Abstract
Background:
The current state of evaluating patients with peripheral artery disease and more specifically of evaluating medical devices used for peripheral vascular intervention (PVI) remains challenging because of the heterogeneity of the disease process, the multiple physician specialties that perform PVI, the multitude of devices available to treat peripheral artery disease, and the lack of consensus about the best treatment approaches. Because PVI core data elements are not standardized across clinical care, clinical trials, and registries, aggregation of data across different data sources and physician specialties is currently not feasible.
Methods and Results:
Under the auspices of the U.S. Food and Drug Administration’s Medical Device Epidemiology Network initiative—and its PASSION (Predictable and Sustainable Implementation of the National Registries) program, in conjunction with other efforts to align clinical data standards—the Registry Assessment of Peripheral Interventional Devices (RAPID) workgroup was convened. RAPID is a collaborative, multidisciplinary effort to develop a consensus lexicon and to promote interoperability across clinical care, clinical trials, and national and international registries of PVI. The current manuscript presents the initial work from RAPID to standardize clinical data elements and definitions, to establish a framework within electronic health records and health information technology procedural reporting systems, and to implement an informatics-based approach to promote the conduct of pragmatic clinical trials and registry efforts in PVI.
Conclusions:
Ultimately, we hope this work will facilitate and improve device evaluation and surveillance for patients, clinicians, health outcomes researchers, industry, policymakers, and regulators.
Peripheral artery disease (PAD) has a high morbidity and mortality, and the prevalence and costs associated with the diagnosis and management of PAD continually increase.1–3
Despite the increasing burden on the health care system, relatively few comparative effectiveness studies are being performed of patients with PAD in the United States.4
Multiple efforts by professional societies, academia, industry partners, and the U.S. Food and Drug Administration (FDA) have recently been undertaken to better define the nomenclature and classification of how patients with PAD are evaluated and treated in clinical practice.5–8
Despite these efforts, the current state of evaluating patients with PAD and more specifically of evaluating medical devices used for peripheral vascular intervention (PVI) remain challenging because of the heterogeneity of the disease process (eg, symptom classification, anatomic location of disease, severity of disease), the multiple physician specialties that care for these patients and perform PVI, the multitude of devices available to treat PAD globally, the lack of consensus about best treatment approaches, and the absence of a consensus regarding a PAD-PVI lexicon. Furthermore, the medical device technologies for treating PAD have evolved at a face pace with relatively little comparative effectiveness or safety data to assess device performance and patient outcomes or to inform clinical guidelines. Finally, in real-world practice, many approved or cleared devices are used “off-label” in anatomic locations or clinical conditions that have not been rigorously studied.
As PVI core data elements are not standardized across clinical care, clinical trials, or registries, aggregation of data across different data sources and physician specialties is currently not feasible. Under the auspices of the FDA Medical Device Epidemiology Network (MDEpiNet) initiative and its PASSION (Predictable and Sustainable Implementation of the National Registries) program in conjunction with other efforts to align clinical data standards,8
the Registry Assessment of Peripheral Interventional Devices (RAPID) workgroup was convened to develop a consensus lexicon to promote interoperability across clinical care, clinical trials, and national and international registries of PVI.
