Abstract
Background:
It has been shown that carvedilol and its non β-blocking analog, VK-II-86, inhibit spontaneous Ca2+ release from the sarcoplasmic reticulum (SR). The aim of this study is to determine whether carvedilol and VK-II-86 suppress ouabain-induced arrhythmogenic Ca2+ waves and apoptosis in cardiac myocytes.
Methods and Results:
Rat cardiac myocytes were exposed to toxic doses of ouabain (50 µmol/L). Cell length (contraction) was monitored in electrically stimulated and non-stimulated conditions. Ouabain treatment increased contractility, frequency of spontaneous contractions and apoptosis compared to control cells. Carvedilol (1 µmol/L) or VK-II-86 (1 µmol/L) did not affect ouabain-induced inotropy, but significantly reduced the frequency of Ca2+ waves, spontaneous contractions and cell death evoked by ouabain treatment. This antiarrhythmic effect was not associated with a reduction in Ca2+ calmodulin-dependent protein kinase II (CaMKII) activity, phospholamban and ryanodine receptor phosphorylation or SR Ca2+ load. Similar results could be replicated in human cardiomyocytes derived from stem cells and in a mathematical model of human myocytes.
Conclusions:
Carvedilol and VK-II-86 are effective to prevent ouabain-induced apoptosis and spontaneous contractions indicative of arrhythmogenic activity without affecting inotropy and demonstrated to be effective in human models, thus emerging as a therapeutic tool for the prevention of digitalis-induced arrhythmias and cardiac toxicity.
Cardiotonic glycosides (CG) or digitalis are positive inotropes used in clinical practice and also exist as endogenous ligands of the Na+/K+-ATPase (NKA).1 These compounds selectively bind to the NKA and cause an increase in intracellular [Na+], which reduces Ca2+ extrusion via the sarcolemmal Na+/Ca2+ exchanger (NCX), resulting in an increase in sarcoplasmic reticulum (SR) Ca2+ load and systolic Ca2+ release. This produces a positive inotropic effect, which has motivated their use for the treatment of heart failure.2 However, CG have toxic effects that limit their clinical use.3
In the heart, they have been associated with arrhythmias and loss of cardiac myocytes due to apoptosis.4–7 The pro-arrhythmic effects have been proposed to occur when the SR Ca2+ storage capacity is exceeded, resulting in diastolic SR Ca2+ waves and delayed after depolarizations (DADs), in a process termed “store overload-induced Ca2+ release” (SOICR).8 Additionally, SOICR has been associated with apoptosis of cardiomyocytes mediated by mitochondrial Ca2+ overload or activation of Ca2+-dependent kinases.9–12 In addition to the role played by SR Ca2+ overload in digitalis-induced arrhythmias, we and others demonstrated that ryanodine receptor (RyR2) site Ser2814 phosphorylation by Ca2+ calmodulin-dependent protein kinase II (CaMKII) is a key determinant of digitalis-induced Ca2+
waves and arrhythmias.5,13
Indeed, pharmacological and genetic interventions that prevent CaMKII-dependent RyR2 phosphorylation have been effective in reducing digitalis-induced SOICR.5,14
Interestingly, beyond its arrhythmogenic effect, CaMKII has been shown to mediate digitalis-induced apoptosis.6
Unfortunately, cardiac-specific CaMKII inhibition is not currently available. However, the pharmacological modulation of RyR2 has emerged as a therapeutic option to prevent arrhythmogenic SR Ca2+
release.15–17
Indeed, carvedilol has been shown to have a unique property, not shared by other β-blockers, which is to suppress SOICR and prevent arrhythmias in mice.16
The antiarrhythmic mechanism of carvedilol has been attributed to its action on RyR2 gating, particularly the reduction in the duration of RyR2 openings.16
Although at present, the specific RyR2 binding site for carvedilol has not been established, we speculate that a reduction in RyR2 open time could be an effective approach to reduce the cardio-toxic effects of digitalis.
Carvedilol is the β-blocker of choice for patients with heart failure.18–20
Nevertheless, the concentrations required to suppress SOICR are higher than those required for β-blockade (~1 nmol/L).8
Therefore, SOICR inhibition would require high doses of carvedilol, inducing excessive β-blockade and bradycardia.21
In contrast, VK-II-86, a structural analog of carvedilol has minimum β-blocking activity, but retains its capacity to reduce SOICR.16
Thus, we hypothesized that VK-II-86 could reduce the incidence of arrhythmogenic SR Ca2+ waves in cardiac myocytes treated with digitalis. Another aim of this work was to test the efficacy of VK-II-86 to reduce digitalis-induced cell death.
Methods
All experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No.85-23, revised 1996) and approved by the Institutional Animal Care and Use Committee of La Plata University.
