Editorials
How Is Uric Acid Related to Atrial Fibrillation?
2019 Volume 83 Issue 4 Pages 705-706


Details
2019 Volume 83 Issue 4 Pages 705-706
Uric acid (UA) is an end product of purine metabolism in humans, produced in the liver, muscles, and intestines. Xanthine oxidoreductase (XO) is the enzyme responsible for UA production. Under normal conditions its serum level is lower than 6 mg/dL in women and 7 mg/dL in men because of homeostatic regulation carried out mostly by the kidney. Dietary factors may influence serum UA, increasing its levels (meat, seafood, alcohol etc.) or decreasing them (coffee, ascorbic acid etc.). In addition, high cellular turnover conditions, such as in neoplastic disease, may increase the UA concentration. When the serum UA concentration is higher, the condition is defined as hyperuricemia. UA may have an opposite role to oxidative stress, according to its intracellular (antioxidant) and extracellular (pro-oxidant) localization.1 UA acts as an antioxidant and accounts for 50% of the total antioxidant capacity of biological fluids in humans. When present in the cytoplasm of cells or in the acidic/hydrophobic milieu of atherosclerotic plaques, UA converts to a pro-oxidant and promotes oxidative stress, participating in the pathophysiology of human disease, including cardiovascular disease (CVD), through this mechanism. Most epidemiological studies suggest the existence of an association between elevated serum UA level and CVD, including coronary artery disease, stroke, congestive heart failure, hypertension and arrhythmias, including atrial fibrillation (AF) (Figure 1).2–4
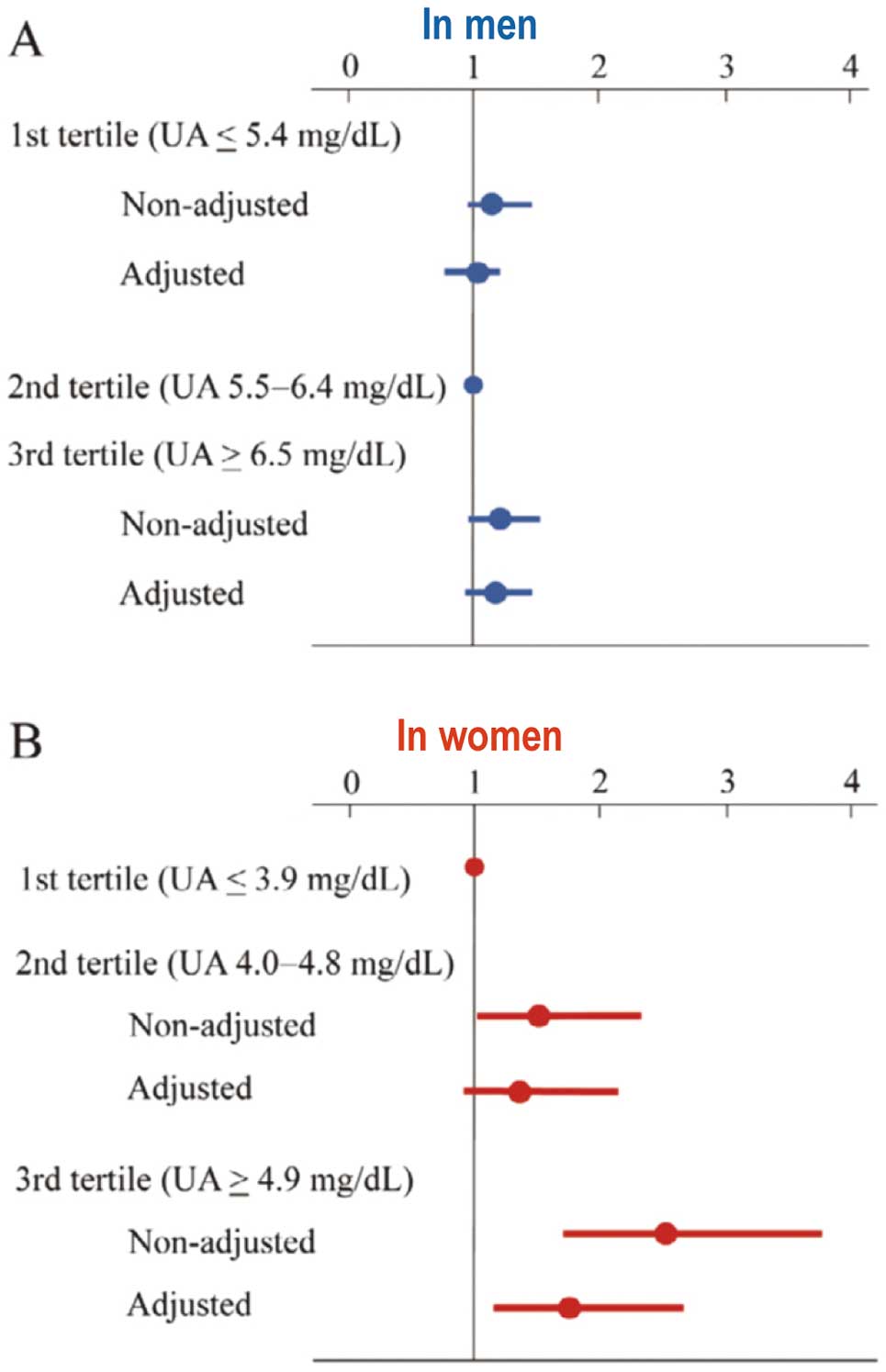
Hazard ratios for new-onset atrial fibrillation according to serum uric acid (UA) tertiles in men (A) and in women (B). Reproduced with permission from reference 4.
Article p 718
Although the relationship between hyperuricemia and AF is well recognized,5,6 the mechanism of AF onset/maintenance caused by excessive UA has not been clarified adequately. Oxidative stress may influence the development of AF, in addition to neurohormonal and inflammatory activation. Both inflammation and neurohormones may activate XO and increase serum UA. In experimental studies, oxidative stress has induced atrial electric remodeling, thus favoring the reentry mechanism,7 as well as decreasing NO, shortening the duration of the action potential (AP) plateau, increasing the repolarization velocity and AF.8,9 The relationship between the increase in oxidative stress markers and AF has been recognized in patients.10 Nevertheless, because the mechanisms responsible for the initiation and maintenance of AF are multifactorial and dynamic, the interaction between UA and AF substrates remains to be elucidated.
In this issue of the Journal, Taufiq et al11 try to clarify the link between UA and AF, focusing on its inflammation-independent effects. Previously, the same group demonstrated a possible mechanism involving soluble UA through the urate transporter to induce AF; that is, activation of the potassium voltage-gated Kv1.5 channel, also known as KCNA5, is one of the main factors in the provocation and recurrence of AF. The study concluded that the accumulation of intracellular UA via activation of urate transporters enhanced Kv1.5 channel expression to shorten the atrial AP, which contributes to the reentry circuit for AF. The current investigation by Taufiq et al,11 together with their previous publication identifies the molecular mechanism for the enhancement of Kv1.5 channel. Namely, the study demonstrates upregulation of the Kv1.5 channel protein by slowing degradation of the proteins without altering its mRNA level. During this process, UA enhanced phosphorylation of Akt and HSF1, and thereby increased both transcription and translation of Hsp70. So far the most acceptable mechanisms of the association of elevated UA with cardiovascular risk factors, particularly hypertension, and UA-induced oxidative stress, endothelial dysfunction and inflammation have also been recognized as factors increasing the risk for AF. In addition, an electrophysiological hypothesis suggesting that UA may increase susceptibility to AF has also been proposed though this study.11,12 According to this hypothesis, UA enters atrial cells via UA transporters and induces the expression of Kv1.5 protein leading to increased activity of Kv1.5 ion and the channel/IKur current, which shortens the AP duration of atrial cardiomyocytes predisposing to AF independently of UA-induced inflammation of cardiomyocytes (Figure 2).13,14 Thus the available evidence suggests an association between elevated UA and the risk for AF, but many aspects of this association, including the ubiquitin–proteasome proteolytic pathway, remain unclear.