Goals of RAPID
In 2010, the FDA launched the MDEpiNet initiative with the goal of advancing national and international infrastructure, novel methodologies, and partnerships for evaluation of medical devices throughout the product’s life cycle. In 2014, MDEpiNet matured to a public-private partnership that contributed to the construction and demonstration of a National Evaluation System for Health Technology. Key components being addressed by MDEpiNet include the need for integrated infrastructure for real-world data, the development of analytic methodologies, and the conduct of demonstration studies that generate high-quality data at lower overall cost than in current approaches that support regulatory and best practice decisions throughout the total device product life cycle.9–11
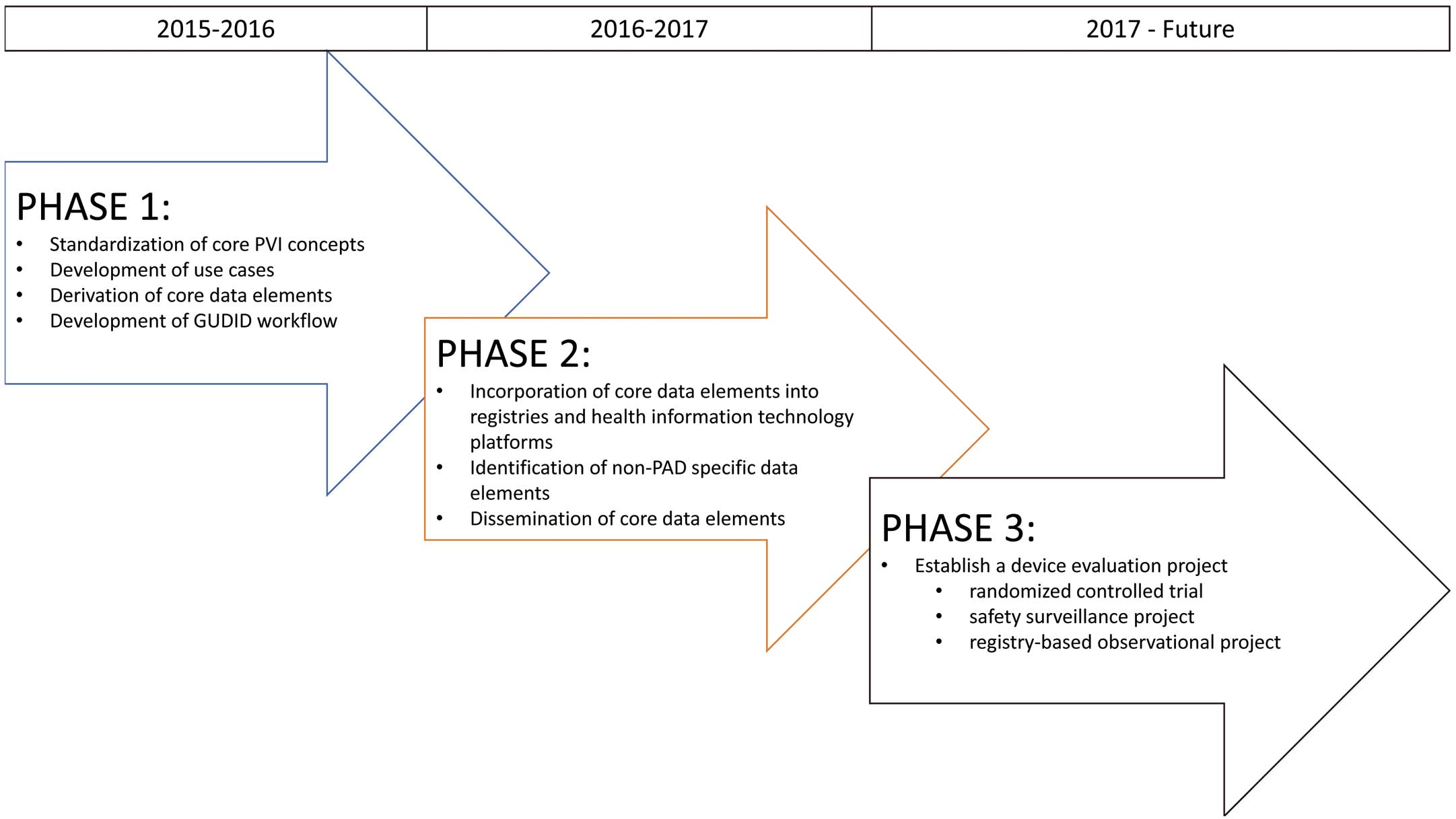
The RAPID project was conceived by MDEpiNet to address the nomenclature and infrastructure gaps of the PVI space. The goals of RAPID are shown in the Fig and include the following:
• Phase 1. Standardization of core PVI clinical concepts and development of the concepts into common data elements to allow utilization of data from multiple sources in both premarket and postmarket assessments of PAD interventional devices.