Rat Cardiac Myocytes Isolation and Culture
Male Wistar rats (200–300 g) were anaesthetized by an intra-peritoneal injection of ketamine (79 mg/kg) and diazepam (5 mg/kg), and hearts were excised when plane three of phase III of anaesthesia was reached. Plane three of phase III of anesthesia was verified by the presence of slow deep diaphragmatic breathing, loss of the corneal reflex and the absence of tongue retraction. Cardiac myocytes were isolated by collagenase-based enzymatic digestion, as previously described.21
For culture, isolated cells were re-suspended in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with (in g/L) 0.017 ascorbic acid, 2 BSA, 0.4 L-carnitine, 0.66 creatine, 0.62 taurine, 50 U/mL penicillin, and 50 U/mL streptomycin, and counted. Myocytes were plated at a density of ∼2×104
rod-shaped cells/mL into culture dishes for 1 h to allow cell attachment. After this period, the culture media was changed for a fresh one with or without ouabain and the presence or absence of pharmacological compounds according to the protocols performed (see Results). After 24 h of culture, the cells were photographed to assess viability and apoptosis.
Pharmacological Agents
Ouabain, Atenolol and Carvedilol were purchased from Sigma-Aldrich; VK-II-86 was synthetized as reported in Zhou et al,16
and Ru360 was purchased from Calbiochem, USA.
Cytosolic Calcium and Cell Shortening Measurements
Myocytes were loaded with 10 μmol/L Fura-2 AM (ThermoFisher Scientific) for 12 min. Fura-2 fluorescence was measured on an inverted microscope adapted for epifluorescence by Ion Optix hardware. The ratio of the Fura-2 fluorescence (510 nm) obtained after exciting the dye at 340 and 380 nm was taken as an index of Ca2+i. The amplitude of the Ca2+ transient induced by rapid application of 15 mmol/L caffeine was used to estimate SR Ca2+ content.9
Cell shortening was registered by video-detection of cell length.9
Detection of Spontaneous Ca2+ Release by Confocal Microscopy
Myocytes were loaded with 10 µmol/L Fluo-3 AM (ThermoFisher Scientific) and examined using a Zeiss 410 inverted confocal microscope (LSMTech, Pennsylvania, USA). Cells were imaged in line-scan mode along their long axis, exciting with the 488 nm line of an argon laser and collecting emission at >515 nm.5
Data were analysed using the ‘Sparkmaster’ plugin for ImageJ.22
Immunodetection by Western Blot Analysis
Homogenates were prepared from the pulverized ventricular tissue of Langendorff-perfused rat hearts, as previously described.9
For details, refer to the supplementary material.
Human Cardiomyocytes Derived From Induced Pluripotent Stem Cells
Induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) were obtained by using the monolayer differentiation protocol described by Burridge et al.23
For this purpose, the FH2.1 induced pluripotent cells were used.24
The assessment of the efficiency of cardiac differentiation was performed by using standard flow cytometry to determine the percentage of Troponin C+
cells. The cells were maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with B27 at 37℃ and 5% CO2
until 35 days after the start of differentiation. Cells were used between days 35 and 37.
Human Cardiac Myocyte Mathematical Model
A modified ten Tusscher-Panfilov human ventricular myocyte model (TP)25
was used to test the effects promoted by VK-II-86, Ouabain and their combination. This model has been validated by us and others.26,27
For details, refer to the supplementary material.
Assessment of Cell Viability and Apoptosis
Cells were evaluated morphologically, being classified as viable or non-viable according to their length-to-width ratio (≥3 were considered viable).21
From each culture, which was considered as an n equal to 1, at least 8 photographs per group were taken to count and classify the cells. Caspase-3 activity, used as an index of apoptosis, was determined using the caspase-3 fluorescent substrate, Phiphilux (OncoImmunin; Gaithersburg, MD, USA). Briefly, 2.5 µmol/L Phiphilux was added to each culture dish at the end of the incubation period and allowed to incubate for 1 h at 37℃. Myocytes were gently washed once and imaged under a fluorescence microscope.21
Statistical Analysis
Unpaired Student’s t-test and one-way ANOVA followed by the Tukey-Cramer post hoc test were used for statistical comparisons when appropriate. Data are expressed as mean±SEM. Differences were considered significant at P≤0.05.