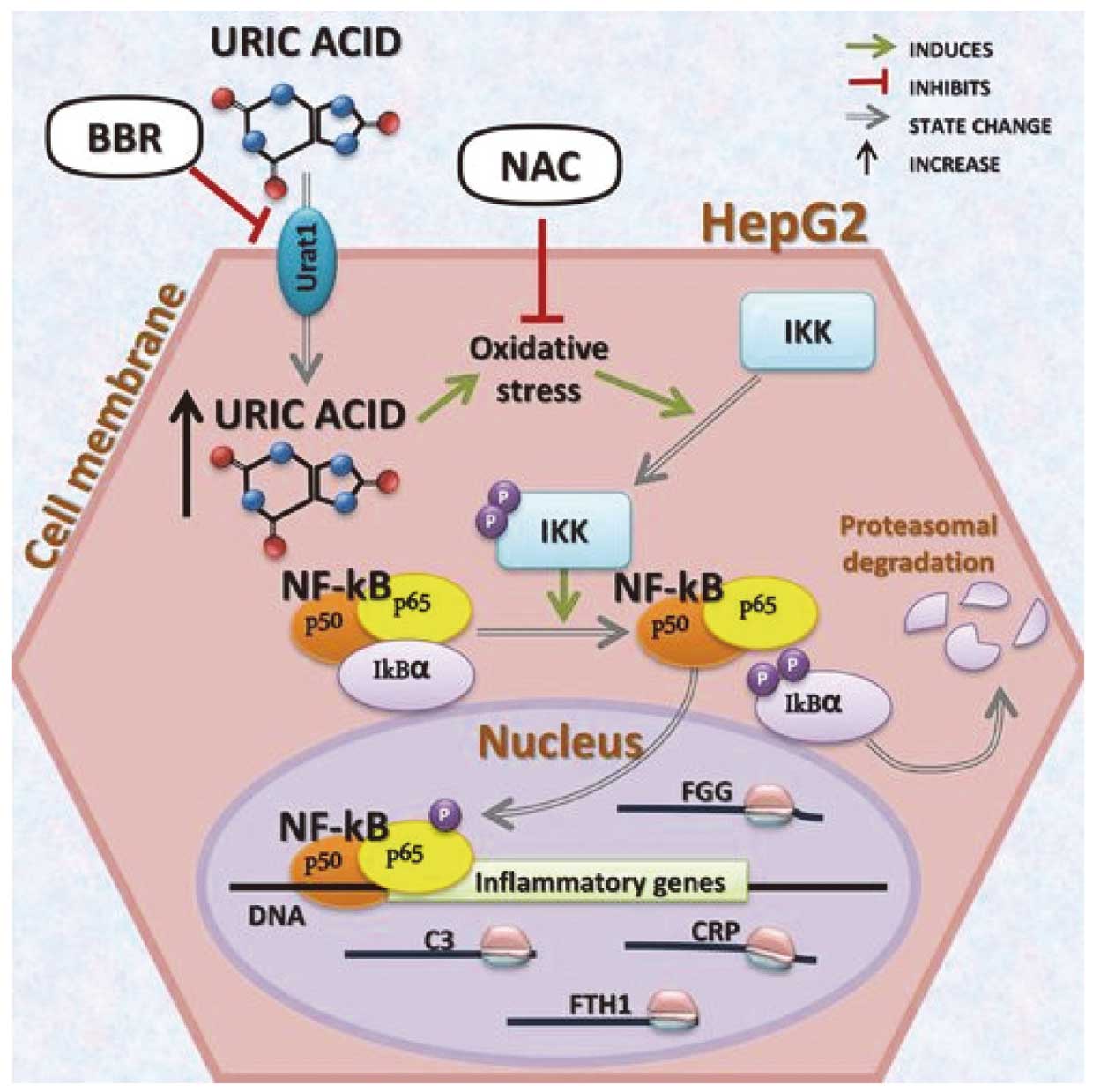
Mechanisms of uric acid-induced inflammation of cardiomyocytes possibly related to atrial fibrillation. Excessive uric acid might induce the expression of inflammatory molecules by activating the proinflammatory NF-κB signaling cascade. Reproduced with permission from reference 14.
None.
The author has declared that no conflict of interest exists.
The author has nothing to disclose.