• Phase 2. Incorporation of the standardized data elements into two major existing registries, the Society for Vascular Surgery Vascular Quality Initiative (SVS VQI) and the American College of Cardiology National Cardiovascular Disease Registry Peripheral Vascular Intervention (ACC NCDR PVI), as well as into the procedure documentation systems of at least one health information technology (HIT) vendor.
• Phase 3. Establishment of a device evaluation project (eg, randomized clinical trial, device surveillance registry) using these data sources to demonstrate the benefit of interoperable device data collection for academia, clinicians, industry, payers, U.S. Centers for Medicare and Medicaid Services, FDA, and other global regulatory agencies.
With these considerations in mind, RAPID is aligned with the current FDA Center for Devices and Radiological Health initiative to improve the total product life cycle assessment of medical devices and to enhance the use of real-world data for regulatory decisions.9,10
RAPID Collaborators
RAPID was developed by forming a multi-stakeholder collaboration of academic members from various specialties that perform PVI, professional societies and their registries, U.S. federal agencies, international partners, HIT vendors, clinical research organizations, and medical device manufacturers. These collaborators are listed in
Table S1. Outreach continues to expand the list of active participants who will add value to the RAPID project.
Operational Aspects of RAPID
The initial in-person meeting that formalized RAPID occurred on June 5, 2015, at the FDA White Oak Campus (Figure). The Duke Clinical Research Institute, which serves as the MDEpiNet Coordinating Center, was selected to provide administrative and informatics support for RAPID. Three workgroups were organized: a clinical group charged with identifying the core clinical concepts; an informatics group responsible for developing the clinical concepts into data elements; and a Global Unique Device Identifier Database (GUDID) workgroup to facilitate the use of device data in the FDA GUDID reference database. At this initial meeting, industry partners committed to financial support of RAPID through unrestricted research grants that were held at the Duke Clinical Research Institute.
Weekly teleconferences and a face-to-face meeting during the MDEpiNet annual meeting (October 1, 2015, Silver Springs, Maryland) were used to ensure participation by stakeholders and to organize the various working groups and their lead participants. On November 6, 2015, all RAPID stakeholders met at the American College of Cardiology Heart House in Washington, D.C., for a kickoff meeting to assign tasks and to introduce the clinical and informatics working group approaches that had begun at the Duke Clinical Research Institute. Following the kickoff meeting, the three working groups convened teleconferences every 1 to 2 weeks, and a near-final product for phase 1 was delivered to the entire group for review in Silver Springs, Maryland, on April 13, 2016. The content was then further edited and improved, with release of the artifacts to the general public occurring on September 14, 2016, on www.mdepinet.org.
Clinical Use Cases
To guide the clinical, informatics, and GUDID working groups, three potential clinical use cases were articulated as reference points and boundaries for the scope of the work. These included a premarket approval use case, a postmarket approval use case, and a randomized clinical trial use case. These use cases exemplify, at a high level, clinical care and regulatory contexts in which the core data elements
could
be successfully leveraged but do not represent an existing registry or project under development. Whereas many aspects of the clinical use cases were hypothetical (rather than factual), this exercise allowed working group members to think broadly about the PAD population being studied. Details about these use cases are in
Table.
Table.
