Abbreviations
| AAV |
ANCA-associated vasculitis |
| ACR |
American College of Rheumatology |
| ADA |
adalimumab |
| ANCA |
anti-neutrophil cytoplasmic antibody |
| AZA |
azathioprine |
| bDMARDs |
biologic disease-modifying anti-rheumatic drugs |
| c-ANCA |
cytoplasmic ANCA |
| CG |
cryoglobulin |
| CHCC |
Chapel Hill Consensus Conference |
| Cr |
creatinine |
| CT |
computed tomography |
| CRP |
C-reactive protein |
| CyA |
cyclosporine |
| CV |
cryoglobulinemic vasculitis |
| CY |
cyclophosphamide |
| DMARDs |
disease-modifying anti-rheumatic drugs |
| eGFR |
estimated glomerular filtration rate |
| EGPA |
eosinophilic granulomatosis with polyangiitis |
| ESR |
erythrocyte sedimentation rate |
| ETN |
etanercept |
| EULAR |
European League Against Rheumatism |
| EVG |
Elastica van Gieson |
| FDG |
fluorodeoxyglucose |
| GC |
glucocorticoid |
| GCA |
giant cell arteritis |
| GBM |
glomerular basement membrane |
| GPA |
granulomatosis with polyangiitis |
| GRADE |
Grading of Recommendations, Assessment, Development and Evaluation |
| GWAS |
genome-wide association study |
| HBV |
hepatitis B virus |
| HCV |
hepatitis C virus |
| HE |
hematoxylin and eosin |
| HLA |
human leukocyte antigen |
| HUV |
hypocomplementemic urticarial vasculitis |
| HUVS |
hypocomplementemic urticarial vasculitis syndrome |
| IFX |
infliximab |
| IL |
interleukin |
| IVCY |
intravenous cyclophosphamide |
| IVIG |
intravenous high-dose immunoglobulin |
| LV-GCA |
large-vessel giant cell arteritis |
| MMF |
mycophenolate mofetil |
| MPA |
microscopic polyangiitis |
| MPO |
myeloperoxidase |
| mPSL |
methylprednisolone |
| MRA |
malignant rheumatoid arthritis |
| MHLW |
Ministry of Health, Labour and Welfare |
| MRI |
magnetic resonance imaging |
| MTX |
methotrexate |
| PAN |
polyarteritis nodosa |
| p-ANCA |
perinuclear ANCA |
| PET |
positron emission tomography |
| PMR |
polymyalgia rheumatica |
| PR3 |
proteinase 3 |
| PSL |
prednisolone/prednisone |
| RA |
rheumatoid arthritis |
| RF |
rheumatoid factor |
| RPGN |
rapidly progressive glomerulonephritis |
| RTX |
rituximab |
| RV |
rheumatoid vasculitis |
| SLE |
systemic lupus erythematosus |
| SNP |
single nucleotide polymorphism |
| TCZ |
tocilizumab |
| TNF |
tumor necrosis factor |
I. Notes on the Revision
1. Background of the Guidelines
Vasculitis is a general term for diseases that cause inflammation in the blood vessels themselves.
Table 1
shows the 2012 revised version of Chapel Hill Consensus Conference (CHCC) classification (CHCC2012).1
In 2007, the Guidelines for Management of Vasculitis Syndrome were published by a joint working group staffed primarily by the Japanese Circulation Society and the Research Committee on Intractable Vasculitis of the MHLW of Japan.2
This time, based on the subsequent development of basic and clinical research, the 2017 totally revised version of the Guidelines for Management of Vasculitis Syndrome and the digest version is published 9 years after the publication of its first version. This digest version plainly describes important points and is compiled for quick reference in daily clinical practice. We hope that it is of use in clinics.
Table 1.
Categories and Diseases of Vasculitis Adopted by CHCC2012
1
| Large vessel vasculitis, LVV |
| Takayasu arteritis, TAK* |
| Giant cell arteritis, GCA* |
| Medium vessel vasculitis, MVV |
| Polyarteritis nodosa, PAN* |
| Kawasaki disease, KD |
| Small vessel vasculitis, SVV |
| Antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis, AAV |
| Microscopic polyangiitis, MPA* |
| Granulomatosis with polyangiitis (Wegener’s), GPA* |
| Eosinophilic granulomatosis with polyangiitis (Churg-Strauss), EGPA* |
| Immune complex SVV |
| Anti-glomerular basement membrane (anti-GBM) disease* |
| Cryoglobulinemic vasculitis, CV* |
| IgA vasculitis (Henoch-Schönlein), IgAV* |
| Hypocomplementemic urticarial vasculitis, HUV (anti-C1q vasculitis)* |
| Variable vessel vasculitis, VVV |
| Behçet’s disease, BD* |
| Cogan’s syndrome, CS |
| Single-organ vasculitis, SOV |
| Cutaneous leukocytoclastic angiitis |
| Cutaneous arteritis |
| Primary central nervous system vasculitis |
| Isolated aortitis |
| Others |
| Vasculitis associated with systemic disease |
| Lupus vasculitis |
| Rheumatoid vasculitis* |
| Sarcoid vasculitis |
| Others |
| Vasculitis associated with probable etiology |
| Hepatitis C virus-associated cryoglobulinemic vasculitis* |
| Hepatitis B virus-associated vasculitis |
| Syphilis-associated aortitis |
| Drug-associated immune complex vasculitis |
| Drug-associated ANCA-associated vasculitis |
| Cancer-associated vasculitis |
| Others |
(Jennette JC, Falk RJ, Bacon PA, et al.,
Arthritis Rheum
2013,1 John Wiley and Sons. (c) 2013, American College of Rheumatology.) *Diseases covered by the present guidelines.
2.1 Target Diseases
Vasculitis is commonly referred to CHCC2012 (Table 1),1
and is classified into large, medium, and small vessel vasculitides based on the size of the affected blood vessel. The present guidelines cover large vessel vasculitis (Takayasu arteritis and giant cell arteritis), Buerger disease, a medium-vessel vasculitis (polyarteritis nodosa) and small vessel vasculitides [anti-neutrophil cytoplasmic antibody (ANCA) associated vasculitis {microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis (EGPA)}, immune complex small vessel vasculitis (anti-GBM disease, cryoglobulinemic vasculitis, IgA vasculitis and hypocomplementemic urticarial vasculitis)].1
Kawasaki disease, which is a medium vessel vasculitis, was excluded from target diseases in consideration of the clinical fields of the guidelines users. CHCC2012 covers variable vessel vasculitis, single-organ vasculitis, vasculitis associated with systemic disease, and vasculitis associated with probable etiology in addition to the above vasculitides.1
Behçet’s disease and rheumatoid vasculitis were selected from them as target diseases in consideration of the frequency of encounters in clinical settings in Japan and diagnostic importance.
2.2 Common Procedures for the Development of Clinical Practice Guidelines
The definition of clinical practice guidelines and the procedure for their preparation change with time. The World Health Organization (WHO) defines clinical practice guidelines as “any document developed by the World Health Organization containing recommendations for clinical practice or public health policy”3
and adopts the GRADE system for their preparation. The Minds Guidelines Center of Japan defines clinical practice guidelines as “a document that presents appropriate recommendations to assist patients and practitioners in making decisions regarding clinical practice of high importance, based on the body of evidence evaluated and integrated by systematic reviews and the balance between benefits and harms”.4
2.3 Methods for the Development of the Present Guidelines
Although clinical practice guidelines are recommended to be developed using the GRADE or Minds system, it is extremely difficult to completely apply these methods to each of rare vasculitides. For these reasons, recommendations for the treatment of MPA and GPA were developed using the GRADE system, but the conventional review style was adopted for the other target diseases. The classification of recommendation and level of evidence for other target diseases were determined using the criteria shown in
Tables 2
and
3.
Table 2.
Classification of Recommendations
| Class I |
Treatment should be administered |
| Class IIa |
It is reasonable to administer treatment |
| Class IIb |
Treatment may be considered |
| Class III |
Treatment should not be administered since it is not helpful and may be harmful |
Table 3.
Levels of Evidence
| Level A |
Data derived from multiple randomized clinical trials or meta-analyses |
| Level B |
Data derived from a single randomized trial or nonrandomized studies |
| Level C |
Only consensus opinion of experts |
II. Takayasu Arteritis
1. Definition/Epidemiology/Subclassification
1.1 Definition
Takayasu arteritis is a large vessel vasculitis that affects the aorta and its primary branches, coronary arteries, and pulmonary arteries. Its principal manifestations are systemic inflammation, pain due to vasculitis, and vascular stenosis, occlusion, and dilatation, and poses problems including disorders of various organs due to disturbance of the blood flow and aneurysms even after remission of inflammation. Its symptoms are diverse and non-specific, but its early diagnosis has become possible, and the prognosis has been improved.5,6
1.2 History
In 1908, Mikito Takayasu reported a 22-year-old woman as a case of “Peculiar changes of the central retinal vessels” (Figure 1).7
Absence of the pulses of the radial artery was pointed out. In 1951, Kentaro Shimizu, et al., amassed 25 cases and reported them under the name of “pulseless disease”.8
Hideo Ueda, et al., renamed the disease “aortitis syndrome.9,10
1.3 Epidemiology
1.3.1 Incidence by Age and Sex
In Japan, more than 6,000 patients with this disease have been registered, and about 300 new patients are counted annually (Figure 2).11
The male/female ratio is about 1:9, and the age of onset in women peaks around 20 years. The symptoms are not only diverse but also often non-specific, and many cases are still considered to be left undiagnosed.
1.3.2 Geographic Differences
Takayasu arteritis is prevalent in Asia and Middle East, and females tend to be more often affected in both regions. While carotid artery lesions are characteristic in Japan and South America, hypertension due to lesions primarily affecting the abdominal aorta is frequently observed in Asian countries.
1.4 Classification
The classification based on the distribution of vascular lesions proposed by Numano et al. is used (Figure 3).12–14
The cases are divided into those with lesions in the 3 branches of the aortic arch and those with lesions also below the diaphragm. This classification is based on angiographic findings, but it should be noted that inflammatory thickening is diffusely observed also in regions with no marked changes in the vascular lumen.15
2. Pathogenesis
Takayasu arteritis is estimated to be caused by destruction of elastic arteries, particularly, the aorta, by autoimmune mechanisms triggered by environmental factors including infection based on genetic factors.
2.1 Genetic Factors
HLA-B*52 has been reported to be related to the pathogenesis of this disease.16,17
In addition, SNP of the
IL12B
gene has recently been identified as a susceptibility factor.18
2.2 Environmental Factors
Although an involvement of viral infection is suspected, viruses that may induce Takayasu arteritis have not been identified.
2.3 Mechanism of Vascular Damage
Cells including T cells, macrophages, and NK cells along with cytokine abnormalities are considered to damage the vascular wall, but detailed mechanism has not been elucidated.
3. Pathological Findings
Takayasu arteritis, which is classified as a large vessel vasculitis, often affects the aorta and its primary branches. Macroscopically, all layers of the vascular wall are thickened in the affected arteries, and the luminal surfaces of the lesions show irregularities and macular or granular changes. Normal areas are often present between lesions (Figure 4). In the adventitia, vaguely circumscribed fibrotic thickening is notable. In the scar stage, the arterial wall develops plate-like calcification and presents a lead-pipe-like appearance (Figure 5).19,20

Histologically, in an early stage, adventitial mononuclear cell infiltration accompanied by inflammatory cell infiltration to areas around the vasa vasorum is observed, and the media exhibits infarct lesions and granulomatous arteritis mixed with multinucleated giant cells that have phagocytosed fragmented elastic fibers (Figure 6).19–22
As a result of inflammation occurring on the adventitial side of the tunica media, elastic fibers of the media are lost in a moth-eaten pattern. While diffuse fibrosis is caused in the media and adventitia, the intima also develops marked thickening due to cell fibrosis, resulting in luminal stenosis (Figure 7).19,20


In the scar stage, the adventitia is thickened due to marked fibrosis, and vasa vasorum with thickened wall is often observed. The arterial intima shows progressive fibrotic thickening, and major branches of the aorta often develop luminal stenosis. In addition, despite the presence of marked fibrotic lesions, active inflammation accompanied by multinucleated giant cells is often observed in the peripheries of the lesions.19
With the recent increases in long-time survivors due to early detection and improvements in treatments, cases of Takayasu arteritis accompanied by atherosclerosis, aortic aneurysm, or aortic insufficiency are increasing.
4. Symptoms
Initial signs and symptoms include FUO, malaise, neck pain, pain in various regions, and dizziness, which resemble those of upper airway inflammation (Table 4).12
They are followed by signs and symptoms caused by vascular lesions.
Table 4.
Chief Complaints of Takayasu Arteritis at Onset
| Head and neck |
365 (26.6) |
| Dizziness/vertigo |
129 (9.4) |
| Headache |
113 (8.2) |
| Syncope |
36 (2.6) |
| Masseter claudication |
5 (0.4) |
| Neck pain |
133 (9.7) |
| Eyes |
45 (3.3) |
| Sight loss |
2 (0.1) |
| Visual disorder |
37 (2.7) |
| Upper limbs |
238 (17.3) |
| Pulselessness |
67 (4.9) |
| Systolic blood pressure difference (≥10 mmHg) |
53 (3.9) |
| Fatigue |
63 (4.6) |
| Coldness |
23 (1.7) |
| Numbness |
49 (3.6) |
| Heart |
153 (11.1) |
| Breathlessness |
61 (4.4) |
| Palpitation |
29 (2.1) |
| Feeling of chest tightness |
21 (1.5) |
| Lung |
92 (6.7) |
| Hemosputum |
11 (0.8) |
| Sensation of dyspnea |
61 (4.4) |
| Hypertension |
54 (4.0) |
| Lower limbs# |
49 (3.6) |
| Body pain* |
218 (15.9) |
| Systemic manifestation |
562 (41.0) |
| Fever |
476 (34.7) |
| Malaise |
166 (12.1) |
| Fatigue |
23 (1.7) |
Data are expressed as n (% of 1,372 patients). #Leg claudication, fatigue, coldness, numbness, or pain. *Chest, back, or abdominal pain. (Cited from Watanabe Y, et al. Circulation. 2015,12 (c) 2015 by American Heart Association, Inc.)