Results
Carvedilol Prevents Ouabain-Induced Spontaneous Contractile Activity
Previous reports from our and other laboratories demonstrated that 50 µmol/L ouabain promotes SOICR and its associated spontaneous contractions in rat cardiac myocytes.5,28
Thus, we compared in the same experimental conditions the efficacy of carvedilol and atenolol, 2 different β-blockers with and without anti-SOICR properties, to abolish ouabain-induced non-stimulated contractile events (NSE). The effect of 50 µmol/L ouabain on cell shortening and the incidence of NSE were tested in rat cardiomyocytes. Myocytes were field-stimulated at 0.5 Hz and superfused with 50 µmol/L ouabain. After 20 min, field stimulation was stopped and myocyte length was monitored for an additional 10 min. Consistent with previous results,5
the typical tracings depicted in
Figure 1
show that ouabain superfusion produces a positive inotropic effect and enhances the incidence of NSE during the non-stimulated period compared to untreated control cells. Overall, ouabain promoted a 51.9±13.3% increase in contractility and increased the number of NSE from 11±2.6 in control cells to 71.4±12.1 events/10 min in ouabain-treated cells. The tracings further show that pretreatment of cells with 1 µmol/L of atenolol does not affect either the ouabain-induced positive inotropic effect or the associated increase in NSE. In contrast, pretreatment with 1µmol/L carvedilol significantly reduced the ouabain-induced increase of NSE without affecting inotropy. The bars graphs in
Figure 1
shows the overall data.
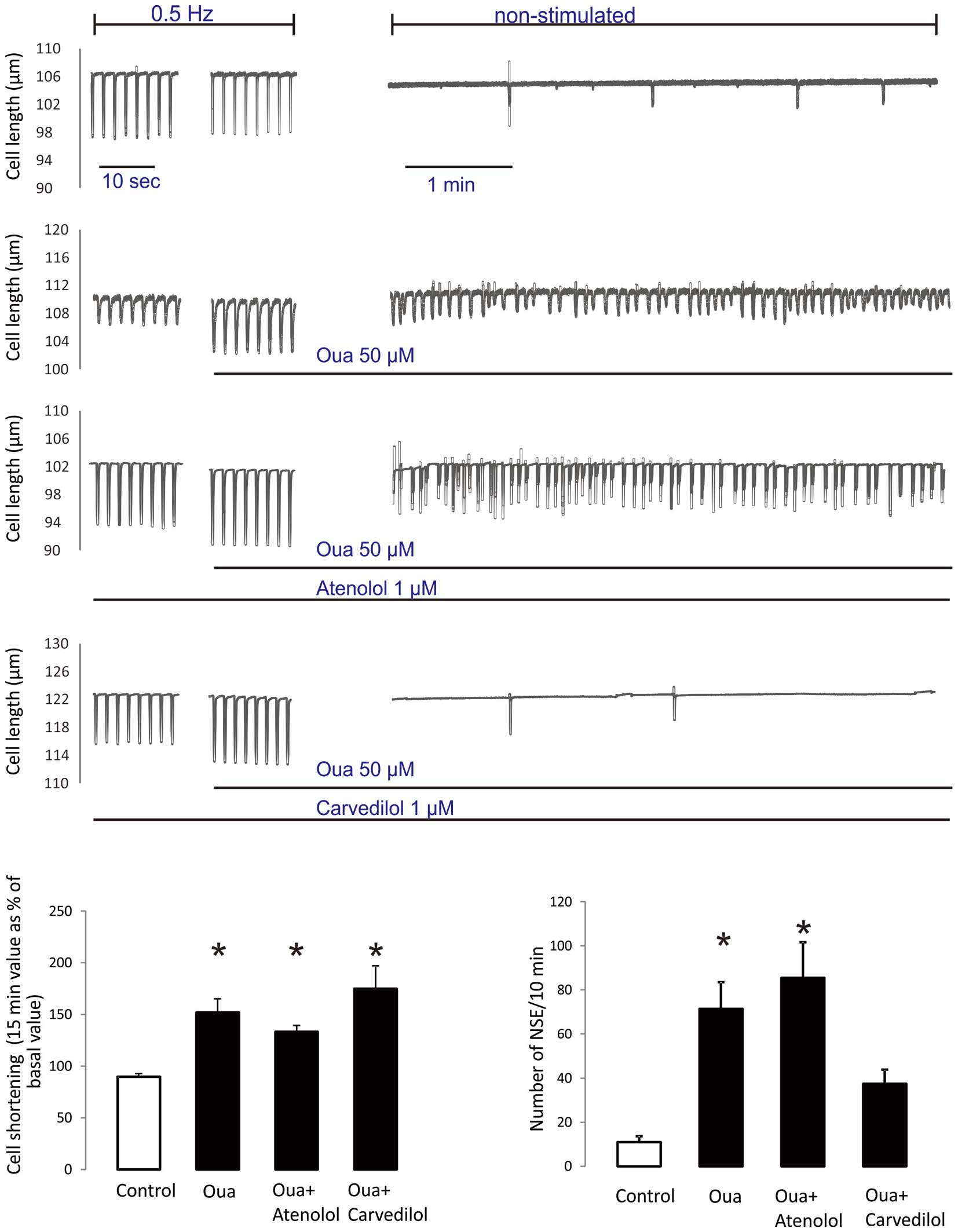
We next evaluated the effects of VK-II-86 on ouabain inotropy and the propensity for NSE. As can be observed in the typical tracings and overall results depicted in
Figure 2, treating cells with 1 µmol/L VK-II-86 did not affect the ouabain-induced positive inotropic effect but significantly reduced the incidence of NSE.