Registry Assessment of Peripheral Interventional Devices (RAPID) Clinical Use Cases
Clinical use case
characteristics |
Premarket approval |
Postmarket approval or
surveillance |
Randomized controlled trial |
| Project summary |
Registry-embedded trial or pragmatic
clinical trial |
Longitudinal study embedded within
registry or EHR system |
Registry-embedded trial or pragmatic
clinical trial |
| Goal |
Comparative effectiveness and safety
assessment of new device technology |
Longitudinal effectiveness and safety
assessment of approved device in
clinical practice |
Comparative effectiveness and safety
assessment of pharmacologic therapy
after PVI |
| Comparators |
Device X (new technology) vs device
Y (currently approved technology) or
objective performance criteria |
None; all patients receive treatment
with an approved device (device Z) |
1 month vs 6 months of dual antiplatelet
therapy (aspirin+P2Y12 inhibitor) |
| Study design |
Single-blind, 1:1 randomized
controlled trial |
Single-arm, nonrandomized study |
Double-blind, 1:1 randomized
controlled trial |
| Study population |
All symptomatic patients with PAD
undergoing PVI for intermittent
claudication and critical limb ischemia;
patients must have disease suitable
for use of device X and device Y |
All symptomatic patients with PAD
undergoing PVI for intermittent
claudication and critical limb ischemia |
All symptomatic patients with PAD
undergoing PVI for intermittent
claudication and critical limb ischemia |
| Enrollment |
Example: All symptomatic patients
(Rutherford 2–6) undergoing planned
infrainguinal endovascular
revascularization |
Example: All symptomatic PAD
patients who have undergone
successful PVI with device Z |
Example: All symptomatic PAD
patients who have undergone
successful PVI |
| Follow-up |
1 month, 3 months, 6 months, 12
months |
1 month, 3 months, 6 months, 12
months |
1 month, 3 months, 6 months, 12
months |
| Clinical outcomes |
Acute, intermediate, and long‐term
cardiovascular end points; limb end
points; functional measures; and
quality of life measures |
Acute, intermediate, and long‐term
cardiovascular end points; limb end
points; functional measures; and
quality of life measures |
Acute, intermediate, and long‐term
cardiovascular end points; limb end
points; functional measures; and
quality of life measures |
EHR, electronic health record; PAD, peripheral artery disease; PVI, peripheral vascular intervention.
Core Data Element Selection
The Duke Clinical Research Institute informatics group evaluated, aggregated, and anonymized data elements from three U.S. professional society registry data forms, three international registry data forms (Germany, Japan, Australia), and seven device company case report forms. A total of 3904 data elements were abstracted, and 2021 of these were deemed specific to PAD; the remaining data elements were deemed to be not specific to PAD. These 2021 PAD-specific data elements were then shared with the clinical working group to derive the core data elements for RAPID. The remaining general data elements, such as the patient’s age, sex, and medical comorbidities and other general concepts, were not included because these elements will be collected as discrete data by electronic health record (EHR) and other clinical documentation systems and have also been defined and specified in many nationally used data models.12,13
Domains and Clinical Concepts
The 2021 PAD-specific data elements were grouped into five domains: condition, test, procedure, device, and event/outcome. Through a series of interactive web conferences and face-to-face meetings during several months, a multi-stakeholder clinical working group reached consensus on the selection of exactly 100 key data elements (shown in
Table S2), prioritized on the basis of their availability in existing data sources, applicability to most PVI devices, and applicability for use across the total product life cycle of PVI interventional devices (eg, early-phase clinical trials, pivotal clinical trials, device surveillance studies). These data elements can be applied at multiple time points during data collection for a clinical trial or registry (eg, at baseline, during the index PVI, during follow-up), and although it is expected that a large proportion of the data extraction will be accomplished using these core data elements, specific clinical trials and device evaluation studies may need to collect an additional set of variables, depending on how a specific device functions.