When objective findings are included, signs of ischemia of the upper limbs such as a difference in blood pressure between the left and right arms and the absence of pulses in the upper limb are observed most frequently in about 66% of the patients, followed by dizziness and headache observed in about 48%. About 14% of the patients had visual disorders, and about 40% of the patients were hypertensive (Table 5).12
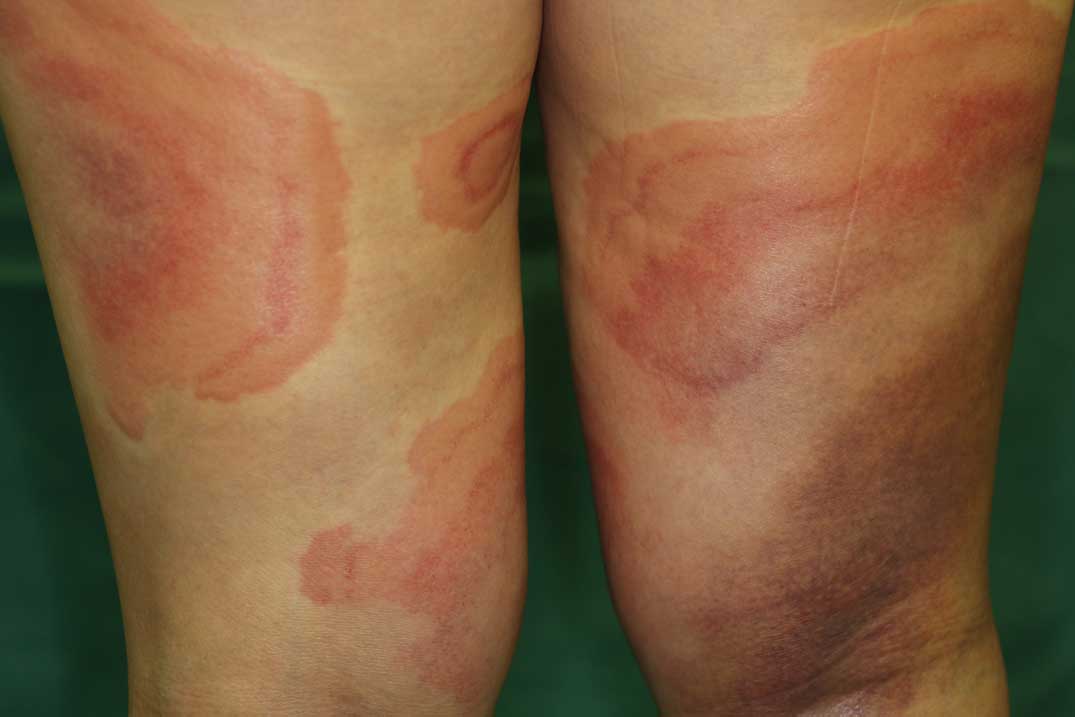
In Takayasu arteritis, many skin lesions (erythema nodosum) are often noted in the lower limbs, particularly, on the anterior aspect of the tibial region (Figure 8).
Table 5.
Clinical Features of Takayasu Arteritis
| |
Prevalence (%) |
| Head and neck |
47.5 |
| Eyes |
14.3 |
| Upper limbs |
66.4 |
| Heart |
37.8 |
| Lung |
10.8 |
| Hypertension |
38.0 |
| Intermittent claudication in the legs |
9.5 |
| Systemic manifestation |
74.9 |
Data are expressed as % of 1,372 patients.
Head and neck ................ Dizziness/vertigo, Headache, Syncope, Hemiplegia, Masseter claudication
Eyes ................................ Sight loss, Visual disorder, Aphose
Upper limbs ..................... Pulselessness, systolic blood pressure difference (≥10 mmHg), Fatigue, Coldness, Numbness
Heart ............................... Breathlessness, Palpitation, Feeling of chest tightness
Lung ................................Hemosputum, Cough, Sensation of dyspnea
Systemic manifestation ... Fever, Malaise, Fatigue
(Excerpts from Watanabe Y, et al. 201512)

Complications include aortic insufficiency, aortic aneurysm, aortic dissection, ischemic attacks of the brain, pulmonary infarction, angina pectoris, subclavian steal syndrome, atypical coarctation of the aorta, and renovascular hypertension (Table 6).12
As observed in
Table 6, complications vary widely including those caused by stenotic lesions and dilated lesions and tend to occur in large numbers. Symptoms are also diverse with some patients remaining asymptomatic and others developing various symptoms in an early stage. Moreover, Takayasu arteritis may be complicated by symptoms of autoimmune diseases such as inflammatory bowel disease, erythema nodosum, and arthritis.
Table 6.
Complications of Takayasu Arteritis
| |
Prevalence (%) |
| Aortic valve regurgitation |
33.2 |
| Grade I* |
14.1 |
| Grade II* |
8.6 |
| Grade III* |
9.8 |
| Grade IV* |
6.2 |
| Ischemic heart disease |
10.6 |
| Eyes |
14.0 |
| Cataract |
7.6 |
| Funduscopic findings |
8.7 |
| Aortic aneurysm |
15.0 |
| Aortic dissection |
1.9 |
| Renal disorder |
11.2 |
| Hypertension |
39.4 |
| Renal artery stenosis |
13.1 |
| Brain ischemia |
13.2 |
| Thrombosis |
3.7 |
| Hemorrhage |
0.9 |
Data are expressed as % of 1,372 patients. *Regurgitation grade measured by color Doppler ultrasound. (Excerpts from Watanabe Y, et al. 201512)
5.1 Laboratory Findings
Currently, there are no specific findings on blood or biological tests for the diagnosis of Takayasu arteritis. Therefore, non-specific indices of inflammation including CRP and ESR are used as the biomarkers for the diagnosis of Takayasu arteritis. Increasing lines of evidence suggest that the presence of specific human leucocyte antigen (HLA) class I molecules, such as HLA-B*52 and HLA-B*67,23,24
are related to clinical manifestations and genetic predisposition of Takayasu arteritis. Thus, the presence of HLA-B*52 and HLA-B*67 are useful as adjunct to the diagnosis of Takayasu arteritis.
Laboratory findings useful for the evaluation of the activity of Takayasu arteritis include an increase in the number of leukocytes, exacerbation of anemia, and abnormalities of immunological indices such as immunoglobulin and complement levels as well as the elevation of CRP and ESR. Particularly, CRP is useful for the follow-up of patients who have received GC therapy,25,26
but it should be noted that CRP cannot be used for the evaluation of the activity of Takayasu arteritis in patients administered TCZ, a humanized antihuman IL-6 receptor monoclonal antibody.
Although serum amyloid A,27
Pentraxin-3,28–30
IL-6,31
IL-18,32
sRAGE,33
soluble ICAM-1,34
and matrix metaprotease35
have also been reported to be useful for the evaluation of the activity of Takayasu arteritis, these biomarkers have not been used for the actual clinical diagnosis of Takayasu arteritis and are not covered by health insurance in Japan as of 2018.
5.2 Imaging Findings
CT and MRI are recommended imaging modalities for the initial work-up of Takayasu arteritis (Figure 9).
5.2.1 Chest Radiography
Irregular contour of the descending aorta is noted in the margin of the descending aorta (Figure 9A, arrows).36
5.2.2 CT
By CT, large vessels, coronary arteries, and various organs are evaluated (Figure 9B).
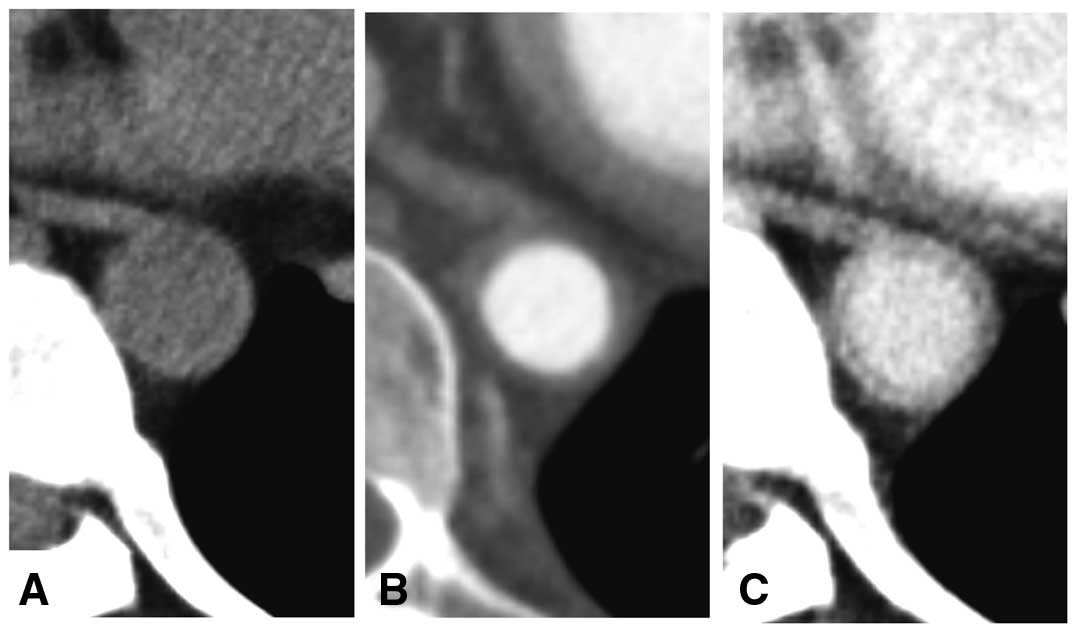
Dynamic CT angiography protocol consisted of a set of non-contrast, arterial-phase and late-phase scans is recommended for the initial examination, but a reduction of radiation exposure must be considered on follow-up examinations. On non-contrast images, the density of the arterial wall appears higher than the lumen (Figure 10A).37
Arterial phase images demonstrates clear contrast between the lumen and wall (Figure 10B). In the late-phase images, the “double ring-like pattern” is observed in the aortic wall in the early stage of Takayasu arteritis (Figure 10C).
In the chronic stage, stenosis or dilatation of the lumen, circumferential calcification of the wall (Figure 9B), and development of collaterals are noted.36,38
5.2.3 MRI
By contrast-enhanced MR angiography, the vascular lumen, wall thickening, and contrast enhancement of the wall are evaluated (Figures 11
and
12).15,39,40
Morphological information of large vessels is also available by non-contrast MR angiography. For long-term follow-up, MRI is recommended because of no risk for radiation exposure.41
5.2.4 Angiography
Angiography is indicated for endovascular treatment and evaluation of coronary and small arteries.42
5.2.5 Ultrasonography
In Takayasu arteritis, a hyperechoic area is observed in the thickened wall of the common carotid artery (macaroni sign) (Figure 9C).43,44
5.2.6 18F-FDG PET/PET-CT
Accumulation of 18F-FDG in the wall of large vessels is a finding useful for the diagnosis of Takayasu arteritis (Figure 13).45
Since April 2018, it has become possible to perform 18F-FDG PET/PET-CT under health insurance at some PET facilities in Japan for patients with large vessel vasculitis when the localization or activity of the lesion is difficult to determine by other examinations.
5.3 Ophthalmological Examinations
5.3.1 Manifestations Related to Ocular Ischemia
18–30% of patients with Takayasu arteritis have ocular symptoms such as blurred vision, transient visual loss, and eye pain,46,47
and most of these symptoms come from circulatory disturbance to the eye. Ophthalmological examinations can reveal manifestations related to ocular ischemia including Takayasu retinopathy (hypoperfusion retinopathy), ischemic optic neuropathy, and rubeosis iridis caused by type I, II, and V Takayasu arteritis (Numano’s classification by angiography)13
(Figure 3).12–14
In Takayasu retinopathy, tortuosity of retinal vessels, dilated retinal veins, retinal microaneurysms, hemorrhage, and cotton-wool spots can be seen in the fundus (Figure 14). Multiple microaneurysms scattered in the retina and wreath-like vascular anastomoses around the optic disc indicating prolonged retinal ischemia are characteristic findings (Figure 1).7
On fluorescein fundus angiography, prolonged arm-to-retina circulation time, occlusion of capillaries, microaneurysms, and arteriovenous anastomoses can be observed, and neovascularization in the retina and optic disc can be seen in the severe cases (Figure 15).

5.3.2 Ocular Manifestations Due to Systemic Hypertension
In Type III, IV, or V Takayasu arteritis, which may be complicated by hypertension due to abdominal aorta/renal artery stenosis, hypertensive retinopathy may be observed. In this condition, narrowing of the retinal artery, retinal edema, white spots, hemorrhage, and optic disc edema develop. Although reports are few, hypertensive choroidopathy may also develop.
6. Diagnostic Methods and Criteria
6.1 Diagnostic Criteria
For the present revision of the guidelines, the Subcommittee of Large Vessel Vasculitis of the MHLW Research Committee for Intractable Vasculitis prepared new diagnostic criteria for Takayasu arteritis with partial modifications of those in the Guidelines 2008 reflecting progress in medicine without changing their basic principles (Table 7). The diagnosis is made primarily by imaging (CT, MRI, ultrasonography, PET-CT, chest radiography, and angiography). According to the diagnostic criteria in
Table 7, Takayasu arteritis is diagnosed when there is 1 or more of the symptoms listed in A), when there are either multiple or diffuse hypertrophic lesions, stenotic lesions (including occlusions), or dilated lesions (including aneurysms) in the aorta or its primary branches or both as in B), and when the differential diagnoses shown in C) can be excluded.
Table 7.
