by AlamandineCoronary flow decreased to 48.8% of the control value by postischemic reperfusion and remained constant throughout the experiments (44.9±1.4% at 50 min). Postischemic coronary flow was improved by alamandine treatment (53.2±2.3% at 50 min), and this effect was attenuated by pretreatment with D-pro or PD123319 (Figure 2A-a). The LDH levels during the postischemic period in the alamandine-treated group were significantly lower than in the control I/R group (125.1±6.3% vs. 244.6±60.9% at 50 min), and this effect was attenuated by pretreatment with D-pro (215.7±47.6%) or PD123319 (182.9±43.0%), but not with losartan (132.0±11.4%; Figure 2A-b). Figure 2A-c shows changes in the total amounts of ANP in the coronary effluent during the pre- and postischemic periods with and without alamandine. In the control I/R group, the total amount of ANP was relatively constant before and after ischemia. However, the total amount of ANP in the I/R+alamandine group was higher (306.4±77.5%) 5 min after the start of reperfusion than in the control I/R group (126.9±13.9%; Figure 2A-c). This effect was completely blocked by pretreatment with D-pro (154.1±25.2%) but not with PD123319 and losartan. To evaluate whether the improvement of postischemic cardiac function by alamandine is related to morphological findings, the infarcted areas were measured 120 min after reperfusion. Infarct sizes in the control I/R group were more extensive compared to the I/R+alamandine group (Figure 2B-a). Quantitative analyses of the infarct size showed that alamandine significantly decreased infarct sizes induced by reperfusion injuries from 48.6±2.2% to 32.7±3.7% IS/AAR, and that pretreatment with D-pro or PD123319 attenuated this effect (45.4±1.7% and 41.6±3.1% respectively; Figure 2B-b).

Changes in ischemia-reperfusion (I/R)-induced coronary flow, lactate dehydrogenase (LDH) activity, atrial natriuretic peptide (ANP) amount and ventricular infarct area by alamandine. (A) Time course of changes in coronary flow (a), and LDH activity (b) and the amount of ANP (c) in the coronary effluent during reperfusion. (B) The infarct area is expressed as a percentage of infarct size relative to the area at risk (AAR). Values are presented as means±SEM of 4–8 rats per group. *vs. the I/R+Veh group, P<0.05, **P<0.01. #vs. the I/R+Ala group, P<0.05.
Figure 3 shows the expression of Bax, Bcl-2, caspase-3, and caspase-9 proteins in the left ventricular tissues after reperfusion with and without alamandine. The expressions of Bax, caspase-3, and caspase-9 proteins were lower, and the expression of the Bcl-2 protein was higher in the I/R+alamandine group than in the I/R group. These effects were attenuated by pretreatment with D-pro or PD123319, but not with losartan. Mn-SOD, catalase, and HO-1 proteins were highly expressed in ventricular tissues of the alamandine-treated group, and these effects were partially attenuated by pretreatment with D-pro or PD123319, but not with losartan (Figure 4).

Changes in ischemia-reperfusion (I/R)-induced expression of apoptotic-related proteins by alamandine. Representative western blots (A) and densitometric analysis (B) of Bax, Bcl-2, caspase-3, and caspase-9 protein expression compared to GAPDH expression in ventricles after reperfusion. Values are presented as means±SEM of 8 rats per group. *vs. vehicle-treated group, P<0.05, **P<0.01.

Changes in ischemia-reperfusion (I/R)-induced expressions of antioxidant-related proteins by alamandine. Representative western blots (A) and densitometric analysis (B) of Mn-superoxide dismutase (Mn-SOD), catalase, and heme oxygenase (HO)-1 protein expression compared to GAPDH in ventricles after reperfusion. Values are presented as means±SEM of 8 rats per group. *vs. vehicle-treated group, P<0.05, **P<0.01.
We also tested the cardioprotective effects of alamandine against I/R injury in an in vivo study. Rats receiving alamandine were subjected to 45 min of LAD coronary artery ischemia followed by 1 day of reperfusion. Both LDH and CK activities in the plasma of I/R rats were higher than those in sham rats (Figure 5A,B). Alamandine-treated I/R rats showed lower LDH and CK activities in the plasma compared to vehicle-treated I/R rats. The plasma concentration of ANP in alamandine-treated I/R rats was higher than in vehicle-treated I/R rats (Figure 5C). Quantitative analyses of the infarct size showed that alamandine also significantly decreased infarct sizes induced by reperfusion injuries (Figure 5D).