Informatics Workflow
After the selection of this minimal set of core data elements, the informatics working group convened to develop the metadata (ie, data that serve to provide context or definition about other data) sufficient for the clinical concepts to be implemented as data elements in electronic HIT systems. The informatics working group followed a permissive approach to be as compatible as possible with the data models currently in use today (including Observational Medical Outcomes Partnership [OMOP], Sentinel, Patient-Centered Outcomes Research Network [PCORnet], Informatics for Integrating Biology & the Bedside [i2b2], Open Clinical Information Modeling Initiative [OpenCIMI], and others) and to support workflows such as the following:
• Federated and distributed research networks (eg, Sentinel, PCORnet)12,14
• Electronic data capture systems (clinical trial-specific databases)
• Multisource, multidata type research (or device evaluation) repositories
• Point-of-care clinical, procedural, and EHR systems with downstream flows to quality improvement registries and other systems used for outcomes analysis
• Data collection systems not set up to receive or to exchange data with EHR systems
In other words, the identification of the core clinical lexicon anticipates clinical care processes that capture data integrated into clinical workflows and the interoperable use of the corresponding data element representations by clinical registries and in the aggregation of rich data sets for analytic purposes. Future work is under way to determine how to procure the data elements from a specific EHR system or electronic data capture system. As this process is developed and tested, it will be imperative to validate the veracity and security of the data extraction process. Ultimately, demonstration projects will be performed that test the value of using the RAPID common data elements for PAD-specific projects, and much of the future workflow (eg, where the data are stored, access to data) will be determined by the specific project (eg, randomized controlled trial, clinical registry, safety surveillance project).
GUDID Workflow
Another novel unmet medical need addressed in this RAPID program is that electronic HITs do not currently have a consistent, efficient mechanism to capture and to link device identification information to the patient at the point of care or across data sources. In 2013, the FDA established a unique device identifier (UDI) system to adequately identify medical devices through their distribution and use. UDI requirements are being phased in over several years on the basis of the risk of the device. When the system is fully implemented, the label of most devices will include a UDI in both human- and machine-readable forms. Device labelers must also submit certain information about each device to the FDA’s GUDID. At the time of writing, mechanisms to integrate UDI into workflows or to associate implantable device UDI with patients over time and across venues are not widely available. In the future, the ability to link device UDI with the patient will facilitate data entry into registries such as the ACC NCDR PVI and SVS VQI that began recording device information in 2016.
The RAPID GUDID working group was formed to explore and to understand opportunities for incorporating UDI and the FDA GUDID reference database into the core clinical data for RAPID. The GUDID integration working group sought to promote knowledge sharing across the FDA, device manufacturers, clinicians, and registries related to using the device identifier of the UDI to extract data from GUDID and to facilitate the transition from existing nonstandardized device identification in device registries to one based on the device identifier portion of the UDI.
The evaluation of GUDID data spawned the creation of additional initiatives. These address augmenting the GUDID system with additional clinically useful device attributes; understanding and improving device categorization; and improving the quality of data in GUDID by decreasing discrepancies, inconsistencies, and errors in the entries into the GUDID system for clinically relevant values, such as size and diameter of the device. Additional workgroups formed under the auspices of MDEpiNet and the Association for Healthcare and Resource Management Learning UDI community will provide the mechanisms to delineate and to suggest approaches to resolving these issues.
Data Elements and Definitions
The RAPID clinical data elements are listed in
Table S2, and a few points warrant highlighting. In addition to deriving core data elements from existing sources, the clinical workgroup provided additional specificity and definition for several key variables. A modified Rutherford classification for PAD symptoms was developed to increase specificity regarding delineation of the activity limitations for patients with mild, moderate, and severe claudication. Whereas many registries and prior studies described patients with critical limb ischemia using a Rutherford classification, the clinical working group recommended use of the now validated Wound, Ischemia, and foot Infection classification system for grading the severity of critical limb ischemia in the RAPID core data elements.7
Definitions for lesion length, degree of lesion stenosis, and degree of vascular calcification were actively debated among stakeholders, as these data elements have been inconsistently documented across typical data sources. A consensus was reached as to how best to classify these in RAPID.
A decision was made to include each target lesion treated during a procedure, to assign a lesion number, and to track the outcome of this target lesion longitudinally. In addition, it was decided to include each access site location, sheath size, and guidance used for access (eg, palpation only, fluoroscopy, ultrasound, open exposure). The included events and outcomes pertain to periprocedural events and the management of these events; however, more generic end points, such as myocardial infarction, stroke, and death, were not included in the RAPID core data elements that focus on PVI-specific outcomes. These events may require identification and longitudinal tracking outside of the EHR and HIT procedural reporting systems (eg, administrative claims data, National Death Index).