Diagnostic Criteria for Takayasu Arteritis
| A. Signs and symptoms |
1. Systemic signs and symptoms: Fever, generalized malaise, easy fatigability, lymphadenopathy (cervical), hypertension in younger
patients (≥140/90 mmHg) |
| 2. Pain: Carotidynia, chest pain, back pain, lower back pain, shoulder pain, upper limb pain, lower limb pain |
3. Visual signs and symptoms: Transient or persistent visual impairment, preocular bright or dark sensation, loss of vision, fundus changes
(hypotensive changes, hypertensive changes) |
4. Head and neck signs and symptoms: Headache, toothache, jaw claudication,*a dizziness, hearing impairment, tinnitus, syncope, cervical
vascular murmurs, hemiplegia |
5. Upper limb signs and symptoms: Numbness, cold sensation, difficulty in arm raising, claudication,*b abnormal pulse and blood pressure
(weakness or loss of radial artery pulse or left-right difference of ≥10 mmHg), increased pulse pressure (related to aortic insufficiency) |
6. Lower limb signs and symptoms: Numbness, cold sensation, weakness, claudication, abnormal pulse and blood pressure (augmented
or weakened pulse of lower limb arteries, reduced blood pressure, blood pressure difference between upper and lower limbs*c) |
7. Chest symptoms: Shortness of breath, palpitation, dyspnea, bloody sputum, sensation of chest compression, anginal symptoms,
arrhythmia, cardiac murmur, back vascular murmur |
| 8. Abdominal signs and symptoms: Abdominal vascular murmur, complication by ulcerative colitis |
| 9. Skin signs and symptoms: Erythema nodosum |
| *a Intermittent mastication due to pain caused by mastication |
| *b Intermittent exertion of the upper limbs due to pain and weakness caused by exertion |
| *c Instances other than “10–30 mmHg higher in the lower limb than upper limb” |
| B. Examination findings |
Imaging examination findings: In the aorta or its primary branches*a or both, multiple*b or diffuse hypertrophic lesions,*c stenotic lesions
(including occlusions),*d or dilated lesions (including aneurysms)*d detected. |
*a The aorta and its primary branches corresond to the aorta (ascending, arch, thoracic descending, abdominal descending), primary
branches of the aorta (including the coronary artery), and pulmonary artery. |
| *b Multiple lesions are defined as those that involve two or more of the above arteries or sites or two or more segments of the aorta. |
*c Hypertrophic lesions are detected by ultrasonography (macaroni sign of the common carotid artery), contrast-enhanced CT, contrast-
enhanced MRI (circumferential contrast enhancement of the arterial wall), and PET-CT (circumferential FDG uptake of the arterial wall). |
*d Stenotic lesions and dilated lesions are detected by chest radiography (wave-like deformation of the descending aorta), CT angiography,
MR angiography, echocardiography (aortic insufficiency), and angiography. They are accompanied by dilatation of the ascending aorta
and frequently also by aortic insufficiency. In the chronic stage, circumferential calcification of the arterial wall is visualized by CT, and
the development of collateral circulation is detected by CT angiography and MR angiography |
Points of attention in imaging diagnosis: Contrast-enhanced CT is performed in the late phase of contrast enhancement. CT angiography is
performed in the early phase of contrast enhancement with 3-dimensional image processing. Angiography is usually performed when other
procedures such as endovascular treatment and coronary artery angiography or left ventriculography are simultaneously intended. |
| C. Conditions to be included in the differential diagnoses of Takayasu arteritis |
Arteriosclerosis, congenital vascular anomaly, inflammatory abdominal aortic aneurysm, infectious aneurysm, syphilitic mesaortitis, giant cell
arteritis (temporal arteritis), vascular Behçet’s disease, IgG4-related diseases. |
| <Diagnostic categories> |
| Definite: At least 1 of the items in A + any of the conditions in B are observed, and conditions in C can be excluded. |
| (Suggestive findings) |
| 1. Hematological/biological findings: Increased ESR or CRP, leukocytosis, anemia |
| 2. Genetic examinations: Presence of HLA-B*52 or HLA-B*67 |
Since findings suggestive of inflammation are exacerbated on blood tests in most patients with active Takayasu arteritis, they are useful as references for the diagnosis. However, inflammatory signs are negative in some patients at the initial examination, and it must be noted that there are usually no signs of inflammation in patients with inactive Takayasu arteritis. Epidemiological characteristics (a predilection for young women and symptoms usually occur at ages under 40 years) and presence of HLA-B*52 or HLA-B*67 are suggestive findings.
Since April 2018, 18F-FDG PET has begun to be performed under health insurance at some PET facilities in Japan for patients with large vessel vasculitis in which the localization or activity of the lesions is difficult to determine by other examinations.
6.2 Differential Diagnosis
Takayasu arteritis should be differentially diagnosed from (1) arteriosclerosis, (2) congenital vascular anomalies, (3) inflammatory abdominal aortic aneurysm, (4) infectious aneurysm, (5) syphilitic mesaortitis, (6) giant cell arteritis (temporal arteritis), (7) vascular Behçet’s disease, and (8) IgG4-related periaortitis. It can be differentiated from arteriosclerosis to an extent by the age of onset and distribution of the affected vessels. Mid-aortic syndrome is a possible congenital vascular anomaly that can be mistaken for Takayasu arteritis, but it can be differentiated, because the aortic wall is smooth despite stenosis. Inflammatory abdominal aortic aneurysm shows signs of inflammation, is often accompanied by hydronephrosis, and exhibits the characteristic mantle sign on CT. Infectious aneurysm often presents as a saccular aneurysm, can be multiple, but lacks other lesions, and it can be differentiated by checking other findings. Syphilitic mesaortitis is rarely encountered recently and is differentiated serologically and bacteriologically. GCA frequently affects older individuals and is often complicated by myalgia (polymyalgia rheumatica). Vascular Behçet’s disease occasionally presents with saccular aneurysms but can be differentiated by checking other findings. IgG4-related periaortitis can be differentiated by the serum IgG4 concentration and IgG4-related lesions in other organs.
7. Policies and Guidelines of Treatment
Table 8
shows recommendations about treatment for Takayasu arteritis, and
Figure 16
shows a flow chart of treatment for Takayasu arteritis.
Table 8.
Recommendations and Levels of Evidence About Treatments for Takayasu Arteritis
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
I |
B |
| Steroid pulse therapy |
IIb |
C |
| Methotrexate (MTX)* |
IIa |
B |
| Azathioprine (AZA) |
IIa |
B |
| Cyclophosphamide (CY) |
IIb |
B |
| Mycophenolate mofetil (MMF)* |
IIb |
B |
| Tacrolimus (TAC)* |
IIb |
C |
| Cyclosporine (CyA)* |
IIb |
C |
| Tocilizumab (TCZ) |
I |
B |
| TNF-inhibitors* |
IIa |
B |
| Antiplatelet drugs |
IIa |
B |
| Vascular bypass surgery |
IIa |
C |
| Endovascular treatment |
IIb |
C |
*Uncovered by health insurance in Japan.
7.1 Glucocorticoid/Immunosuppressant
7.1.1 Glucocorticoid
a. Indications
In previous cohort analyses, PSL was administered to 79–94% of the patients6,48–50
based on the disease activity (judged according to Kerr’s criteria50a).
b. Initial Dose and Period of Its Continuation
According to overseas reports, regimens such as PSL at 1 mg/kg/day×1 month48,50–52
have been used. The Guidelines 2008 recommend a low dose regimen of PSL 20–30 mg/day×2 weeks but has the additional statement, “increased to a maximum of 60 mg/day depending on the case”, and the efficacy rate of PSL alone by this regimen was about 50%.53
In a Japanese retrospective cohort of 106 cases,6
the initial dose of PSL was 38.9±14.6 mg/day and approximately corresponded to 0.5–1 mg/kg/day.
c. Steroid Pulse Therapy (recommendation class IIb, evidence level C)
This regimen is performed in emergent cases and at relapse of refractory cases.52,54,55
d. Dose-Reduction Rate and Maintenance Dose
The dose of PSL should be reduced by confirming remission according to (1) symptoms, (2) blood inflammation markers, and (3) imaging findings. In a Japanese retrospective cohort study of 106 cases,49
the PSL dose-reduction speed was the most important factor that contributed to relapse, and the relapse rate differed significantly between dose-reduction speeds faster and slower than 1.2 mg/month. By the protocol of the Guidelines 2008,56
the dose was reduced by 5 mg every 2 weeks to 10 mg/day and by 2.5 mg every 2 weeks thereafter to the maintenance dose. However, considering that the PSL dose at relapse was 13.3±7.5 mg/day in the Japanese cohort,6
the dose should be reduced very carefully after it has been reduced to less than 20 mg/day. The maintenance dose is PSL 5–10 mg/day in many protocols.49,52,57
e. GC-Free Remission
GC-free remission was maintained in 5 (17%) of the 30 patients by a protocol of the United States aimed to discontinue PSL.51
According to the surveillance of 150 cases in Japan reported in 1975, the GC discontinuation rate was 37.4%.56
f. Dose Increase at Relapse
Whether the remission-inducing therapy is repeated or minor dose increases are attempted should be judged according to the severity.
7.1.2 Immunosuppressants
a. MTX (recommendation class IIa, evidence level B) (Uncovered by Insurance in Japan)
In a single-arm study in the United States, MTX (gradually increased from 0.3 mg/kg/week) and PSL (gradually reduced from 1 mg/kg/day) were administered to those in whom the effectiveness of PSL alone was insufficient or relapse was observed, resulting in remission attained in 81% and remission maintained after 7–18 months in 50%.58
MTX is the immunosuppressant that has been used most frequently for Takayasu arteritis. However, its problems are that the progression of vascular lesions may not be prevented and that it is often discontinued due to the lack of effect or adverse effects.6,50,59
b. AZA (recommendation class IIa, evidence level B) (Covered by Health Insurance in Japan Since February 23, 2011)
In a single-arm study in India, AZA (2 mg/kg/day) and PSL (gradually reduced from 1 mg/kg/day) were administered to newly-onset patients. While remission was attained and maintained until after 12 months in all patients, CRP or ESR rose after 12 months in 40%, and progression of vascular lesions was observed in some.57
AZA is highly evaluated in protocols of various countries and is often administered to patients not responding to MTX.
c. CY (recommendation class IIb, evidence level B) (Covered by Health Insurance in Japan Since February 23, 2011)
In a prospective cohort of 20 cases in the United States, CY was added at 2 mg/kg/day (dose was modified to adjust the WBC to >3,000/μL) to 7 patients who responded insufficiently to PSL alone. The PSL dose-reduction effect was observed in all patients, and further progression was prevented in 4 of the 6 patients who had shown progression of vascular lesions.48
In many protocols, CY is substituted for MTX or AZA after 3 months in consideration of the possibility of adverse effects.
d. MMF (recommendation class IIb, evidence level B) (Uncovered by Health Insurance in Japan)
According to reports of 21 cases from India55
and 10 cases from Brazil,60
MMF (2 g/day) was administered to patients first treated for Takayasu arteritis or those who showed poor treatment responses, and reduced disease activity, decreases in CRP/ESR, and PSL-reducing effect were observed in most cases.
e. Calcineurin inhibitors (recommendation class IIb, evidence level C) (Uncovered by Health Insurance in Japan)
There have been case reports using tacrolimus at 1.5–4 mg/day61,62
and CyA at 150 mg/day or 4.3 mg/kg/day.63,64
7.2 Biological Agents, Antiplatelets
7.2.1 Biological Agents
The efficacy of biological agents for Takayasu arteritis has been evaluated primarily regarding two groups of biological agents, namely, TNF inhibitors and anti-IL-6 receptor antibody.
a. TCZ (recommendation class I, evidence level B) (Covered by Health Insurance in Japan Since August 25, 2017)
In the serum of patients with Takayasu arteritis, the IL-6 concentration has been reported to increase in association with the disease activity.31,32
Since Nishimoto et al. first reported TCZ treatment for patients with refractory Takayasu arteritis,65
similar reports have appeared in Japan, Europe, and the United States suggesting the efficacy of TCZ against Takayasu arteritis.66–70
In a retrospective registry study of biological agents (TNF inhibitors and TCZ) in France, 49 patients with Takayasu arteritis were treated with biological agents (35 with a TNF-inhibitor, 14 with TCZ) with remission rates after 6 and 12 months of 75 and 83%, respectively. The 3-year relapse-free rate was 90.9% in those treated with biological agents and 58.7% in those treated with conventional synthetic DMARDs, showing a significant difference (P=0.0025).71
In Japan, a clinical trial of TCZ was carried out in patients with refractory Takayasu arteritis (TAKT study).72
By the double-blind design, GC was forcedly tapered, and the time until relapse was evaluated as a primary endpoint. The hazard ratio in the TCZ group relative to the placebo group was 0.41 (decrease rate of relative risk: 59%) (P=0.0596).
b. TNF Inhibitors (recommendation class IIa, evidence level B) (Uncovered by Health Insurance in Japan)
Hoffman et al. reported that TNF inhibitor therapy was effective for Takayasu arteritis resisting GC therapy.73
However, according to the report by Schmidt et al. in 20 patients with refractory Takayasu arteritis, the remission induction rate was also satisfactory, but relapse was noted in 33% while TNF inhibitor therapy was continued, and the administration was discontinued due to adverse effects in 20%.74
No clear conclusion has been reached regarding the efficacy of TNF inhibitor therapy against Takayasu arteritis.
7.2.2 Antiplatelets (recommendation class IIa, evidence level B)
In patients with Takayasu arteritis, the occurrence of acute ischemic events such as acute myocardial infarction, unstable angina, transient ischemic attacks, stroke, acute lower limb ischemia, and acute intestinal ischemia has been reported to be significantly suppressed by oral administration of an antiplatelet (aspirin).75
7.3 Surgical Treatment (Open Surgery and Endovascular Treatment)
7.3.1 Principles of Surgical Treatment
In both open surgery and endovascular treatment (EVT) for patients with Takayasu arteritis, pre- and post-operative medical management of disease activity is crucial to obtain good surgical outcomes. The evaluation of indications for surgery, selection of strategies and the procedures, and postoperative management should be conducted by a multidisciplinary team. As for occlusive lesions, vascular restenosis occurs more frequently after EVT than after bypass surgery. Therefore, careful consideration is required to select EVT as the first-line treatment. After surgical treatment, all patients should have lifelong follow-up care.
7.3.2 Disease Activity and Surgical Outcomes
The purpose of surgical treatment is to improve organ blood flow or to prevent rupture of aneurysms. Surgical treatment is best performed during a period of remission (non-active period) in which the patient needs neither GC nor immunosuppressants for maintaining the condition, if possible. However, even in patients in active phase, urgent surgery is often unavoidable to prevent aneurysm rupture or permanent ischemic organ damage, and performed under every effort to manage the inflammation.76–79
After surgery, maintaining remission leads to favorable long-term outcomes.76,79,80
The selection of surgical treatment strategy, open surgery or EVT, is still based on experts’ experiences. The strategy should be selected considering the long-term prognosis of the patient, since many patients with Takayasu arteritis have a potential of favorable life prognosis.