Changes in the plasma levels of lactate dehydrogenase (LDH), creatine kinase (CK), atrial natriuretic peptide (ANP), and ventricular infarction in ischemia-reperfusion (I/R) rats by alamandine. Values are presented as means±SEM of 5 rats per group. *vs. vehicle-treated group, P<0.05, **P<0.01.
In both in vitro and in vivo studies, the amounts of ANP in the coronary effluent and plasma were higher in the alamandine-treated group than in the control group. In order to define whether alamandine directly stimulates ANP secretion, alamandine was perfused into isolated beating rat atria. Figure 6 shows the effects of alamandine on high-stretch-induced atrial contractility and ANP secretion with time. By high atrial stretch, atrial contractility and the secretion of atrial ANP increased significantly and remained constant throughout the experiments (Figure 6A). When different doses of alamandine were perfused into atria during high atrial stretch, atrial contractility and ANP secretion increased. To quantitatively compare the effects of alamandine on high-stretch-induced atrial parameters, data were recalculated by the percent change from the mean of the control periods (fractions 1 to 5) and the peak periods (fractions 21 to 25). Alamandine, 1, 3, and 10 μmol/L, increased high-stretch-induced ANP secretion from 60.6±4.4% to 54.6±6.5%, 112.4±8.0%, and 158.5±14.4% respectively (Figure 6B-c). The ANP concentration was also increased in a dose-dependent manner by alamandine. Atrial PP and ECF translocation did not change significantly (Figure 6B-a,b).

Effects of alamandine on atrial parameters in isolated perfused atria. (A) Effects of alamandine (1, 3, and 10 μmol/L) on pulse pressure (PP), extracellular fluid (ECF) translocation, atrial natriuretic peptide (ANP) secretion and concentration as a function of time in high-stretched atria. (B) Relative percent changes in atrial parameters by alamandine. Values are presented as means±SEM of 10 rats per group. Transloc, translocation; conc, concentration. *vs. vehicle, P<0.05, **P<0.01.
In order to define Ang receptor subtypes related to alamandine-induced stimulation of ANP secretion, losartan, PD123319, and D-pro were used as pretreatments 20 min before the administration of alamandine or the vehicle. Alamandine (3μmol/L) increased ANP secretion and concentration but did not change atrial contractility and ECF translocation. Pretreatment with losartan did not block