Medications of interest that were included in the RAPID core data elements include the following:
• Antithrombotic medications (eg, aspirin, P2Y12
receptor inhibitors, unfractionated heparin, low-molecular-weight heparin, glycoprotein IIb/IIIa inhibitors, bivalirudin, oral anticoagulants)
• Other cardioprotective medications (eg, angiotensin-converting enzyme inhibitors, angiotensin receptor blockers, hydroxymethylglutaryl-coenzyme A reductase inhibitors [statins])
• Cilostazol, pentoxifylline
RAPID Phase 2
The second phase of RAPID involves the dissemination and implementation of the RAPID core data elements. The priorities of phase 2 include
• Identification of non-PVI-specific data elements from other data models
• Introduction of the RAPID clinical data elements into the OpenCIMI data modeling process (ie, develop the formal informatics of the RAPID clinical data elements)
• Implementation of the RAPID clinical data elements in HIT procedural documentation systems (eg, the Veterans Affairs system procedure documentation module, Clinical Assessment, Reporting, and Tracking System for Cardiac Catheterization Laboratories)
• Implementation of the RAPID clinical data elements in three different U.S. professional society registries
◦ ACC NCDR PVI
◦ Society of Interventional Radiology National Interventional Radiology Quality Registry
◦ SVS VQI
• Development of an augmented UDI data set of clinical data for all classes of PAD interventional devices
• Dissemination of RAPID clinical data elements along with prototype history and physical examination data, structured reports, and structured reporting approaches for PVI (to all HIT procedure documentation systems)
• Continued work on an augmented unique device identification database
• Recommendations for structured reporting systems and documents
RAPID Phase 3
The third phase of RAPID will be able to demonstrate a significantly improved approach to PVI device research in the United States and internationally. For example, RAPID will be able to facilitate the International Consortium of Vascular Registries effort to harmonize the SVS VQI and European Vascunet registries. The SVS VQI and ACC NCDR PVI steering committees have already approved the addition and consolidation of RAPID data elements into their individual workflow. Although differences exist in how data are entered into each registry (ie, real-time data entry at the point of care vs data abstraction after a care episode), the standardization and use of core data elements, informatics workflow, and GUDID implementation from RAPID make the possibilities almost limitless. However, as in all projects, the approach to phase 3 requires intense focus and strategic planning. Concepts from the use cases (Table) or alternative research proposals will be used to design a study or studies to be performed, but it will be imperative for government, industry, or other funding agencies to operationalize the data elements and data collection approach. Similar pragmatic trials or surveillance programs, such as studies using the RAPID core data elements and infrastructure, will have the potential to be more efficient, more comprehensive, and less costly than traditional methods of data collection.15
Society registries have demonstrated the feasibility of such standardized approaches (eg, American College of Cardiology National Cardiovascular Data Registry Transcatheter Valve Therapy registry), but the RAPID approach will be the first comprehensive effort in the complex PAD space.
Conclusions
RAPID is a demonstration project for the MDEpiNet public-private partnership, and as such, it is designed to improve device evaluation and surveillance for patients, clinicians, researchers, industry, policymakers, and regulators. Similarly, RAPID is one project in a series initiated to advance and to demonstrate the interoperable flow of data and information across electronic health information systems as a precursor to the National Evaluation System for Health Technology articulated by Shuren and Califf.9
RAPID will serve as a collaborative, multidisciplinary model for the standardization of clinical data elements and definitions, establishment of a framework within EHR and HIT procedural reporting systems, and implementation of an informatics-based approach to promote the conduct of pragmatic clinical trials nested in the national and international registries of PVI. In doing so, RAPID has laid the groundwork to promote more efficient data collection and more streamlined clinical trial processes. As a demonstration project under the MDEpiNet’s PASSION program, RAPID is designed to maximize the efficiencies of clinical trials nested in the registries. These efforts have just begun, and it will require continued focus and collaboration to complete the second and third phases of work.