7.3.3 Postoperative Management
If GC has been used preoperatively, GC therapy should be resumed immediately after surgery. Even in patients in remission phase, low dose GC therapy is recommended.76
The operated sites and residual lesions should be followed up routinely to prevent late complications including pseudoaneurysm at the anastomosis.
7.3.4 Treatment for Vascular Lesions
a. Surgical Revascularization
While Numano’s angiographic classification (Figure 3)13
is appropriate for representing epidemiological case profiles, the following Ueno’s clinical classification is useful to consider the target lesions of surgical treatment.81
i. Treatment for Type I Lesions (Occlusive Lesions of Aortic Arch Branches)
The patient can be treated conservatively if they have few problems in their daily life. Concerning visual impairment, stage 2 and early stage 3 by Uyama’s classification82
or a fundus blood pressure of around 50 mmHg are considered to be indications for revascularization.83
Bypass surgery is often selected.51,84
If there are severely diseased lesions in the bilateral carotid arteries, unilateral revascularization is recommended to avoid cerebral hyperperfusion syndrome.83–85
ii. Treatment for Type II Lesions (Occlusive Lesions of Thoracoabdominal Aorta)
If the lesion does not involve the orifice of major branches of abdominal organs, extra-anatomical bypass surgeries such as aorto-aortic bypass grafting or replacement of the diseased aorta can be indicated. If there are symptoms of organ ischemia, surgical treatment is indispensable. Particularly the refractory renovascular hypertension should be managed for improving life prognosis by adopting renal artery bypass surgery, autologous renal transplantation, or even nephrectomy.86
For the patients with abdominal angina, mesenteric artery revascularization should be considered.
iii. Treatment for Type III Lesions (Extensive Occlusive Lesions; Type I+Type II)
In patients with both severe brain ischemia and severe hypertension, to prevent the brain from being exposed to high blood pressure after cerebral arterial reconstruction, simultaneous surgical treatment for renovascular hypertension should be considered. If brain ischemia is not severe, the initial target for surgery should be renovascular hypertension. Then, if hypoperfusion-associated brain disorders appear due to normalized blood pressure, carotid revascularization is performed in the second stage.83
iv. Treatment for Type IV Lesions (Aneurysmal Lesions)
True aneurysms are the majority of the aneurysm. The Surgical indications for aneurysms in Takayasu arteritis are similar to those for common degenerative aortic and peripheral artery aneurysms.
Vascular occlusion and restenosis are may occur frequently after surgical revascularization. On the contrary, there is a report that the long-term patency and survival in Japan are relatively satisfactory.87
b. EVT
i. EVT for Occlusive Lesions
Even though EVT is usually selected restrictively for localized lesions,80,88,89
restenosis occurs more frequently after EVT than after bypass surgery.51,79,80,88–90
Since the target artery wall is occasionally scarred and fibrosed in the whole layers, a deliberate decision should be made to select EVT as the first-line treatment. The effect of stenting on the patency remains unknown.
ii. Stent-Graft Implantation for Dilated Lesions
Long-term outcomes have not been revealed yet.
7.3.5 Treatments for Cardiac Lesions
a. Aortic Insufficiency
Indications for aortic valve replacement should be evaluated based on patient’s condition and left ventricular function.91,92
Biological valves are used for elderly patients and women who wish pregnancy. Reports that positively recommend aortic root replacement (Bentall procedure) have increased. In addition, if the patient seems difficult to achieve remission of the inflammation even after long-term medical treatment, aortic root replacement is recommended even without aortic root dilatation.93–95
b. Aortic Root Dilatation
Composite graft replacement of aortic valve, aortic root, and ascending aorta, combined with re-implantation of the coronary arteries into the graft (Bentall procedure) is the most common procedure. Valve-sparing aortic root replacement is not recommended as the aortic valve tends to degenerate in many patients in a long period after surgery.96
The suture usually needs additional reinforcements in the surgical procedures.97
c. Coronary Artery Stenosis
In coronary artery bypass surgery, appropriate graft selection is essential. If there are lesions in the aortic arch branches, the internal thoracic artery graft is inappropriate. The proximal end of a free graft should not be anastomosed to the diseased aorta. If coronary artery bypass grafting is performed simultaneously with procedures such as ascending aortic replacement, the proximal anastomosis should be made at the prosthetic aortic graft. Ostial endarterectomy and ostial patch angioplasty are effective in some patients.98
7.3.6 Treatment for Pulmonary Artery Lesions
A pressure overload of the right ventricle due to pulmonary hypertension is an indication for surgery and is treated by patch angioplasty using the pericardium or prosthetic graft replacement of the pulmonary artery. Favorable results of plain old balloon angioplasty (POBA) have also been reported.99
8. Prognosis
The 15-year survival rate of patients with Takayasu arteritis has been reported to be around 80%,100,101
but it can be modified by some poor prognostic factors, which include retinopathy, hypertension, aortic insufficiency, aortic aneurysm, progressive course, onset at a young age, and increased ESR.100
The disease activity may temporarily subside, but there have been many reports of high relapse rates.
The percentage of patients who require surgical treatment varies from 12 to 50%, but the percentage of those who receive early surgical intervention including percutaneous procedures is increasing.96
Heart failure is a more frequent cause of death than anastomotic complications, and heart failure is caused by aortic valve disease or ischemic heart disease.87
Late detachment of the aortic prosthetic valve and coronary bypass graft insufficiency are observed in about 10% of the patients who have undergone surgery,97
and the incidence of surgery-related late complication in Takayasu arteritis is high.
For the future, the cure rate is expected to be improved by the development of diagnostic techniques, particularly imaging modalities such as PET and MRI, and the use of biological agents.
9. Takayasu Arteritis in Children
According to the database for national research of intractable diseases run by the MHLW of Japan, there were 140 patients with childhood-onset Takayasu arteritis in Japan in 2016. The list included patients who had survived to adulthood as well as 70 children who were <16 years of age. The male/female ratio was 1:7, and the median age at the onset was 10–11 years.
The initial symptoms of Takayasu arteritis are fever, malaise, abdominal pain, chest pain, arthralgia, and lymph node enlargement, with fever noted in about 80% of the patients.102,103
Children are now increasingly being diagnosed at an early stage of the disease because of improved diagnostic techniques and the spread of knowledge about Takayasu arteritis. In more than half the patients with childhood-onset Takayasu arteritis, the lesions are first seen in the abdominal aorta and its branches. In adult patients, the lesions occur most frequently in the head and neck arteries, ascending aorta, and aortic arch.102,103
As there are no diagnostic criteria for childhood-onset Takayasu arteritis, the diagnosis depends on the criteria established for adults. Nevertheless, specific diagnostic criteria have been established for those applying for medical fee subsidies for children with specific chronic diseases.104
In such cases, inflammatory markers such as CRP and the ESR are generally elevated during the acute period. Imaging modalities, including ultrasonography, contrast-enhanced CT, CT angiography, MRI, and positron emission tomography (PET)-CT, are used for diagnosis. In addition, as lesions in children with Takayasu arteritis are more often located in the abdominal aorta and its branches than in adults, ultrasonography is a useful, convenient, non-invasive tool for diagnosis and follow-up. PET-CT is also a powerful emerging modality not only for the early diagnosis but for evaluating the distribution of lesions and monitoring disease activity. Since April 2018, PET-CT has become available under health insurance coverage at some PET facilities in Japan for patients with large-vessel vasculitis in whom the localization or activity of the lesions is difficult to determine by other modalities.
Treatment for children with Takayasu arteritis is commonly initiated with two to three courses of steroid pulse therapy followed by daily PSL (0.8–1.0 mg/kg). MTX (not covered by insurance in Japan) or AZA is generally used to prevent relapse and to accelerate GC tapering from the onset.58
IVCY and TCZ could be considered for those who have severe visceral lesions, who resist the initial treatment, who face difficulty reducing their GC dose, and/or who have repeated relapse. As children are less likely than adults to develop gonadal dysfunction from the CY, early treatment with IVCY may be positively considered.105
Because GCs — including pulsed therapy for patients whose status is complicated with renovascular hypertension — could increase the risk of reversible posterior leukoencephalopathy and hypertensive brain hemorrhage, the blood pressure should be tightly controlled according to the age-matched standard value. Growth impairment is also a serious adverse effect of GC in children. Therefore, if inflammatory markers have been negative for 6–12 months, the PSL dose should be reduced to a level that does not affect growth. As CY has exhibited both carcinogenicity and gonadal toxicity, excessive use should be avoided. Intravenous TCZ therapy has been reported effective for treating childhood-onset Takayasu arteritis. The reports regarding subcutaneous TCZ therapy are limited, however, and future evaluation is awaited. Antiplatelets and anticoagulants are administered to patients who have vascular stenosis or marked vascular dilatation and are at high risk of thrombosis. More than half the children with Takayasu arteritis experience relapse and require treatment over a long period, although some can eventually discontinue all treatment.
III. Giant Cell Arteritis
1. Definition/Epidemiology/Subclassification
1.1 Definition
Giant cell arteritis (GCA) was defined by the 2012 CHCC as “arteritis, often granulomatous, usually affecting the aorta and/or its major branches, with a predilection for the branches of the carotid and vertebral arteries. Often involves the temporal artery. Onset usually in patients older than 50 years and often associated with polymyalgia rheumatica.”1
Since the temporal artery is not affected in all patients with GCA, and since temporal artery may be affected in other vasculitides, the term “giant cell arteritis” was adopted.1
1.2 Epidemiology
The age at onset is usually 50 years or above, and the disease has a slight predilection for females. According to the nationwide epidemiological survey by the Ministry of Health and Welfare of Japan, the estimated number of treated patients was about 690 (0.65/100,000 persons), and the mean age at onset was 71.5 years.106
1.3 Subclassification
GCA is subclassified into cranial GCA (C-GCA) localized in intracranial arteries and large-vessel GCA (LV-GCA), arteritis not localized intracranially but is present extracranially. However, the definition or clinical significance of LV-GCA has not been fully established.
2. Pathogenic Mechanism
Although the etiology of GCA is still unclear, it is known to be correlated with HLA-DRB1*0401, HLA-DRB1*0404, HLA-DQA1*0301, and HLA-DQB1*0302 as genetic factors.107,108
In addition, infection by microorganisms such as
Mycoplasma pneumoniae,
Chlamydia pneumoniae, and
parvovirus 19
and environmental factors including smoking have been also reported to be involved in its pathogenesis.109
In GCA, inflammatory cell infiltration in all layers is observed as in Takayasu arteritis. Especially, giant cells in the media and intima and occlusion of vascular lumen due to intimal thickening are characterized. CD83-positive dendritic cells and CD4-positive T cells in the adventitia play an important role for this process. Immature dendritic cells in the adventitia are matured through Toll-like receptor (TLR) and their matured dendritic cells activate CD4-positive T cells. Moreover, cytokines secreted by these cells, IFN-α and IL-17, contribute to activation of macrophages and formation of giant cells. These macrophages and giant cells infiltrate into the intima, and secrete vascular endothelial growth factor, resulting in proliferation of vascular smooth muscle cells and remodeling of the vascular wall.
3. Pathological Findings
3.1 Affected Vessels
In GCA, middle-sized muscular arteries, particularly branches of the carotid artery and vertebral artery, are likely to be affected. Also, large elastic arteries such as the aorta, subclavian artery, and common iliac artery are often affected. Because the lesions are localized and occasionally segmented, temporal artery should be sampled over 2–4 cm and examined carefully.
3.2 Histological Findings
In muscular arteries such as the temporal artery, the lesions are formed primarily around the internal elastic lamina and the intimal side of the media (Figure 17).110
Inflammation is granulomatous; histiocyte proliferation, lymphocyte, plasma cell, and macrophage infiltration, and Langhans and foreign body giant cells are observed (Figure 18).110
The internal elastic lamina is ruptured and lost, multinucleated giant cells are likely to appear near it, and they occasionally seen to phagocytose the elastic lamina. Fibrinoid necrosis or neutrophil infiltration is rarely observed. The intima shows non-specific fibrotic thickening and may cause stenosis, but it is considered a secondary change associated with inflammation of the media.


In the aorta (elastic artery), granulomatous inflammation is formed primarily in the middle layer of the media. Elastic fibers are lost locally in a moth-eaten pattern (Figure 19),110
and histiocyte and lymphocyte infiltration and multinucleated giant cells are observed at such sites (Figure 20).110
Inflammation is also observed along the vasa vasorum that enters from the adventitia. In addition, loss of smooth muscle cells considered an ischemic change may develop at sites not affected by granulomatous inflammation, but elastic fibers remain at such sites. Thus, many infiltrating cells are observed with loss of elastic fibers at inflamed areas, whereas elastic fibers are observed without inflammatory cells at non-inflamed areas; these contrasting observations are mixed (Figure 19, inserted figure).110
3.3 Differential Diagnoses and Similar Diseases
Takayasu arteritis: Since the frequent age at onset and sex difference in prevalence differ, age and sex are important information. In fact, differential diagnosis of GCA from Takayasu arteritis is considered difficult by pathological findings alone. However, inflammation occurs primarily in the inner and middle layers of the media in GCA, but the outer layer of the media to the adventitia are affected in Takayasu arteritis. Giant cells and granulomatous change are more notable in GCA than in Takayasu arteritis.
Buerger disease-like arteritis: Cases in which marked intimal thickening is observed in the temporal artery not accompanied by destruction of the internal elastic lamina or the media have been reported as Buerger disease.
Localized GCA: Granulomatous arteritis with giant cell infiltration occasionally occurs locally in small arteries of the urogenital organs including the uterus, ovary, and ureter and mammary glands, but their sites and the size of the affected arteries differ from those in GCA.
4. Symptoms
Generalized symptoms of GCA include fever associated with inflammation (often mild fever, occasionally remittent fever), malaise, easy fatigability, body weight loss, polymyalgia, and polyarthralgia. Local symptoms vary according to the site of the affected vessel. In the nationwide survey of 66 cases of GCA in Japan, symptoms including headache of the temporal region (80.3%), scalp pain (63.3%), tenderness of the temporal artery (52.6%), weakened pulse of the temporal artery (40.0%), visual impairment (43.9%), and ischemic optic neuropathy (21.2%).106
External carotid artery lesions may cause temporal pain, swelling/tenderness of the temporal artery, jaw claudication, tongue claudication, and mandibular pain occur. These symptoms may occur both bilaterally and unilaterally. Scalp pain is often complained of when the patient combs or brushes hair, and jaw claudication is characterized by jaw pain during mastication.