alamandine-induced stimulation of ANP secretion and concentration (Figure 7C,7D). However, pretreatment with PD123319 or D-pro blocked alamandine-induced stimulation of ANP secretion and concentration (Figure 7C,7D). Losartan, PD123319, or D-pro alone did not show any significant changes in atrial parameters. To define the downstream signaling pathway of alamandine-induced stimulation of ANP secretion through the MrgD receptor, several inhibitors such as L-NAME, U73122, and SB239063 were perfused 20 min before the administration of alamandine or the vehicle. Pretreatment with L-NAME, U73122, or SB239063 attenuated alamandine-induced stimulation of ANP secretion and concentration (Figure 8C,8D). And L-NAME, U73122, or SB239063 alone did not show any significant changes in atrial parameters.14–16,22
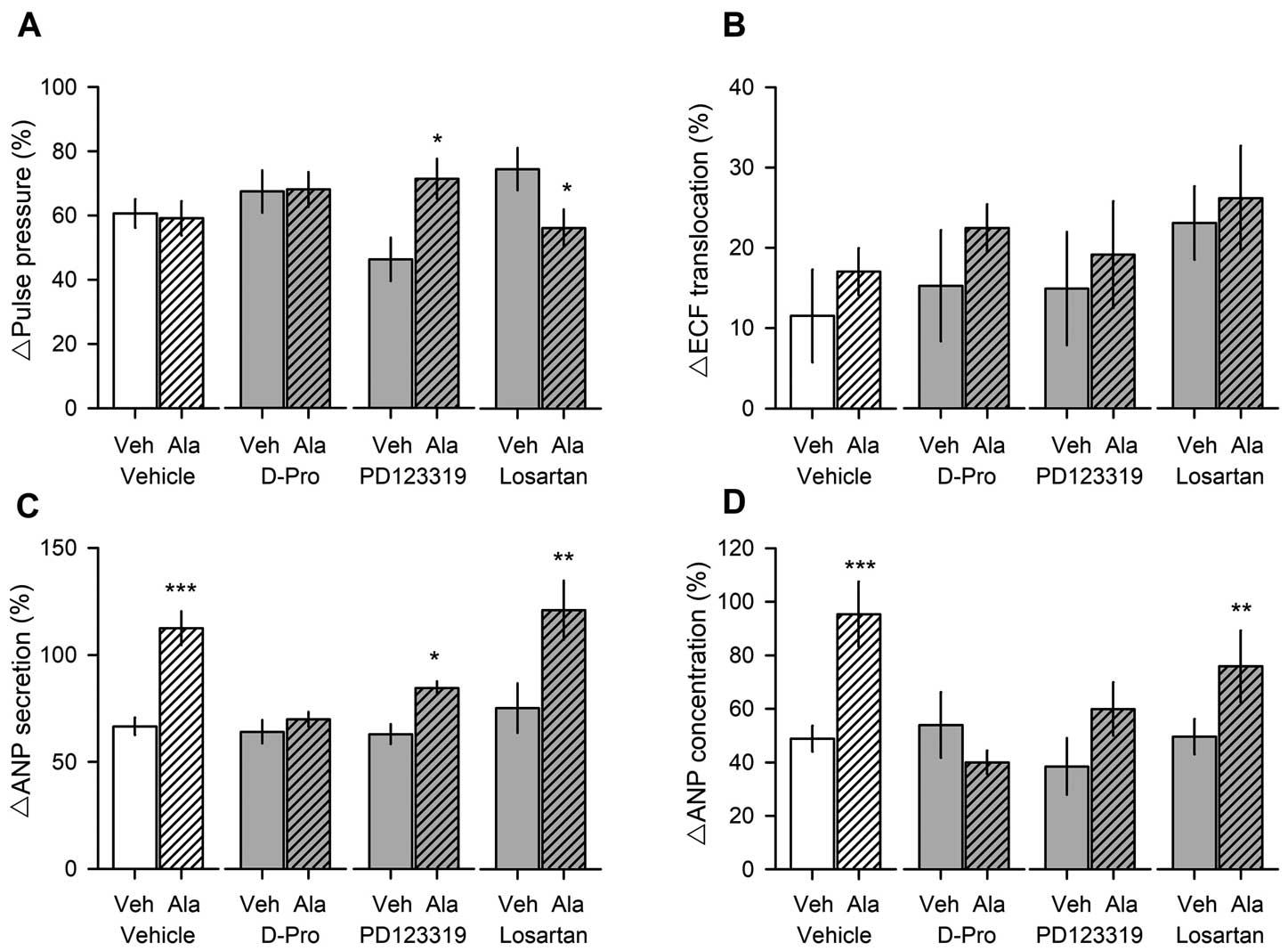
Effects of alamandine on atrial parameters in the presence of receptor antagonists. Twenty minutes before sample collection, atria were pretreated with losartan, PD123319, or D-pro, and then alamandine (3 μmol/L) was perfused into the atria with a high stretch. Values are presented as means±SEM of 10 rats per group. *vs. vehicle, P<0.05, **P<0.01, ***P<0.001.

Effects of alamandine on atrial parameters in the presence of signaling inhibitors. Twenty minutes before sample collection, atria were pretreated with L-NAME, SB239063, or U73122, and then alamandine (3 μmol/L) was perfused into the atria with a high stretch. Values are presented as means±SEM of 10 rats per group. L-NAME, NG-nitro-L-arginine methyl ester. *vs. vehicle, P<0.05, **P<0.01.
The present study demonstrates that alamandine improved reperfusion-induced ventricular hemodynamics with a high ANP level, and that it also attenuated I/R-induced increases in the LDH level and infarct size. Alamandine suppressed the expression of apoptotic proteins and enhanced the expression of antioxidant proteins in ventricles exposed to I/R. These effects were attenuated by cotreatment with the MrgD receptor antagonist or PD123319. Similar cardioprotective effects of alamandine with an elevated plasma ANP level were also observed in an in vivo study. Alamandine directly stimulated ANP secretion via the MrgD receptor. These results suggest that the cardioprotective effects of alamandine against I/R injury may be partially related to activating the antioxidant and antiapoptotic enzymes and the ANP system via the MrgD receptor.