VK-II-86 Does Not Affect Ouabain-Induced RyR2 Phosphorylation or SR Ca2+ Load
Previous reports (by us and others) showed that ouabain-induced arrhythmias result from SR Ca2+ overload and a CaMKII-dependent phosphorylation of the RyR2.5,14
To assess whether the antiarrhythmic effects of VK-II-86 are due to reversal of these alterations, we examined the effect of VK-II-86 on ouabain-induced CaMKII activity, RyR2 phosphorylation and SR Ca2+ load.
Figure 3
shows typical blots and average results indicating that ouabain-induced CaMKII activity (p-CaMKII), oxidation (OxiCaMKII); phosphorylation of phospholamban (PLN; residues Serine 16 and Threonine 17) and the RyR2 (residues Serine 2808 and 2814) are preserved in the presence of VK-II-86.
The traces in
Figure 4A
show the effect of ouabain, in the absence and presence of VK-II-86, on the caffeine-induced Ca2+ transients, performed to evaluate SR Ca2+ content. As can be observed in the typical traces and average results, VK-II-86 fails to affect caffeine-induced SR Ca2+ release in ouabain-treated cells.
Consistent with the observation that VK-II-86 prevents spontaneous contractions that arises from Ca2+
waves,
Figure 4B
depicts typical confocal images and average results showing that ouabain significantly increases Ca2+
wave frequency compared to untreated cells, and that this increase is prevented when ouabain is administered in the presence of VK-II-86. In addition, the overall results show that VK-II-86 prevents the ouabain-induced increase in Ca2+ spark frequency without affecting sparks morphology. This suggests that the effect of VK-II-86 on RyR2 gating directly has an effect on the probability of Ca2+ spark formation, which ultimately reduces Ca2+ waves frequency.
Finally, as SR Ca2+ waves represent propagated Ca2+ release triggered by cytosolic Ca2+ released from neighboring RyR2 clusters, we examined the effect of VK-II-86 on the velocity of SR Ca2+ waves as a measure of RyR2 response to cytosolic Ca2+. As shown in Figure 4C, VK-II-86 also fails to affect the velocity of Ca2+ waves.
Taken together, these results suggest that the ability of VK-II-86 to reduce ouabain-induced arrhythmic activity is due to its capacity to reduce RyR2 open time rather than to a non-specific reduction in CaMKII activity, SR Ca2+ load or the sensitivity of the RyR2 to cytosolic Ca2+.
Human Cardiomyocytes Derived From Induced Pluripotent Stem Cells and a Human Computational Cardiomyocyte Model Reproduce the Arrhythmic and Inotropic Effects of Ouabain and the Antiarrhythmic Action of VK-II-86
Cell shortening was assessed in human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) between days 35 and 37 of culture. Control cells showed stable contraction cycles during a 15-min period, and a similar pattern was observed in cells treated with 1 µmol/L VK-II-86 (not shown). Treatment with 50 µmol/L ouabain produced a typical positive inotropic effect that was not affected by co-treatment with VK-II-86 (Figure S2). As can be observed in
Figure 5A, ouabain promoted an increase in the frequency of spontaneous beats, whereas VK-II-86 was effective in preventing this alteration. To further characterize the anti-arrhythmic efficacy of VK-II-86 in a human model, we examined the effect of ouabain in the presence and absence of VK-II-86 in a mathematical human ventricular myocyte model.
Figure 5B
shows myocyte shortening simulation in the presence and absence of ouabain with and without VK-II-86. The model predicts that ouabain application produces an increase in sarcomere shortening and elicits spontaneous contractions during the unstimulated phase.
Figure 5B
further shows that pre-treatment with VK-II-86 prevents the occurrence of ouabain-induced spontaneous contractions. Similarly, simulating CaMKII inhibition significantly reduced the number of spontaneous contractions, as shown in the typical traces and the bar graphs of overall results. The bar graphs further show the inotropic response to ouabain (25% increase in sarcomere shortening) and its preservation in the presence of either VK-II-86 (22%) or CaMKII inhibition (25%). This is consistent with the preserved ouabain-induced increase in SR Ca2+ load in the presence of VK-II-86 or CaMKII inhibition.