Registry Assessment of Peripheral Interventional Devices (RAPID) working group members included representatives
from academic research groups from the United States, Japan, and Europe; representatives from vascular medicine,
vascular surgery, interventional radiology, and cardiology; industry representatives; the U.S. Food and Drug
Administration; and the Pharmaceuticals and Medical Devices Agency regulatory authorities from Japan. |
Author Contributions
Conception and Design: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Analysis and Interpretation: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Data Collection: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Writing the Article: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Critical Revision of the Article: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Final Approval of the Article: W.J., M.K., P.M., R.W., A.H., M.W., J.T., J.M.D., M.M., T.R., R.F., R.L., N.H., H.A., D.B., M.J., T.T., J.S., M.Z., R.T., J.C.
Statistical Analysis: Not applicable
Obtained Funding: W.J., M.K., P.M., R.W., R.T., J.C.
Overall Responsibility: W.J.
Grants
Unrestricted research grants were provided from industry sponsors (Abbott Vascular, Bard Peripheral Vascular, Boston Scientific Corp, Medtronic, Cardiovascular Systems Inc, Cook Medical, Terumo Medical Corp, Volcano Corp, and W. L. Gore) listed to the Duke Clinical Research Institute to cover the costs of meeting expenses and partially fund the informatics work involved in phase 1.
Author Conflict of Interest
W.S.J. has received research grants from the Agency for Healthcare Research and Quality, AstraZeneca, American Heart Association, Bristol-Myers Squibb, Doris Duke Charitable Foundation, and Patient-Centered Outcomes Research Institute; honorarium/other from American College of Radiology, Bayer, Bristol-Myers Squibb, Daiichi Sankyo, and Janssen Pharmaceuticals. M.W.K. has received funding from Abbott Vascular, ACIST Medical Systems, AngelMed Inc, Bellerophon Therapeutics, Boston Scientific, Epi-EP, Medtronic Inc, Micelle Technologies Inc, OrbusNeich, St. Jude Medical Inc, Terumo Corp, and Philips Volcano Corp. R.W.W., A.H.H., and M.F.W. received unrestricted research grants during the conduct of the study. J.E.T. received grants from the U.S. Food and Drug Administration during the conduct of the study; personal fees from Medstreaming Inc; personal fees from Cardiovascular Systems Inc outside the submitted work; American College of Cardiology National Cardiovascular Data Registry Management Board member, Informatics and Health Information Technology Task Force chair. M.R.J. received nonfinancial support from Abbott Vascular, Boston Scientific, Cordis Corporation, and Medtronic Vascular; other from PQ Bypass; other from VIVA Physicians, a 501(c)(3) not-for-profit education and research organization; other from Vascular Therapies; other from Embolitech; and personal fees from Philips Volcano Corp, outside the submitted work. J.A.S. is an employee of Bard Peripheral Vascular, Inc. M.J.Z. is an employee of Abbott Vascular. R.B.T. is a prior employee of Cardiovascular Systems, Inc. J.L.C. receives financial support from Medstreaming.
Disclosures
The editors and reviewers of this article have no relevant financial relationships to disclose per the JVS policy that requires reviewers to decline review of any manuscript for which they may have a conflict of interest.
Supplementary Files
Supplementary File 1
Table S1.
Registry Assessment of Peripheral Interventional Devices (RAPID) collaborators
Table S2.
Registry Assessment of Peripheral Interventional Devices (RAPID) data elements
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-17-1156
References
- 1.
Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation 2015; 131: e29–e322.
- 2.
Rooke TW, Hirsch AT, Misra S, Sidawy AN, Beckman JA, Findeiss LK, et al. 2011 ACCF/AHA Focused Update of the Guideline for the Management of Patients With Peripheral Artery Disease (updating the 2005 guideline): A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 58: 2020–2045.