If the ophthalmic artery is affected, symptoms including impairment of visual acuity, loss of vision, misty vision, and diplopia may occur. Abnormalities of visual acuity/visual field appear early after the onset, and caution is necessary, because about 23–44% of the patients show reduced visual acuity, and 4.4–6.5% show complete loss of vision.106,111
Sudden loss of vision is often first noted on awakening in the morning.
As mentioned in the definition of CHCC2012, lesions of GCA appear in the aorta and/or its major branches. The presence of subclavian artery lesions is considered a characteristic of LV-GCA.112
Common carotid artery lesions cause headache, dizziness, syncopal attacks, and hemiplegia. Subclavian artery lesions cause pain, cold sensation, and easy fatigability of the upper extremities and present attenuation or loss of the pulse of the radial artery and a difference in the blood pressure between the left and right limbs (≥10 mmHg). Aortic lesions cause disorders including chest pain, back pain, aortic aneurysm/dissecting aortic aneurysm, and aortic insufficiency.
Caution is needed as polymyalgia rheumatic (PMR) is observed in 30–60% of the patients with GCA, and as PMR is complicated by GCA in 16–21% of the patients.106,113
5. Laboratory/Imaging Findings
5.1 Laboratory Findings
There are no laboratory findings specific to GCA. On blood tests, high ESR, increased CRP, chronic inflammatory anemia, and hypoalbuminemia are observed. An ESR of ≥50 mm/h is included in criteria for the classification of GCA, and GCA with normal CRP and ESR is rare. CRP and ESR are also used as markers of the flare of disease activity, and elevations of CRP and ESR may be the only findings at the relapse.114–116
The IL-6 concentration in peripheral blood (uninsured) has been reported to change with the disease activity.117,118
5.2 Imaging Findings
Diagnostic imaging modalities for GCA include CT, MRI, ultrasonography, and FDG-PET.
On MRI, thickening of the vascular wall and contrast enhancement by contrast agents are observed in 81% of the patients. MRI is also useful for the evaluation of extracranial and intracranial arteries (Figure 21).119
On CT, wall thickening and contrast enhancement are observed in 45% of the patients,119
and the modality is superior to MRI in that a broader area can be imaged more quickly and that image quality is relatively high.
On ultrasonography, the “dark halo” is detected around the superficial temporal artery due to edema of the arterial wall (Figure 22).120
It may not be observed early after the onset, and the sensitivity is reported to be about 40%.121
The diagnostic ability of FDG-PET is high as abnormal accumulation has been reported to be observed in the blood vessel in 80% of the patients.119,122
It has become possible to perform FDG-PET under health insurance at some PET facilities in Japan for patients with large vessel arteritis in whom the localization or activity of lesions is difficult to determine by other examinations since April 2018. However, there are limitations such as that it is expensive and that it can be performed at relatively few facilities.
5.3 Ophthalmological Examinations
20–30% of the GCA patients have blurred vision or visual deterioration,111,123,124
and some patients with GCA have diplopia, transient visual impairment, and the visual field defects. Blurred vision, visual deterioration and visual field defects can be caused by anterior ischemic optic neuropathy, occlusion of the central retinal artery, or occlusion of the cilioretinal artery,111,125
and they can be revealed by fundus examination. Diplopia is caused by restriction of ocular movements associated with ischemic external ophthalmoplegia. In anterior ischemic ocular neuropathy, which is the most frequent cause of visual impairment in GCA, pale edema of the optic disc is observed (Figure 23).126

In silent GCA, which presents with systemic symptoms such as fever, easy fatigability, and loss of body weight rather than conventionally known symptoms (headache, tenderness of the temporal artery, weakened pulse, blindness), visual disorders are rare, but fundus lesions such as cotton-wool spots may be observed.124,127,128
6. Diagnostic Methods and Criteria
6.1 Diagnostic Criteria
The classification criteria by ACR of 1990 (Table 9),129
which are widely used for the diagnosis of GCA, are also adopted as the criteria of the MHLW of Japan. Tenderness or reduced pulse along the temporal artery is evaluated by palpation. While conditions can be classified as GCA even without pathological demonstration of GCA, biopsy of the unilateral temporal artery is recommended for the diagnosis. Vascular ultrasonography for wall thickening of the temporal artery is useful as an adjuvant diagnostic method.130,131
Table 9.
ACR Criteria for the Classification of Giant Cell Arteritis (Proposed by the ACR in 1990)
| Criterion |
Definition |
1. Age at disease onset
≥50 years |
Development of initial symptoms or findings beginning at age 50 or older |
| 2. New headache |
New onset of, or new type of, localized headache |
3. Temporal artery
abnormality |
Tenderness to palpation or decreased pulsation of temporal artery, un-related to arteriosclerosis of cervical
arteries |
4. Elevated erythrocyte
sedimentation rate |
Elevated ESR: ESR ≥50 mm/hr by Westergren method |
| 5. Abnormal artery biopsy |
Biopsy specimen of artery revealing vasculitis characterized by predominant mononuclear or granulomatous
inflammation with multinucleated giant cells |
(From Hunder GG, Bloch DA, Michel BA, et al., Arthritis Rheum 1990,129 John Wiley and Sons. (c) 1990, American College of Rheumatology.) GCA is diagnosed if at least 3 of these 5 criteria are present (sensitivity: 93.5%, specificity: 91.2%)
Jaw claudication has low diagnostic sensitivity but is a clinical feature highly suggestive of vasculitis, which can be confirmed by temporal artery biopsy.132
Ischemic optic neuropathy is poorly sensitive but highly specific to GCA,129
and prompt diagnosis in cooperation with an ophthalmologist is recommended if the patient is aware of reduced visual acuity.
6.2 Points of Attention in Diagnosing GCA
Since the 1990 ACR classification criteria are not prepared as diagnostic criteria, diagnosis by exclusion is necessary concerning unidentified fever, chronic inflammatory disease, and headache. Special attention to differential diagnoses must be paid if GCA cannot be pathologically demonstrated.133,134
Temporal artery lesions may be observed in ANCA-associated vasculitis,129,135
PAN,129,136
and systemic amyloidosis.137
Patients in whom both CRP and ESR are normal are rare.138,139
In patients who have developed ophthalmic lesions and show reduced visual acuity, treatment must be initiated before biopsy because of the risk of blindness as delay of treatment may result in irreversible loss of vision. It is recommended to perform biopsy of the temporal artery within 1–2 weeks after the initiation of GC therapy.140,141
6.3 Evaluation of Aortic Lesions
Aortic lesions are considered to complicate GCA in about 20–30% of the patients. Aortic lesions such as stenosis of branches of the aorta and aortic aneurysm appear at the diagnosis of GCA in some patients, but they appear 5 or more years after the diagnosis in others.142–144
The relationship between aortic lesions and disease activity is unclear, but, epidemiologically, the risk of the occurrence of aortic aneurysm is higher than in those with non-GCA,145
and aortic lesions are related to the prognosis of GCA.146
Therefore, aortic lesions should be evaluated at the diagnosis or relapse of GCA by the same method as in Takayasu arteritis. FDG-PET is useful (it has become possible to perform the examination under health insurance at some PET facilities in Japan since April 2018 for patients with large vessel arteritis in which the localization or activity of the lesion is difficult to determine by other examinations).
7. Policies of Treatment
Recommendation classes and evidence levels of treatments for GCA are shown in
Table 10.
Figure 24
shows a flow chart of treatments for GCA.
Table 10.
Recommendations and Evidence Level of Treatments for GCA
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
I |
B |
| Steroid pulse therapy |
I |
B |
| Methotrexate (MTX)* |
IIa |
A |
| Cyclophosphamide (CY) |
IIb |
B |
| Azathioprine (AZA) |
IIb |
B |
| Cyclosporine (CyA)* |
III |
B |
| Tocilizumab (TCZ) |
I |
A |
| Infliximab (IFX)* |
III |
B |
| Adalimumab (ADA)* |
III |
B |
| Etanercept (ETN)* |
IIb |
B |
| Antiplatelets |
IIa |
B |
7.1 Glucocorticoid/Immunosuppressants
7.1.1 Glucocorticoid
GC is effective and is the first-line treatment for GCA (recommendation class: I, evidence level: B). Since a delay in the initiation of treatment increases the risk of blindness and irreversible neurological damage, a high dose of GC should be initiated promptly if GCA is suspected.
a. Initial Treatment
1) In patients without ophthalmic or neurological symptoms, PSL at 0.5–1 mg/kg/day (60 mg/day at the maximum) is recommended (recommendation class: I, evidence level: B).
2) In patients with rapidly developed ophthalmic or neurological symptoms, steroid pulse therapy (mPSL at 0.5–1 g/day, 3 days) is recommended to be initiated and followed by PSL at 1 mg/kg/day (60 mg/day at the maximum) (recommendation class: I, evidence level: B).
GC reduction should be considered only in the absence of clinical symptoms and normalized inflammatory markers such as CRP and ESR after sustaining the initial dose for 2–4 weeks. Most patients can be weaned from GC in 1–2 years, but GCA recurs in about half the patients.147,148
b. Treatment of Relapse
1) An increase of 5–10 mg/day of PSL is usually sufficient to treat a relapse in the absence of ophthalmic or neurological symptoms.
2) An increase to the initial dose of PSL should be considered in the presence of ophthalmic or neurological symptoms.
7.1.2 Immunosuppressants
Immunosuppressants are administered with GC to those who resist GC, those who have developed relapse with gradual reduction of GC, and those in whom rapid reductions of GC are needed due to adverse reactions. However, the therapeutic effect of the concomitant use of immunosuppressants on GCA is considered limited.149
GCA is not treated with immunosuppressants alone. MTX (uninsured)116
(recommendation class: IIa, evidence level: A), CY150–153
(recommendation class: IIb, evidence level: B), and AZA154,155
(recommendation class: IIb, evidence level: B) have been reported to be effective as concomitant immunosuppressants.
7.2 Biological Preparations/Antiplatelets
7.2.1 Biological Preparations
GCA responds well to moderate or high dose of GC therapy. While early tapering of its dosage is necessary to reduce adverse events, GCA often relapses during tapering. To examine whether the concomitant use of biologics prevents flare-up of the disease, and reduce cumulative dose of GC, RCTs concerning the efficacy of the concomitant use of TCZ, ADA, or IFX were carried out in Europe and the United States.115,156,157
7.2.2 TCZ
The efficacy of a protocol of initiating the administration of TCZ, which is an anti-IL-6 receptor antibody, with GC therapy has been confirmed by phase II and phase III trials. The phase II trial showed that the remission rate at week 12 was significantly higher, and the relapse rate until after week 52 was significantly lower, when TCZ was used concomitantly with early tapering schedule of PSL.157
In the phase III trial, the remission maintenance rates at weeks 12–52 were significantly higher in the TCZ group even when PSL was discontinued at week 26, and cumulative dose of GC was significantly decreased in the TCZ group.158
Concerning refractory cases with a history of treatment using immunosuppressants and TNF inhibitors, there have been case reports and small case series reporting that PSL could be reduced to a low dose or discontinued by the concomitant use of TCZ.67,69,159,160
The evidence level of TCZ is higher than those of immunosuppressants or TNF inhibitors. It should be administered to cases that flare during tapering of GCs or require early tapering of GC, with attention to the safety and in consideration of the risk-benefit balance (recommendation class: I, evidence level: A, covered by health insurance in Japan).
7.2.3 TNF Inhibitors
Since the efficacy of ADA and IFX, among the TNF inhibitors (uncovered by health insurance in Japan), has not been demonstrated by the results of clinical trials,115,156
the concomitant use of TNF inhibitors with GC from an early stage of treatment is not recommended (recommendation class: III, evidence level: B) despite the problem that the evaluation method for the activity of GCA has not been sufficiently established. The efficacy of TNF inhibitors against relapse during GC therapy or the use of immunosuppressants is unclear. Etanercept (ETN) is expected to have a GC-sparing effect, and its reevaluation is awaited (recommendation class: IIb, evidence level: B).
7.2.4 Antiplatelets (recommendation class: IIa, evidence level: B)
GCA is occasionally complicated by cerebrovascular disorders and cardiovascular events, although vasculitis rarely develops in intracranial arteries except for the ophthalmic artery.161
The concomitant use of low-dose aspirin (81–100 mg/day) has been reported to reduce the risk of cerebrovascular disorders by retrospective observational studies,162–164
and EULAR recommends the concomitant use of a small dose of aspirin without contraindications.140
7.3 Invasive Treatments
The evidence concerning surgical/endovascular treatments for large vessel lesions associated with GCA is meager. Please refer to Chapter II 7-3 (Invasive Treatments for Takayasu Arteritis).
8. Prognosis
Factors that affect the prognosis include (1) sequelae of vascular ischemia (blindness, cerebral infarction, myocardial infarction), (2) aneurysm (dissecting/ruptured), and (3) treatment-related complications (infections, and morbid fracture due to immunosuppressive therapy such as long-term GC therapy). According to the national epidemiological survey by the Ministry of Health and Welfare of Japan in 1998, cure/recovery was reported in 81.8%, cerebral infarction in 12.1%, blindness in 6.5%, and death in 4.5% as short-term outcomes.106
GCA as well as PMR is a disease that recurs repeatedly, and GC therapy was continued in 43% and 25% of the patients 5 and 9 years, respectively, after its initiation according to a report from Sweden.122
Analysis of causes of death in a cohort study in Minnesota showed that the mortality rate due to ischemic enteritis was 4.9 times higher in patients with GCA, and 5.1 times higher in those with GCA complicated by aortic aneurysm or dissection, compared with the general population.143
In a single facility cohort in northwestern Spain, aortic aneurysm occurred secondarily in 9.5% of the patients with GCA, and hypertension and marked acute inflammatory reaction at the time of first diagnosis were risk factors.165
IV. Buerger Disease
1. Definition and Epidemiology
1.1 Definition
Buerger disease causes segmental lesions in arteries and veins of the extremities and is frequently observed in males in their 20–40 s. It is closely related to smoking and is occasionally complicated by migrating thrombophlebitis. Lesions mainly affect the arteries of the forearm to the hand and the infrapopliteal artery to arteries of the foot.