In previous reports, we and others demonstrated that chronic exposure (24 h) to sub-arrhythmogenic concentrations of ouabain (2 µmol/L) promote cardiomyocyte apoptosis.6,29
Although the mechanisms underlying ouabain-induced cardiomyocyte death are not fully understood, previous reports from our and other laboratories have suggested that under pathological conditions, Ca2+ leaking from the SR can be shuttled into the mitochondria via the Ca2+ uniporter, resulting in mitochondrial Ca2+ overload, which triggers the apoptotic cascade.11,30 Figure 6A depicts representative images and overall results, showing that 10 µmol/L of the mitochondrial Ca2+ uniporter inhibitor, Ru360, is able to prevent ouabain-induced apoptosis, supporting that mitochondrial Ca2+ overload is involved in ouabain cytotoxicity. In addition, Figure 6B shows that ouabain-induced cell death can be prevented by carvedilol and VK-II-86. These results indicate that reducing RyR2-mediated Ca2+ release is effective to prevent ouabain-induced cell death. Control experiments showed that 10 µmol/L Ru360, 1 µmol/L carvedilol or 1 µmol/L VK-II-86 did not, per se, affect cell viability (not shown).

Discussion
Major findings of this study are: (1) carvedilol and its analog, VK-II-86, reduce the propensity for arrhythmogenic Ca2+
waves and the associated spontaneous contractions in rat cardiac myocytes treated with toxic doses of ouabain; (2) based on the use of 2 human cardiomyocyte models, our results may be extrapolated to human ventricular cardiomyocytes; (3) VK-II-86 does not affect the ouabain-induced increase in CaMKII activity, PLN and RyR2 phosphorylation or SR Ca2+ load, suggesting that its antiarrythmic effect is due to modulation of RyR2 gating properties; (4) VK-II-86 does not blunt the ouabain-induced positive inotropic effect; and (5) VK-II-86 is able to prevent ouabain-induced cardiomyocyte death.
Diastolic SR Ca2+ waves activate the inward NCX current (INCX) causing DADs. Threshold-surpassing DADs have been proposed to underlie arrhythmias in catecholaminergic polymorphic ventricular tachycardia (CPVT), heart failure and digitalis toxicity.31
Indeed, previous reports from our and others have shown that an increase in SR Ca2+ load and in CaMKII-dependent RyR2 phosphorylation contributes to the enhanced diastolic SR Ca2+ release that generates the DADs associated with digitalis treatment.5,14
These arrhythmias and toxic effects such as cardiomyocyte death have limited the therapeutic use of digitalis.3
In an attempt to enhance the safety of these compounds, we previously suggested that CaMKII inhibition could prevent digitalis-induced arrhythmias.5
However, the ubiquitous nature of CaMKII expression and its effects on different targets precludes its inhibition as a therapeutic tool. Albeit not impossible, pharmacological CaMKII inhibition would require cardiac-specific CaMKII inhibitors, which are at present unavailable. The involvement of RyR2 in the development of arrhythmias during digitalis toxicity allows us to postulate an alternative, which is to directly target RyR2 function. We are aware of one report that tested this approach in the setting of digitalis treatment.32
Using the multi-channel blocker, JTV-519, which stabilizes RyR2-FK506 binding protein (FKBP) interaction, Sacherer et al were able to reduce SR Ca2+
leak. However, JTV-519 impaired the positive inotropic response to digitalis due to inhibitory effect on the L-type Ca2+
current.32
Recently, carvedilol has been shown to reduce the RyR2 mean open time, thus suppressing spontaneous SR Ca2+
release in a mouse model of CPVT.16
Moreover, the antiarrhythmic effects of carvedilol were shared by its non-β-blocking analog, VK-II-86.16
These results, prompted us to evaluate whether carvedilol and VK-II-86 were able to prevent arrhythmogenic SR Ca2+ release during digitalis toxicity. Using the number of NSE as an index of diastolic SR Ca2+ release, we observed that although not affecting basal contractility (not shown), carvedilol and VK-II-86 significantly reduce the arrhythmogenic substrate induced by ouabain without blunting its positive inotropic effect (Figures 1,2).
Mechanisms Underlying VK-II-86-Dependent Ca2+ Waves Inhibition
Recently, Zhou et al reported that VK-II-86 shortens the duration but increases the frequency of RyR2 channel opening.16
This could affect how Ca2+ leaks out the SR by reducing intra-cluster RyR2 recruitment, inter-cluster RyR2 recruitment or both. In light of our results, it seems that VK-II-86 reduces intra-cluster RyR2 recruitment as it prevented ouabain-induced increase in spark frequency and secondarily reduced Ca2+ wave incidence. This interpretation is compatible with a reduction in spark-mediated SR Ca2+ leak without significant changes in SR Ca2+ content. Consistently, we showed that VK-II-86 was able to reduce the ouabain-induced increase in wave frequency without affecting SR Ca2+ content (Figure 4A,B).