Alamandine is generated from Ang-(1–7) by decarboxylation of aspartate into alanine2 or Ang A by ACE2.1 Alamandine level can be detected in human plasma1 and is increased in patients with chronic renal failure.2 The ratio of alamandine/Ang-(1–7) under normal conditions is expected to be ~0.2, whereas it is ~0.7 during renal failure.2 There are some similarities in the cardiovascular effects between alamandine and Ang-(1–7),6 but they differ in cellular proliferation and angiogenesis.3 However, it is unknown whether alamandine has cardioprotective effects against I/R injury. In order to define this, in vitro and in vivo I/R injury rat models were used. Alamandine improved reperfusion-induced changes in LVDP, LVEDP, ±dP/dt, and coronary flow. Alamandine also decreased the LDH activity and infarct size induced by reperfusion. A similar beneficial effect of alamandine was observed in an in vivo study. These protective effects were attenuated by the MrgD receptor antagonist and PD123319. These results are consistent with previous studies.1,24,25 Losartan primarily blocks the AT1 receptor,26 whereas PD123319 blocks the AT2 receptor and MrgD receptor,1 and D-pro can block the MrgD receptor.1 Therefore, we suggest that alamandine improved reperfusion-induced cardiac dysfunction via the MrgD receptor.1,10,27
It has been reported that postischemic cardiac dysfunction is associated with oxidative stress28,29 and apoptosis.30,31 Several cardioprotective compounds used against reperfusion injury such as urotensin II,23 sulforaphane,17,30 and Ang III16 potentiate antiapoptotic signals related to reactive oxygen species, whereas Ang II and the related STAT1 transcription factor enhanced apoptotic cell death31,32 and limited the recovery of contractile function following I/R injury. Ang-(1–7) alleviates reversible and/or irreversible I/R injury.14 In this study, we also demonstrated that alamandine enhanced the expression of antioxidant enzyme levels such as Mn-SOD, catalase, and HO-1. It also attenuated changes of Bax, caspase-3, and caspase-9 protein expressions in I/R ventricles. These results support earlier reports showing cardioprotection with reduced apoptosis after I/R.16,17,23,33
Interestingly, the amounts of ANP in the coronary effluent of in vitro I/R hearts and in the plasma of in vivo I/R rats were significantly higher in alamandine-treated groups than in the control groups. These results suggest that alamandine may increase ANP secretion. We have previously reported that Ang-(1–7) is cardioprotective,14 which is partially related to the ANP system.7,13 In order to further understand the roles of alamandine in ANP secretion, alamandine was directly perfused into isolated perfused rat atria. Alamandine stimulated ANP secretion, which was attenuated by D-pro or PD123319, but not by losartan. It has been reported that alamandine binds to the MrgD receptor to induce NO production by increasing eNOS activity,1,12 and reduces leptin expression through the PLC/cSrc/p38 MAPK pathway in adipose tissue.12 Our results showing an attenuation of alamandine-induced ANP secretion by pretreatment with L-NAME, U73122, or SB239063 support published studies showing that the alamandine pathway functions via the MrgD receptor and PLC/p38 MAPK/NO-dependent mechanisms.1,3,12 Even though we did not show direct evidence that ANP is responsible for the cardioprotection in this study, ANP has been reported to decrease the cardiac load by decreasing the ECF volume and shifting the intravascular fluid into the interstitial space,34 and improve cardiac functions by vasodilation of the coronary artery in patients with non-ischemic heart failure.35 Therefore, ANP can be one of many possible explanations of the alamandine-induced improvement of cardiac dysfunction against I/R injury.
From these results, we suggest that alamandine has cardioprotective effects against reperfusion injury by activating antiapoptosis via the MrgD receptor. For the consideration with our and other studies,3,27 the alamandine/MrgD receptor can be another beneficial axis similar to the Ang-(1–7)/ACE2/Mas receptor to counterbalance the Ang II/ACE/AT1 receptor.
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (NO 2017-R1A2B-4002214 and NO 2016-R1A6A3A-11930515).