The life prognosis is favorable, but the quality of life (QOL) is sometimes impaired by ischemic pain at rest or ulcer/gangrene which leads to minor limb amputation. The incidence of major amputation increases if the patient fails to stop smoking.
1.2 Epidemiology
As of 2014, about 7,000 patients were certified to have Buerger disease, one of the specific diseases (Figure 25).11
Recently, the number of female patients has been increased to 11–23% of the whole patients, presumably due to an increase in smoking rates in female.166–169
In Japan, the majority of patients are elderly patients who have been grown old after the onset in young age.
The disease is prevalent in South Asia, Ease Asia, and Turkey. In Japan, Buerger disease used to be reported to account for 16% of patients treated for occlusive arterial disease170
but is presently estimated to account for 1% or less due to the decrease in the number of patients with this disease and the increase in that with atherosclerotic diseases.
2. Pathogenesis
It has been suggested that pathogenesis of Buerger disease are related to smoking, infection, activated condition of vascular endothelial cells,171–174
and the occurrence of microcirculation disorder.175,176
The involvement of racial/ethnic factors has also been suspected.176
Many of the patients are heavy smokers.167,177
The disease has been reported to be induced by exposure to cold, trauma, and sympathetic nerve stimulating drugs,178
but patients are considered to have some history of smoking including passive smoking. Many patients have dental root inflammation,179
and the same periodontal pathogens have been detected in the arterial wall and oral cavity.180
Attenuation of electric signals of the sympathetic nerve to the muscles and skin,181
reduced blood catecholamine level due to change in peripheral sympathetic nerve responses to smoking,182
impairment of endothelium-dependent vasodilatation of peripheral arteries, and increased expression of adhesion factors in endothelial cells and inflammatory cells183
have been reported, and activation of platelets via serotonin receptors is speculated.184
3. Pathological Findings
Figure 26
shows pathological images, and
Figure 27
shows a diagram of characteristic features of Buerger disease. In Buerger disease, middle-sized vessels of the lower or upper extremities are affected, and as thrombi cause segmental occlusion of arteries, gangrene is observed in peripheries. The following pathogenic features characteristic of occluded arteries are observed: (1) In the acute period, microabscesses in which neutrophils aggregate and multinucleated giant cells are occasionally observed in the vascular wall, particularly, intima.172,185
(2) The internal elastic lamina is not displaced, remains flexed, and may even be overflexed.186
This contrasts with displacement of the internal elastic lamina by the intima observed in occlusive atherosclerosis and thrombosis, which are major differential diagnoses. (3) Fibrosis of the adventitia with no fibrosis of the media,187
(4) edema immediately inside the external elastic lamina,187
(5) onion-like multi-layering of endothelial cells of recanalized vessels,187
and (6) thickening of the endothelial cells of vasa vasorum187
are also observed frequently. These are features caused by inflammation within the vascular wall or from outside the blood vessel and impairment of microvessels. (7) Immunohistochemically, macrophage/lymphocyte infiltration tightly attached to the internal elastic lamina may be demonstrated.188


4.1 Ischemic Signs/Symptoms in the Upper and Lower Limbs
4.1.1 Ischemic Signs/Symptoms in the Lower Limbs
a. Coldness
Patients feel coldness mainly in their toes.
b. Paresthesia
Numbness often occurs commonly in the fingers, toes, feet, and hands, particularly after exercising or walking.
c. Skin Color Change
The skin color of affected digits turns red (rubor) even when the limbs are located at the same level with the heart. Cyanosis indicates critical digit ischemia.
d. Intermittent Claudication
Ischemic intermittent claudication develops in the foot in an early stage, in the calf when the above-knee arteries are involved, and in the thigh and buttock when the iliac arteries are involved.
e. Rest Pain
Severe and stinging rest pain often disturbs patient’s sleep.
f. Ulcer/Gangrene
Ulcer and gangrene often occur in the fingers and toes, particularly around the nails. Ulcer and gangrene can develop beginning from small injury and extend rapidly.
4.1.2 Ischemic Signs/Symptoms in the Upper Limbs
Ischemic signs/symptoms in the upper limbs are milder than those in the lower limbs and are reported to be primary chief complaints in about 5–14% of the patients.189,190
4.2 Involvement of Visceral Arteries
Although rare, lesions in the cerebral artery,191
coronary artery,192,193
renal artery,194
mesenteric artery,195,196
and internal thoracic artery193
have been reported.
4.3 Migratory Superficial Thrombophlebitis
Recurrent and migratory superficial thrombophlebitis (phlebitis migrans/migrating thrombophlebitis) occurs in the limbs.
4.4 Incidences of Signs/Symptoms
According to a survey in Japan in 1993, signs/symptoms were observed in the upper limbs alone in 5.1% of patients, lower limbs alone in 74.7%, and both upper and lower limbs in 20.2%. The affected arteries were the anterior tibial artery in 41.4%, posterior tibial artery in 40.4%, and ulnar artery in 11.5%.197
According to the analysis conducted at the Department of Vascular Surgery of Nagoya University from 1977 to 1988, the initial symptoms were paresthesia, coldness, and cyanosis in 37% of patients, foot or calf claudication in about 30%, and severe ischemic symptoms such as rest pain and ulcer/gangrene in about 30%. Through the entire course, ulcer/gangrene was observed in 72%, migratory phlebitis in 43%, and upper limb lesions in 90%.189
5. Clinical Features and Laboratory Findings
5.1 Physical Findings (Table 11)
Table 11.
Physical Findings of Buerger Disease
(1) Appearance: Atrophy of the fingers and toes, underdeveloped nails, difference in hair growth between the right and
left extremities, and migratory superficial thrombophlebitis |
| (2) Auscultation: Iliac artery, femoral artery, and popliteal artery |
(3) Palpation: Decreased skin temperature in the extremities, fingers, and toes, and decrease or loss of pulse in peripheral
arteries |
| (4) Provocation tests: Negative Allen’s test (abnormal), positive straight leg raising test |
5.2 Examination Items (Table 12)
Table 12.
Noninvasive Testing for Buerger Disease
| A. Functional diagnosis |
| (1) Blood pressure (upper arm, fingers, ankle, toes): Doppler method, pulse wave method |
| (2) Skin perfusion pressure (SPP): Laser Doppler method |
| (3) Skin oxygen saturation: Transcutaneous oxygen tension (tcPO2) measurement |
| (4) Skin temperature: Thermography |
| (5) Skin blood flow: Laser Doppler method |
| (6) Muscle oxygen saturation: Near-infrared spectroscopy (uninsured) |
| (7) Blood flow: Air plethysmography |
| B. Diagnostic imaging |
| (1) Magnetic resonance angiography |
| (2) Contrast three-dimensional computed tomography (3D-CT) |
| (3) Angiography |
Thermographic evaluation with cold-water stress is useful for the diagnosis of Raynaud’s phenomenon. For patients with intermittent claudication, maximum walking distance as well as recovery time of the ankle-brachial index, which is reduced after walking, can evaluate circulatory reserve of the lower limb. Angiography is indispensable in patients considered to have indication for bypass surgery of the lower leg.
5.3 Findings of Vascular Imaging (Table 13,198 Figures 28–30198–200)
Table 13.
Arteriographic Findings of Buerger Disease
| (1) Arterial occlusive lesions of infrapopliteal or below-elbow segments |
| (2) Chronic arterial occlusion secondary to the extension or thrombus |
| (3) Smooth arterial wall with no evidence for arteriosclerosis such as a moth-eaten appearance and calcification |
| (4) Abrupt or tapering occlusion of affected arteries |
| (5) Collateral arteries with corkscrew, tree-root, or bridging appearance |
(Prepared from the 1976 report by a subgroup of the Study Group on Systemic Vascular Lesions as Specific Diseases of the Ministry of Health and Welfare of Japan, 1977198)
Differentiation from collagen diseases such as SLE, scleroderma, and vascular Behçet’s disease is particularly important. However, the differentiation is difficult by imaging findings alone.201,202
5.4 Hematological Examinations
There is no specific hematological abnormality. ESR and CRP values are usually normal.
6. Diagnostic Methods and Criteria
6.1 Diagnosis
There are various diagnostic criteria.203–205
When patients fulfill all of the 5 criteria of Shionoya’s clinical diagnostic criteria (Table 14),206
the clinical features are typical. However, even without fulfillment of all criteria, Buerger disease can be diagnosed if clinical findings including vascular images or pathological findings are consistent, and differential diagnoses can be excluded. Since young people with hypertension or dyslipidemia have increased, the careful diagnosis based on imaging and histological examinations is necessary, including differentiation from arteriosclerosis obliterans.
Table 14.
Shionoya’s Clinical Diagnostic Criteria for Buerger Disease
| (1) Onset before the age of 50 years |
| (2) History of smoking (including passive smoking) |
| (3) Infrapopliteal arterial occlusive lesions |
| (4) Either upper limb involvement or migratory thrombophlebitis |
| (5) Absence of atherosclerotic risk factors other than smoking |
Buerger disease can be diagnosed without fulfillment of all diagnostic criteria if imaging and pathological findings are consistent, and other diseases can be excluded. See the text. (Prepared as a table from Shionoya S. 1993206)
6.2 Conditions to Be Differentiated From Buerger Disease
Conditions to be differentiated from Buerger disease include arteriosclerosis obliterans, traumatic arterial thrombosis, popliteal artery entrapment syndrome, popliteal artery adventitial cyst, SLE, scleroderma, vascular Behçet’s disease, thoracic outlet syndrome, and atrial fibrillation.
7. Policies and Guidelines of Treatment
Table 15
shows recommendations about treatment for Buerger disease.
Table 15.
Recommendations and Evidence Levels of Treatments for Buerger Disease
| |
Recommendations |
Levels of evidence |
| Smoking cessation |
I |
C |
| Antiplatelets |
I |
C |
| PGE1-CD, lipoPGE1 |
IIa |
C |
| Exercise therapy |
IIa |
C |
| Pain control |
I |
C |
| Sympathetic nerve block |
IIb |
C |
| Sympathectomy |
IIb |
C |
| Vascular bypass surgery |
IIa |
C |
| Endovascular treatment |
III |
C |
7.1 Conservative Treatment
7.1.1 Smoking Cessation/Lifestyle Guidance
Patients should avoid even passive smoking, keep the affected limbs warm and clean, and avoid injury.207
Smoking after the initial visit correlates with increased limb amputation rate.208
Symptoms can be resolved in many patients by conservative therapy centering on smoking cessation.
7.1.2 Drug Treatment
For coldness and numbness, drug treatment should be attempted first. As oral antiplatelet agents, Cilostazol, beraprost, sarpogrelate, limaprost alfadex, ticlopidine, and clopidogrel are used. Alprostadil (a complex of prostaglandin E1
[PGE1]-α-cyclodextrin [CD] complex, lipo-PGE1) is an injection drug used for the treatment.
7.1.3 Exercise Therapy
Supervised exercise therapy is useful for the treatment of intermittent claudication.209
The effects should be evaluated after the program has been carried out for 3–6 months.
7.1.4 Pain Control
In addition to oral medications, fentanyl tape is also available. Continuous epidural anesthesia or toe amputation can be an option.
7.2 Surgical Treatment
7.2.1 Revascularization
Revascularization is indicated particularly in patients with rest pain or ulcer when no improvement has been observed in symptoms by conservative treatment. Using autogenous vein grafts is recommended for bypass surgery. The patency of infrapopliteal bypass in Buerger disease is generally lower than that in arteriosclerosis obliterans. However, major amputation is required in only small number of patients even if the graft occlusion has occurred.
7.2.2 Sympathectomy
Sympathectomy should be considered for painful ulcers of the toes and fingers when revascularization is impossible. Pharmacological sympathetic blockade can also be a choice.
7.2.3 Others
Angiogenesis therapy (gene therapy, autologous bone marrow (monocyte) transplantation,210,211
peripheral blood monocyte transplantation212), remains in the experimental stage.
8. Prognosis
8.1 Prognosis of Ischemic Limbs
It is estimated that 70% of the patient will experience ischemic ulcer or necrosis at least once during the course. Patients tend to repeat exacerbation and remission of symptoms, therefore, not all the patients with Fontaine stage III/IV disease need limb amputation.
On long-term follow-up studies, major limb amputation was performed in 2.7–10.5% of the patients.200,213
According to questionnaire surveys by the MHLW Group Conference, 8.8% resulted in major limb amputation, and 16.7–20.5% of the patients required toe amputation.208,214
Patients who need revascularization of the upper limb are few. While some patients require finger amputation, very few patients need major amputation of the upper limb.
8.2 Factors Contributing to Exacerbation and Improvement
Smoking is considered to shorten the remission period and invite exacerbation. Patients who continue smoking after the appearance of symptoms resist various treatments and often come to need major amputation consequently.208
If patients can avoid foot injury by hospitalization, their symptoms may be alleviated. Hospitalization also leads patients to strict smoking cessation.
8.3 Prognosis of Non-Critical Ischemic Limbs
Almost all patients have two or more affected limbs.200
The disease rarely progresses in non-critical or non-dominant ischemic limbs if patients can continue smoking cessation but may progress if patients continue smoking.
8.4 Concomitant Diseases and Life Prognosis
The life expectancy of patients is considered favorable. The most common cause of death is malignant neoplasm, followed by heart failure, cerebral infarction, and liver cirrhosis.213
Concomitant diseases include diabetes, hypertension, dyslipidemia, cerebrovascular disorders, ischemic heart disease, and arteriosclerosis obliterans.208
Therefore, the disease management including atherosclerotic risk factors is required according to the patients’ age.
V. Polyarteritis Nodosa
1. Definition and Epidemiology
Polyarteritis nodosa (PAN) is defined as “Necrotizing arteritis of medium or small arteries without glomerulonephritis or vasculitis in arterioles, capillaries, or venules, and not associated with ANCAs”.1
Presently, the number of patients is estimated to be about 600, but 3,442 patients held certificates for medical benefits for specific diseases in 2015. The male/female ratio of the patients is 1:1 to 1:2, and the average age at onset is 55 years.215
2. Pathogenic Mechanism
The etiology is unknown, but secondary PAN that follows HBV infection has been recognized.