Previous reports from our laboratory showed that CaMKII-dependent RyR2 phosphorylation is a crucial determinant of digitalis-induced arrhythmias.5,33
More recently, Ho et al proposed that digitalis-induced arrhythmias occur due to oxidative activation of CaMKII, which leads to phosphorylation of the RyR2 at site Ser2814.14
Supporting this conclusion, transgenic mice, in which the Ser2814 site of the RyR2 cannot be phosphorylated (S2814A mice), were resistant to digitoxin-induced arrhythmias, providing unequivocal evidence of the causal role of S2814 phosphorylation in the digitalis-induced RyR2 dysfunction.14
This last observation also suggests that RyR2 oxidation does not contribute to the arrhythmic effect of digitalis. Nevertheless, carvedilol has been shown to have acute antioxidant activity on RyR2,34
and we cannot exclude that this antioxidant activity could explain part of the antiarrhythmic action of this compound and its structurally similar analog. Indeed, our results showing that VK-II-86 does not affect ouabain-induced CaMKII autophosphorylation (p-CaMKII) or its oxidation state (Oxi-CaMKII) (Figure 3) suggests that the antiarrhythmic effect of VK-II-86 is due to a direct action on RyR2.
In addition, although VK-II-86 reduced ouabain-induced Ca2+ wave frequency (Figure 4B), it failed to affect Ca2+ wave velocity (Figure 4C). Ca2+ waves represent SR Ca2+ release triggered by cytosolic Ca2+ released from neighboring RyR2 clusters. Therefore, the velocity of Ca2+ waves can be used as an index of RyR2 sensitivity to cytosolic Ca2+.35 Thus, our results suggest that VK-II-86-antiarrhythmic actions are not due to a reduction in RyR2 sensitivity to cytosolic Ca2+. These results are consistent with previous evidence showing that the antiarrhythmic mechanism of carvedilol and its analogs is mediated by their direct action on RyR2-mediated SOICR.16
Carvedilol and VK-II-86 Do Not Affect the Inotropic Response of Ouabain-Treated Cardiac Myocytes
One of the most important therapeutic benefits of digitalis is their capacity to increase intracellular Ca2+
with the consequent development of a positive inotropic effect. Thus, prevention of arrhythmias during digitalis treatment should not blunt the development of their inotropic effect. Interestingly, carvedilol and VK-II-86 reduced ouabain-induced arrhythmias, without affecting the ouabain-induced positive inotropic effect (Figures 1,2).
The possibility of using an Food and Drug Administration (FDA)-approved drug such as carvedilol to target RyR2 in combination with cardiac glycosides to treat heart failure patients is promising. However, a possible limitation is that the concentration of carvedilol required to reduce RyR2 open time is high, and its β-blocking action would produce undesirable bradycardia. The lack of β-blocking effect of VK-II-86 represents a favorable feature and suggests that it could be used as an adjunct to digitalis therapy.
Human Cellular and Mathematical Models Support Antiarrhythmic Actions of VK-II-86
To enhance the translational relevance of our findings, we performed experiments in hiPSC-CMs, which exhibit molecular and functional properties of human cardiac cells.36,37
Importantly, ouabain-induced arrhythmias have been detected in hiPSC-CMs, suggesting that these cells can be used to analyze clinically relevant outcomes.38
Similar to rat cardiomyocytes, VK-II-86 prevented the ouabain-induced increase in spontaneous contractile activity of hiPSC-CMs without affecting inotropy, suggesting that VK-II-86 may be effective in humans (Figures 5A,S2).
We further corroborated these findings in a mathematical model that simulates human cardiac Excitation-contraction coupling. The model reproduced the positive inotropic effect induced by ouabain, together with the arrhythmic activity (Figure 5B). Furthermore, the model predicts that a reduction of RyR2 open time by VK-II-86 is effective to reduce the ouabain-induced arrhythmic events in human ventricular myocytes. Taken together, these results provide evidence supporting the protective role of VK-II-86 against ouabain-induced arrhythmias in a human model of digitalis toxicity.