- 3.
European Stroke Organisation, Tendera M, Aboyans V, Bartelink ML, Baumgartner I, Clément D, et al. ESC Guidelines on the diagnosis and treatment of peripheral artery diseases: Document covering atherosclerotic disease of extracranial carotid and vertebral, mesenteric, renal, upper and lower extremity arteries: The Task Force on the Diagnosis and Treatment of Peripheral Artery Diseases of the European Society of Cardiology (ESC). Eur Heart J 2011; 32: 2851–2906.
- 4.
Subherwal S, Patel MR, Chiswell K, Tidemann-Miller BA, Jones WS, Conte MS, et al. Clinical trials in peripheral vascular disease: Pipeline and trial designs: An evaluation of the ClinicalTrials.gov database. Circulation 2014; 130: 1812–1819.
- 5.
Diehm N, Pattynama PM, Jaff MR, Cremonesi A, Becker GJ, Hopkins LN, et al. Clinical endpoints in peripheral endovascular revascularization trials: A case for standardized definitions. Eur J Vasc Endovasc Surg 2008; 36: 409–419.
- 6.
Patel MR, Conte MS, Cutlip DE, Dib N, Geraghty P, Gray W, et al. Evaluation and treatment of patients with lower extremity peripheral artery disease: Consensus definitions from Peripheral Academic Research Consortium (PARC). J Am Coll Cardiol 2015; 65: 931–941.
- 7.
Mills JL Sr, Conte MS, Armstrong DG, Pomposelli FB, Schanzer A, Sidawy AN, et al. The Society for Vascular Surgery Lower Extremity Threatened Limb Classification System: Risk stratification based on wound, ischemia, and foot infection (WIfI). J Vasc Surg 2014; 59: 220–234.e1–e2.
- 8.
Hicks KA, Tcheng JE, Bozkurt B, Chaitman BR, Cutlip DE, Farb A, et al. 2014 ACC/AHA key data elements and definitions for cardiovascular endpoint events in clinical trials: A report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Cardiovascular Endpoints Data Standards). Circulation 2015; 132: 302–361.
- 9.
Shuren J, Califf RM. Need for a national evaluation system for health technology. JAMA 2016; 316: 1153–1154.
- 10.
U.S. Food and Drug Administration. Center for Devices and Radiological Health Reports. Strengthening our national system for medical device postmarket surveillance.
September 2012. Available at: http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDRH/CDRHReports/UCM301924.pdf (accessed January 31, 2017).
- 11.
U.S. Food and Drug Administration. Center for Devices and Radiological Health Reports. Strengthening our national system for medical device postmarket surveillance: Update and next steps. April 2013. Available at: http://www.fda.gov/downloads/MedicalDevices/Safety/CDRHPostmarket/UCM348845.pdf (accessed January 31, 2017).
- 12.
Ohno-Machado L. Technical report. Standards in the use of collaborative or distributed data networks in patient centered outcomes research. March 2012. Available at: http://www.pcori.org/assets/pdfs/Standards%20Outcomes%20Research.pdf (accessed January 31, 2017).
- 13.
Davies M, Erickson K, Wyner Z, Malenfant J, Rosen R, Brown J. Software-enabled distributed network governance: The PopMedNet experience. EGEMS (Wash DC) 2016; 4: 1213.
- 14.
Brown J, Lane K, Moore K, Platt R. Defining and evaluating possible database models to implement the FDA Sentinel Initiative.
Final report. May 2009. Available at: https://www.pharmamedtechbi.com/~/media/Images/Publications/Archive/The%20Pink%20Sheet/71/020/00710200015/sentinel_database_models_05_09.pdf (accessed January 31, 2017).
- 15.
Jones WS, Roe MT, Antman EM, Pletcher MJ, Harrington RA, Rothman RL, et al. The changing landscape of randomized clinical trials in cardiovascular disease. J Am Coll Cardiol 2016; 68: 1898–1907.