3. Pathological Findings
In the acute period, severe inflammation and necrosis are observed in the vascular wall of muscular arteries, and lesions are present as segments.216
In the affected blood vessels, fibrinoid necrosis and rupture of the internal/external elastic laminae are observed, often causing aneurysms (Figure 31).
4. Symptoms
Patients with PAN present systemic symptoms and organ symptoms due to ischemia (Table 16).217
PAN in which lesions are localized in the skin is called cutaneous PAN218
or cutaneous arteritis.1
Table 16.
Clinical Symptoms of Polyarteritis Nodosa
| Clinical signs/symptoms |
Prevalence (%) |
| General symptoms |
Fever, Malaise, Weight loss |
80 |
| Neurologic manifestations |
Mononeuritis multiplex, Polyneuropathy |
75 |
| Arthralgia, Myalgia |
Joint pain or diffuse pain of the limbs |
60 |
| Cutaneous signs/symptoms |
Livedo reticularis, Purpura, Ulcer |
50 |
| Renal manifestations |
Increased creatinine, Hematuria, Glomerulonephritis |
50 |
| Gastrointestinal manifestations |
Abdominal pain, Rectal bleeding |
40 |
| Hypertension |
New onset |
35 |
| Pulmonary manifestations |
Lung infiltrates, Nodular shadow, Cavity |
25 |
| Central nervous system manifestations |
Stroke, Confusion |
20 |
| Testicular manifestations |
Orchitis or testicular tenderness |
20 |
| Cardiac manifestations |
Myocarditis, Pericarditis |
10 |
| Peripheral vascular manifestations |
Claudication, Ischemia, Necrosis |
10 |
(Reproduced with modification from Pagnoux C, et al. 2010217)
There is no laboratory finding specific to PAN. ANCA must be confirmed to be negative. Examinations including skin biopsy, angiography, and sural nerve biopsy are performed.
6. Diagnosis and Diagnostic Criteria
Table 17
shows the diagnostic criteria for PAN by the MHLW of Japan.219
Table 17.
MHLW Diagnostic Criteria for Polyarteritis Nodosa
| For Definite and Probable Diagnoses |
| [Diagnostic features] |
| (1) Major clinical findings |
| 1) Fever (≥38℃ continued ≥2 weeks) and weight loss (≥6 kg over ≤6 months) |
| 2) Hypertension |
| 3) Rapidly progressive renal failure, renal infarction |
| 4) Cerebral hemorrhage, cerebral infarction |
| 5) Myocardial infarction, ischemic heart disease, pericarditis, heart failure |
| 6) Pleurisy |
| 7) Gastrointestinal hemorrhage, intestinal obstruction |
| 8) Mononeuritis multiplex |
| 9) Subcutaneous nodule, skin ulcer, gangrene, purpura |
| 10) Polyarthralgia (polyarthritis), myalgia (myositis), muscular weakness |
| (2) Histological finding |
| Fibrinoid necrosis in medium- and small-sized arteries |
| (3) Angiographic findings |
| Multiple microaneurysms, stenoses, and occlusions in branches of the abdominal aorta (characteristically in renal arterioles) |
| (4) Diagnosis |
| 1) Definite |
| Patients with ≥2 major clinical findings and the histological finding |
| 2) Probable |
| (a) Patients with ≥2 major clinical findings and the angiographic findings |
| (b) Patients with ≥6 major clinical findings including item 1) |
| (5) Suggestive laboratory findings |
| 1) Leukocytosis (≥10,000/μL) |
| 2) Thrombocytosis (≥400,000/μL) |
| 3) Elevated sedimentation rate |
| 4) Strongly positive CRP |
| (6) Differential diagnosis |
| 1) Microscopic polyangiitis |
| 2) Granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis) |
| 3) Eosinophilic granulomatosis with polyangiitis (formerly known as allergic granulomatous angiitis) |
| 4) Kawasaki disease |
| 5) Connective tissue disease (SLE, RA) |
| 6) IgA vasculitis (formerly known as Henoch Schönlein purpura) |
| [Notes] |
(1) Histologically, polyarteritis nodosa has been classified into Stage I (degenerative stage), Stage II (inflammatory stage),
Stage III (granulation tissue stage), and Stage IV (scar tissue stage). |
(2) Clinically, patients in Stages I and II exhibit signs and symptoms of severe inflammation of systemic vessels,
while patients in Stages III and IV exhibit signs and symptoms of ischemia of affected organs. |
(3) The diseases to be differentiated from polyarteritis nodosa also demonstrate necrotic vasculitis, but may be clearly
differentiated from it on the basis of characteristic findings and laboratory results. |
(Report of the Intractable Vasculitis Study Group of the MHLW, 2005 Revised219)
Table 18
shows recommendations about treatments for PAN.
Table 18.
Recommendations and Evidence Levels Concerning Treatments for PAN
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
I |
B |
| Steroid pulse therapy |
IIa |
C |
| Cyclophosphamide (CY) |
IIa |
A |
| Methotrexate (MTX)* |
IIb |
B |
| Azathioprine (AZA) |
IIb |
B |
| Plasma exchange therapy* |
IIb |
C |
*Uncovered by medical insurance in Japan.
7.1 Remission Induction Therapy
7.1.1 Moderate/Severe PAN With Lesions in Vital Organs
Four-week initial treatment is performed using PSL at 1 mg/kg/day with IVCY or daily oral CY administration from the beginning of treatment220
(recommendation class: I, evidence level: B). If symptoms are extremely severe, steroid pulse therapy is performed simultaneously (recommendation class: IIa, evidence level: C). If severe renal disorder is noted, plasma exchange (uninsured)221
should also be considered (recommendation class: IIb, evidence level: C).
7.1.2 Mild PAN Not Accompanied by Vital Organ Lesions
Treatment is initiated with PSL alone (0.5–1.0 mg/kg/day)222
(recommendation level: I, evidence level: B). If symptoms are not improved, CY or MTX is used concomitantly.
7.1.3 Symptomatic Treatments/Management of Adverse Reactions
Antihypertensive drugs, circulation improving medications, and treatments for neurological disorders should be used in combination with prophylaxis for infection.
7.2 Remission Maintenance Therapy
PSL is reduced and continued at a maintenance dose of 5–10 mg/day. CY is replaced with AZA at 1–2 mg/kg/day or MTX at 7.5–15 mg/week (uninsured) (recommendation class: IIb, evidence level: B).
8. Prognosis
The 5-year survival rate of patients with PAN is 75–93%.223,224
As prognostic factors, proteinuria of ≥1 g/day, renal insufficiency, cardiomyopathy, severe gastrointestinal manifestation and central nervous system involvement were extracted in 1996,225
and an age of ≥65 years, cardiac symptoms, renal insufficiency, and gastrointestinal involvement were reported in 2011.226
VI. Microscopic Polyangiitis
1. Definition and Epidemiology
1.1 Definition
Microscopic polyangiitis (MPA) is classified as small-vessel angiitis related to ANCA and defined as “a necrotizing vasculitis that primarily affects small vessels (capillaries, venules, arterioles) and shows only little or no immune deposits. Necrotizing arteritis may affect not only small vessels but also middle-sized vessel. Necrotizing glomerulonephritis is observed in most patients, pulmonary capillary angiitis is often observed, and granulomatous inflammation is not observed.”1
1.2 Epidemiology
According to the summary of the number of certificates for recipients of medical care for specific diseases issued at the end of the fiscal year 2015, the number of MPA patients was 8,511 (Figure 25).11
The male/female ratio is 1:1.1 slightly in favor of women, and the mean age of the patients was 70.5 years.227
The annual incidence of MPA per 1 million people has been reported to be 18 in Japan,228
7 in the UK, 8 in Spain, and 3 in Germany.229
Among subtypes of ANCA, myeloperoxidase (MPO) ANCA (MPO-ANCA) is predominant in Japan, accounting for 97.4%, and proteinase 3 (PR3) ANCA (PR3-ANCA) accounts for 2.6%.227
In Western countries, MPO-ANCA and PR3-ANCA occupy 40–58% and 26–50%, respectively, and the PR3-ANCA is more prevalent than in Japan. Also, interstitial pneumonia is prevalent at 45% in Japan.227
2. Pathogenic Mechanism
An autoimmune mechanism, i.e., genetic and environmental factors interact to produce ANCA, which is an autoantibody, and it induces vascular endothelial damage, is suspected to be involved.
2.1 Genetic Factors
HLA haplotype analysis: In Japanese patients with MPA, 50% are reported to be positive for HLA-DRB1*0901, and the percentage is significantly higher than in healthy controls.230
According to the results of GWAS (comprehensive analysis of genetic polymorphisms in whole genome) concerning ANCA-associated vasculitis based on multicenter collaborative research in Europe, MPA and ANCA-associated vasculitis positive for MPO-ANCA were suggested to be significantly related to SNP in the HLA-DQ region.231
In addition, gene polymorphism of ANCA-associated vasculitis is estimated to be more closely related to the specificity of ANCA such as MPO-ANCA and PR3-ANCA than clinical classification of MPA and GPA.231
2.2 Environmental Factors
Agents including silica, drugs, and infection (viruses, Gram-negative rods) are suspected to be involved in MPA. Among drugs, propylthiouracil (anti-thyroid drug) has been reported most frequently, and minocycline (antibiotic) and hydralazine (antihypertensive) have also been reported.
2.3 Pathology
Inflammatory cytokines such as TNF-α, ANCA, MPO, and neutrophil activation have been suggested to be involved (ANCA-cytokine sequence theory). ANCA produced by an autoimmune mechanism excessively activates neutrophils alone with inflammatory cytokines is estimated to release a network of fine fibers (chromatin fibers) called neutrophil extra-cellular traps and damage the vascular endothelium.232–234
3. Pathological Findings
In MPA, necrotizing vasculitis is observed primarily in small vessels (capillaries, venules, arterioles) distributed to various organs including the kidney, lung, skin, and nervous system (Figure 32).235
On immunoglobulin staining by the fluorescent antibody method, little or no immune deposits are observed (pauci-immune).
Typical small-vessel vasculitides due to MPA are focal/segmented necrotizing glomerulonephritis and crescentic glomerulonephritis in the kidney and alveolar hemorrhage due to pulmonary capillaritis in the lungs.
4. Signs and Symptoms
As systemic symptoms, there are fever, malaise, anorexia, and body weight loss. As local lesions/symptoms, there are a wide variety of symptoms due to organ disorders shown below.
Kidney symptoms: Various symptoms due to necrotizing glomerulonephritis (edema, glomerular hematuria, RPGN).
Lung lesions: Bloody sputum, hemoptysis, melanoptysis, cough, and dyspnea due to alveolar hemorrhage and dry cough and dyspnea due to interstitial pneumonia.
Skin symptoms: Purpura, livedo reticularis, subcutaneous nodules
Ophthalmic lesions: Conjunctival congestion, diplopia, reduced visual acuity, ophthalmalgia.
ENT symptoms: Hearing impairment due to otitis media
Peripheral nerve lesions: Paresthesia of the limbs, motor disorders
Central nervous system disorders: Symptoms due to lesions of the Hemiplegia, headache, and disturbance of consciousness due to cerebral infarction or cerebral hemorrhage, symptoms due to lesions of the cranial nerves such as facial nerve palsy, and headache and disturbance of consciousness due to hypertrophic pachymeningitis.
Gastrointestinal lesions: Abdominal pain, melena
5. Laboratory and Imaging Findings
As findings indicating inflammatory reaction, there are increases in CRP/gammaglobulin, ESR, peripheral blood leukocyte count, and platelet count. Laboratory/imaging findings in typical organ disorders are presented.
In patients with kidney lesions, abnormal urinalysis findings (proteinuria, glomerular hematuria, red blood cell cast, and granular cast) indicating nephritis and a decrease in eGFR or increase in BUN/serum creatinine indicating the reduction of renal function may be observed. RPGN is often noted, and serial measurements of eGFR are important.
Concerning lung lesions, non-segmental infiltrative shadow indicating alveolar hemorrhage is a typical finding. Interstitial pneumonia often presents reticular or annular shadows primarily in the bilateral lower lung fields on chest radiography and reticular shadow, honeycomb lung, and infiltrative shadow immediately below the pleura on CT, showing a usual interstitial pneumonia (UIP) pattern.
Ophthalmic lesions (episcleritis) and ENT lesions (neural deafness) are observed, and there are peripheral nerve lesions (findings of mononeuropathy multiplex), central nervous system disorders (headache, disturbance of consciousness due to cerebral infarction, cerebral hemorrhage, or hypertrophic pachymeningitis), and gastrointestinal lesions (abdominal pain, melena).
6. Diagnosis/Diagnostic Criteria
6.1 Diagnosis
Table 19
shows the diagnostic criteria for MPA by the MHLW Research Committee of Intractable Vasculitis.236
Table 19.
Diagnostic Criteria for Microscopic Polyangiitis
| <Diagnostic Criteria> For definite and probable diagnoses |
| [Major items] |
| (1) Major clinical findings |
| 1. Rapidly progressive glomerulonephritis |
| 2. Pulmonary hemorrhage or interstitial pneumonia |
3. Visceral signs/symptoms other than renal/pulmonary manifestations: purpura, subcutaneous hemorrhage,
gastrointestinal hemorrhage, mononeuritis multiplex, etc. |
| (2) Major histological findings |
Necrosis of arterioles, capillaries and postcapillary venules, and infiltration of inflammatory cells into tissues surrounding
vessels |
| (3) Major laboratory findings |
| 1. Positive MPO-ANCA |
| 2. Positive CRP |
| 3. Proteinuria, hematuria, elevation in BUN, elevation in serum creatinine |
| 4. Chest X-ray findings: Infiltrations (alveolar hemorrhage), interstitial pneumonia |
| (4) Diagnosis |
| 1. Definite |
| (a) Patients with ≥2 major clinical findings and positive histological finding |
| (b) Patients with ≥2 major clinical findings including Items 1and 2 and positive MPO-ANCA |
| 2. Probable |
| (a) Patients with 3 major clinical findings |
| (b) Patients with one major clinical finding and positive MPO-ANCA |
| (5) Differential diagnosis |
| 1. Polyarteritis nodosa |
| 2. Granulomatosis with polyangiitis (formerly known as Wegener’s granulomatosis) |
3. Eosinophilic granulomatosis with polyangiitis (formerly known as allergic granulomatous angiitis/Churg Strauss
syndrome) |
| 4. Arteritis of Kawasaki disease |
| 5. Collagen disease (SLE, RA, etc.) |
| 6. IgA vasculitis (formerly known as Henoch Schönlein purpura) |
| [Notes] |
1. In many patients, infection (mainly upper respiratory infection) develops one or two weeks prior to manifestation of major
clinical findings. |
2. Items 1 and 2 of the major clinical findings develop simultaneously in about half of patients, and either of the two
develops first in the remaining patients. |
| 3. In many patients, MPO-ANCA titer is related to disease activity. |
| 4. Early discontinuation of treatment may be associated with relapse of disease |
5. Various diseases to be differentiated exhibit necrotizing vasculitis but can be differentiated according to characteristic
signs and symptoms and laboratory findings. |
(Report of the Intractable Vasculitis Study Group of the MHLW236)
6.2 Procedure of Differential Diagnosis
First, vasculitides due to infection, malignant neoplasm, drugs, and other collagen diseases must be excluded. ANCA may also be positive in infectious endocarditis and collagen diseases such as SLE. For the differentiation of MPA from infectious endocarditis, the history of dental treatment, blood culturing, and echocardiography findings are important.