Carvedilol and VK-II-86 Prevent Ouabain-Induced Cardiac Myocyte Death
Activation of CaMKII is a common intermediate of diverse death stimuli that induce apoptosis in cardiac cells.6,11
Consistently, we have demonstrated that CaMKII activity mediates ouabain-induced myocyte apoptosis.6
Interestingly, we recently reported that rapid pacing-induced apoptosis, also CaMKII dependent, can be prevented by targeting RyR2 with carvedilol and VK-II-86.9
Considering that ouabain-induced cytotoxicity involves SR Ca2+ release trough RyR2, which could drive mitochondrial Ca2+ overload and cell death, we hypothesized that reducing RyR2 open time could prevent ouabain-induced cell death. Confirming the role of mitochondrial Ca2+ overload in ouabain-induced cell death, cells treated with the mitochondrial Ca2+ uniporter inhibitor, Ru360, were protected from ouabain cytotoxicity (Figure 6A). In addition, we show herein that carvedilol and VK-II-86 prevented ouabain-induced apoptosis, suggesting additional benefits of reducing RyR2-mediated spontaneous Ca2+ release during digitalis treatment (Figure 6B). Interestingly, chronic carvedilol incubation has been shown to have antioxidant activity in cardiac myocytes.34
Thus, the antioxidant capacity of carvedilol could be additionally beneficial to prevent ouabain-induced cell death.
In summary, our results demonstrate that carvedilol and VK-II-86 are able to prevent ouabain-induced arrhythmias and myocyte death, indicating that these compounds, in particular VK-II-86, could be a promising approach to enhance the safety of digitalis therapy.
Funding
This study was supported by grant PICT 1678 from Fondo para la Investigación Científica y Tecnológica (FONCYT) to M.V.P.
Acknowledgments
The technical support of Mónica Rando, Omar Castillo and Lucia Pagola are gratefully acknowledged. We also acknowledge the support from the Canadian Institutes of Health Research, the Heart and Stroke Foundation of Canada, and the Heart and Stroke Foundation Chair in Cardiovascular Research to S.R.W.C.
Supplementary Files
Supplementary File 1
Supplementary Methods
Figure S1.
Diagram of the 4-state RyR2 model.
Supplementary Results
Figure S2.
hiPSC-CMs shortening expressed as a percentage of Lmax.
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-18-0247
References
- 1.
Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, et al. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA 1991; 88: 6259–6263.
- 2.
Altamirano J, Li Y, DeSantiago J, Piacentino V 3rd, Houser SR, Bers DM. The inotropic effect of cardioactive glycosides in ventricular myocytes requires Na+-Ca2+ exchanger function. J Physiol 2006; 575: 845–854.
- 3.
Rathore SS, Curtis JP, Wang Y, Bristow MR, Krumholz HM. Association of serum digoxin concentration and outcomes in patients with heart failure. JAMA 2003; 289: 871–878.
- 4.
Ferrier GR. Digitalis arrhythmias: Role of oscillatory afterpotentials. Prog Cardiovasc Dis 1977; 19: 459–474.
- 5.
Gonano LA, Sepulveda M, Rico Y, Kaetzel M, Valverde CA, Dedman J, et al. Calcium-calmodulin kinase II mediates digitalis-induced arrhythmias. Circ Arrhythm Electrophysiol 2011; 4: 947–957.
- 6.
Sapia L, Palomeque J, Mattiazzi A, Petroff MV. Na+/K+-ATPase inhibition by ouabain induces CaMKII-dependent apoptosis in adult rat cardiac myocytes. J Mol Cell Cardiol 2010; 49: 459–468.
- 7.
Ramirez-Ortega M, Zarco G, Maldonado V, Carrillo JF, Ramos P, Ceballos G, et al. Is digitalis compound-induced cardiotoxicity, mediated through guinea-pig cardiomyocytes apoptosis? Eur J Pharmacol 2007; 566: 34–42.
- 8.
Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, et al. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci USA 2004; 101: 13062–13067.
- 9.
Sepulveda M, Gonano LA, Back TG, Chen SR, Vila Petroff M. Role of CaMKII and ROS in rapid pacing-induced apoptosis. J Mol Cell Cardiol 2013; 63: 135–145.
- 10.
Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, et al. CaV1.2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci USA 2010; 107: 4996–5000.
- 11.
Di Carlo MN, Said M, Ling H, Valverde CA, De Giusti VC, Sommese L, et al. CaMKII-dependent phosphorylation of cardiac ryanodine receptors regulates cell death in cardiac ischemia/reperfusion injury. J Mol Cell Cardiol 2014; 74: 274–283.
- 12.
Ruiz-Meana M, Fernandez-Sanz C, Garcia-Dorado D. The SR-mitochondria interaction: A new player in cardiac pathophysiology. Cardiovasc Res 2010; 88: 30–39.
- 13.
Priori SG, Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res 2011; 108: 871–883.
- 14.
Ho HT, Liu B, Snyder JS, Lou Q, Brundage EA, Velez-Cortes F, et al. Ryanodine receptor phosphorylation by oxidized CaMKII contributes to the cardiotoxic effects of cardiac glycosides. Cardiovasc Res 2014; 101: 165–174.
- 15.
Hwang HS, Hasdemir C, Laver D, Mehra D, Turhan K, Faggioni M, et al. Inhibition of cardiac Ca2+ release channels (RyR2) determines efficacy of class I antiarrhythmic drugs in catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol 2011; 4: 128–135.
- 16.
Zhou Q, Xiao J, Jiang D, Wang R, Vembaiyan K, Wang A, et al. Carvedilol and its new analogs suppress arrhythmogenic store overload-induced Ca2+ release. Nat Med 2011; 17: 1003–1009.
- 17.
Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, et al. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science 2004; 304: 292–296.
- 18.
Greenberg B. Nonselective versus selective beta-blockers in the management of chronic heart failure: Clinical implications of the carvedilol or Metoprolol European Trial. Rev Cardiovasc Med 2004; 5(Suppl 1): S10–S17.
- 19.
Stroe AF, Gheorghiade M. Carvedilol: Beta-blockade and beyond. Rev Cardiovasc Med 2004; 5(Suppl 1): S18–S27.
- 20.
Remme WJ. Which beta-blocker is most effective in heart failure? Cardiovasc Drugs Ther 2010; 24: 351–358.
- 21.
Vila-Petroff M, Salas MA, Said M, Valverde CA, Sapia L, Portiansky E, et al. CaMKII inhibition protects against necrosis and apoptosis in irreversible ischemia-reperfusion injury. Cardiovasc Res 2007; 73: 689–698.
- 22.
Picht E, Zima AV, Blatter LA, Bers DM. SparkMaster: Automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 2007; 293: C1073–C1081.
- 23.
Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, et al. Chemically defined generation of human cardiomyocytes. Nat Methods 2014; 11: 855–860.
- 24.
Questa M, Romorini L, Bluguermann C, Solari CM, Neiman G, Luzzani C, et al. Generation of iPSC line iPSC-FH2.1 in hypoxic conditions from human foreskin fibroblasts. Stem Cell Res 2016; 16: 300–303.
- 25.
ten Tusscher KH, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol 2006; 291: H1088–H1100.
- 26.
Mazzocchi G, Sommese L, Palomeque J, Felice JI, Di Carlo MN, Fainstein D, et al. Phospholamban ablation rescues the enhanced propensity to arrhythmias of mice with CaMKII-constitutive phosphorylation of RyR2 at site S2814. J Physiol 2016; 594: 3005–3030.
- 27.
Davis J, Davis LC, Correll RN, Makarewich CA, Schwanekamp JA, Moussavi-Harami F, et al. A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 2016; 165: 1147–1159.
- 28.
Lotan CS, Miller SK, Pohost GM, Elgavish GA. Amiloride in ouabain-induced acidification, inotropy and arrhythmia: 23Na & 31P NMR in perfused hearts. J Mol Cell Cardiol 1992; 24: 243–257.
- 29.
Satoh H, Ginsburg KS, Qing K, Terada H, Hayashi H, Bers DM. KB-R7943 block of Ca(2+) influx via Na(+)/Ca(2+) exchange does not alter twitches or glycoside inotropy but prevents Ca(2+) overload in rat ventricular myocytes. Circulation 2000; 101: 1441–1446.
- 30.
Chen X, Zhang X, Kubo H, Harris DM, Mills GD, Moyer J, et al. Ca2+ influx-induced sarcoplasmic reticulum Ca2+ overload causes mitochondrial-dependent apoptosis in ventricular myocytes. Circ Res 2005; 97: 1009–1017.
- 31.
Pogwizd SM, Bers DM. Cellular basis of triggered arrhythmias in heart failure. Trends Cardiovasc Med 2004; 14: 61–66.
- 32.
Sacherer M, Sedej S, Wakula P, Wallner M, Vos MA, Kockskamper J, et al. JTV519 (K201) reduces sarcoplasmic reticulum Ca(2)(+) leak and improves diastolic function in vitro in murine and human non-failing myocardium. Br J Pharmacol 2012; 167: 493–504.
- 33.
Gonano LA, Petroff MV. Subcellular mechanisms underlying digitalis-induced arrhythmias: Role of calcium/calmodulin-dependent kinase II (CaMKII) in the transition from an inotropic to an arrhythmogenic effect. Heart Lung Circ 2014; 23: 1118–1124.
- 34.
Mochizuki M, Yano M, Oda T, Tateishi H, Kobayashi S, Yamamoto T, et al. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J Am Coll Cardiol 2007; 49: 1722–1732.
- 35.
Swietach P, Spitzer KW, Vaughan-Jones RD. Modeling calcium waves in cardiac myocytes: Importance of calcium diffusion. Front Biosci (Landmark Ed) 2010; 15: 661–680.
- 36.
Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res 2010; 4: 107–116.
- 37.
Kattman SJ, Witty AD, Gagliardi M, Dubois NC, Niapour M, Hotta A, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011; 8: 228–240.
- 38.
Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, et al. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Sci 2011; 123: 281–289.