After the above condition have been excluded, and primary middle-sized/small-vessel vasculitis is considered possible, ANCA-associated vasculitides are classified by following the 3 steps shown in
Figure 33.237
EGPA is excluded first, and GPA is excluded next. If these two diseases have been excluded, urinalysis findings characteristic of glomerulonephritis and findings of small-vessel vasculitis such as purpura are present, and ANCA is positive, the possibility of MPA is high. Even if MPA is initially diagnosed, the diagnosis is changed if lesions due to granulomatous inflammation such as pulmonary nodules are confirmed during the course, and the diagnosis of GPA or EGPA are fulfilled.
In MPA, caution is necessary as impairment of only one organ may be observed at the facility that diagnoses the disease initially, but disorders of various organs may appear successively.
7. Policies and Guidelines of Treatment
The basic treatments for MPA are GC and immunosuppressants (including RTX) (Figure 34).238,239
Plasma exchange therapy may also be included (uncovered by insurance in Japan).
By the Guidelines for the Management of ANCA-associated vasculitis 2017 (jointly prepared by the MHLW Study Groups on Intractable Vasculitides, Intractable Kidney Diseases, and Diffuse Lung Diseases), the following treatments are recommended (the strength of recommendation and reliability of the evidence are presented according to the GRADE system).238
See the recommendations and comments in Part 1 of the same Guidelines. See the section, VIII Treatments in Part 2 of the same Guidelines for the dosage and dose regimen of each drug.
7.1 Remission Induction Therapy
The following CQs and the answers to them are the same for MPA and GPA.
CQ1
What regimens are effective as remission induction therapies for ANCA-associated vasculitis?
(1)-(i) As a remission induction therapy for ANCA-associated vasculitis, GC+IVCY or oral CY is proposed instead of GC alone.
Strength of recommendation: Weak
Reliability of evidence: Very low
(1)-(ii) As a remission induction therapy for ANCA-associated vasculitis, GC+IVCY is proposed instead of GC+oral CY.
Strength of recommendation: Weak
Reliability of evidence: Very low
Oral CY may be used as an alternative for IVCY.
(2) As a remission induction therapy for ANCA-associated vasculitis, GC+CY is proposed instead of GC+RTX.
Strength of recommendation: Weak
Reliability of evidence: Very low*/low**
For patients in whom the use of RTX is judged to be appropriate, GC+RTX may be used as an alternative of GC+CY by a physician with sufficient knowledge and experience in the treatment for ANCA-associated vasculitis.
*Compared with IVCY, **Compared with oral CY
(3) When neither CY nor RTX can be used, GC+MTX* is proposed as a remission induction therapy for patients with ANCA-associated vasculitis with no severe organ disorders and mild impairment of renal function.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Uncovered by insurance in Japan
(4) GC+MMF* is proposed when neither CY nor RTX can be used and when the above recommendation (3) is inappropriate.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Uncovered by insurance in Japan
CQ2
Is plasma exchange useful as a remission induction therapy for ANCA-associated vasculitis with severe kidney disorder?
(1)-(i) As a remission induction therapy for ANCA-associated vasculitis with severe kidney disorder, GC+oral CY+plasma exchange* is proposed instead of GC+oral CY+steroid pulse therapy.
Strength of recommendation: Weak
Reliability of evidence: Very low
(1)-(ii) As a remission induction therapy for ANCA-associated vasculitis with severe kidney disorder, GC+oral CY+plasma exchange* is proposed instead of GC+oral CY.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Covered by insurance in Japan from April 2018
Concerning the recommendations in CQ1 and CQ2, there are the following points of attention. Drugs/treatments indicated by * cannot be recommended in general, because they are uncovered by insurance in Japan. Consultation with nephrologists is recommended concerning patients with severe kidney disorders including RPGN. Plasma exchange should be performed by a physician with sufficient experience.
7.2 Remission Maintenance Therapies
CQ3
What regimens are useful as remission maintenance therapies for ANCA-associated vasculitis?
As a remission maintenance therapy for ANCA-associated vasculitis, the concomitant use of AZA in addition to GC is proposed.
Strength of recommendation: Weak
Reliability of evidence: Very low
RTX, MTX*, and MMF* can be alternative drugs to be used for remission maintenance therapy.
The drugs/treatments indicated by * cannot be recommended in general, because they are uncovered by insurance in Japan.
8. Prognosis
The prognosis of MPA has been markedly improved due to early detection as a result of the increased prevalence of ANCA measurement and knowledge about the disease, modifications of immunosuppressive therapy, improvements in anti-infection measures.
In 2008, EULAR reported that the remission rate of MPA except the renal-limited type was 81–91% and that the 5-year survival rate was 45–76%.240
However, in 2011, the European Vasculitis Study Group evaluated the long-term survival rate of patients with ANCA-associated vasculitis including MPA (mean follow-up period: 5.2 years) and reported a mortality rate of 25%.241
The report mentioned a eGFR of <15 mL/min/1.73 m2, old age, and high disease activity (BVAS) as factors that increased the mortality rate.
According to the Prospective Study of the Severity-Based Treatment Protocol for Japanese Patients with MPO-ANCA-associated Vasculitis (JMAAV), a prospective clinical study to clarify the usefulness of severity-based treatment protocols for MPO-ANCA-associated vasculitis carried out in Japan, the remission induction rate was 89.4%, mortality rate was 10.6%, rate of progression to end-stage renal disease was 2.1%, and relapse rate was 19.0%.242
The mortality rate by the severity of the disease was 50% for severest, 13% for severe, and 4% for mild cases, and the causes of death included vasculitis and infection.
The RemiT-JAV-RPGN, a prospective study of 321 cases of ANCA-associated vasculitis by the Study Group on Intractable Vasculitis and Study Group on Progressive Kidney Diseases of the MHLW of Japan, which included 197 cases of MPA, reported that the survival rate was 92.5%, and the remission induction rate was 76%, half a year after the initiation of remission induction therapy.227
In addition, the condition progressed to end-stage renal disease in 12.7% during the observation period.
VII. Granulomatosis With Polyangiitis
1. Disease Concept, Definition, and Epidemiology
Granulomatosis with polyangiitis (GPA), first reported in 1939 by Wegener, a German pathologist,243
is an intractable vasculitis with the following 3 clinicopathological characteristics: (1) Necrotizing granulomatous inflammation in the upper airway (E; ear and nose) and the lung (L; lung), (2) focal segmental necrotizing glomerulonephritis in the kidney (K; kidney), and (3) generalized necrotizing vasculitis of middle-sized/small vessels. According to a recent clinical research, the disease has a slight predilection to women, and often affects people in their 40 s–60 s, but may also occur in children and older people.244
2. Pathogenic Mechanism
Concerning the action mechanism of ANCA in GPA, the ANCA-cytokine sequence theory was proposed,245
neutrophil extracellular traps released by neutrophils at cell death are considered to be involved in the formation of pathological features.246
3. Pathological Findings
In E and L, parenchymal necrosis and granulomatous inflammation are noted (Figure 35), and necrotizing vasculitis is observed from middle-sized to small arteries, veins and capillaries. Histological findings in K are consistent with focal segmental or crescentic nephritis and are pauci-immune.245
4. Signs and Symptoms
In addition to systemic symptoms such as fever and body weight loss, (1) symptoms of E (purulent rhinorrhea, nasal bleeding, saddle nose, otitis media, reduced visual acuity, laryngopharyngeal ulcer, hoarseness), (2) symptoms of L (bloody sputum, dyspnea, lung infiltration), (3) symptoms of K (hematuria, oliguria, RPGN), and (4) other symptoms of vasculitis (purpura, polyarthralgia, polyneuritis). They often occur in the order of (1)→(2)→(3).
5. Laboratory and Imaging Findings
Typical chest radiography findings are nodular lesions accompanied by multiple or single cavities (Figure 36), and differentiation from malignant neoplasm and tuberculosis is necessary. In Western countries, cytoplasmic ANCA (c-ANCA) (PR3-ANCA) is positive in 80–96% of the patients with previously untreated active GPA, but patients positive for perinuclear ANCA (p-ANCA) (MPO-ANCA) and those positive for c-ANCA (PR3-ANCA) are observed at a ratio of approximately 1:1 in Japan.227
6. Diagnosis and Diagnostic Criteria
A revised version of the diagnostic criteria proposed by the MHLW study group in 1998 is used for the diagnosis of GPA (Table 20).247
Table 20.
Diagnostic Criteria for Granulomatosis With Polyangiitis (Formerly, Wegener’s Granulomatosis)
| Criteria for diagnosis |
1. Upper respiratory tract (E) signs/symptoms |
Nose (Purulent rhinorrhea, bleeding, saddle nose), eyes (eye pain, reduced visual acuity, exophthalmos), ears
(otitis media), oropharyngeal pain (ulcer, hoarse voice, airway obstruction) |
| 2. Pulmonary (L) signs/symptoms |
| Bloody sputum, cough, dyspnea |
| 3. Renal (K) signs/symptoms |
| Hematuria, proteinuria, rapidly progressive renal failure, swelling, hypertension |
| 4. Signs/symptoms of vasculitis |
| (a) Systemic findings: Fever (≥38℃ for ≥2 weeks) and weight loss (≥6 kg in ≤6 months) |
(b) Visceral findings: Purpura, polyarthritis (polyarthralgia), episcleritis, mononeuritis multiplex, ischemic heart
disease (angina pectoris, myocardial infarction), gastrointestinal hemorrhage (hematemesis, melena),
pleuritis, etc. |
| Major histological findings |
1. Necrotizing granulomatous inflammation with infiltration of giant cells in E, L, K lesions |
| 2. Necrotizing crescentic glomerulonephritis without immunoglobulin deposits |
| 3. Necrotizing granulomatous vasculitis in small-sized arteries and arterioles |
| Major laboratory findings |
Positive for PR3-ANCA (c-ANCA using the fluorescent antibody technique) |
| Diagnosis |
1. Definite |
| (a) Patients with ≥3 major clinical findings in E, L, K including visceral findings of E, L, and K |
(b) Patients with ≥2 major clinical findings in E, L, K or vasculitis and at least one of the major histological
findings (1) to (3) |
(c) Patients with ≥1 major clinical findings in E, L, K or vasculitis and at least one of the major histological
findings (1) to (3) and positive c (PR3)-ANCA |
| 2. Probable |
| (a) Patients with ≥2 major clinical findings in E, L, K or vasculitis |
(b) Patients with ≥1 major clinical findings in E, L, K or vasculitis and one of the major histological findings (1)
to (3) |
| (c) Patients with 1 major clinical finding in E, L, K or vasculitis and positive c(PR3)-ANCA |
| Supportive laboratory findings |
1. Leukocyte count, elevation in CRP |
| 2. Elevation in BUN and serum creatinine |
| Differential diagnosis |
1. Granulomatous disorder in E and L due to other causes (such as sarcoidosis) |
2. Other vasculitis syndromes (microscopic polyangiitis, eosinophilic granulomatosis with polyangiitis [Churg-
Strauss syndrome], polyarteritis nodosa, etc.) |
| Notes |
1. Wegener’s granulomatosis with visceral findings for E, L, K organs is referred to as systemic Wegener’s
granulomatosis, while that with findings of one or two of E, L is referred to as localized Wegener’s
granulomatosis. |
| 2. Signs and symptoms of systemic Wegener’s granulomatosis often develop in the order of E, L, then K. |
| 3. Shortly after onset, infection mainly with Staphylococcus aureus often occurs in E and L lesions. |
4. CT, MRI, and scintigraphy are useful in diagnosing space-occupying lesions due to granuloma formation in E
and L. |
| 5. PR3-ANCA titer is often correlated with disease activity. There are some patients with positive MPO-ANCA. |
(Cited with modification from the Study Group on Specific Diseases Systematic Vascular Disorders. 1998247)
Treatments for GPA can be divided into remission induction therapy and remission maintenance therapy. The Guidelines for the Management of ANCA-associated vasculitis 2017 recommends the following based on the GRADE system.238
In ANCA-associated vasculitis, it is difficult to induce remission by inadequate immunosuppressive therapy, and the exacerbation rate is high even if remission has been achieved. However, excessive immunosuppressive therapy increases the risk of severe infection, and treatments dependent on GC is likely to induce complications such as osteoporosis and diabetes, which affects the patient QOL.
The following CQs and related recommendations apply both to MPA (Chapter VI) and GPA. (See
Figure 34
in Chapter VI.)
7.1 Remission Induction Therapy
CQ1
What regimens are useful for remission induction therapy for ANCA-associated vasculitis?
(1)-(i) As a remission induction therapy for ANCA-associated vasculitis, GC+IVCY or oral CY is proposed instead of GC alone.
Strength of recommendation: Weak
Reliability of evidence: Very low
(1)-(ii) As a remission induction therapy for ANCA-associated vasculitis, GC+IVCY is proposed instead of GC+oral CY.
Strength of recommendation: Weak
Reliability of evidence: Very low
Oral CY may be used as an alternative of IVCY.
(2) As a remission induction therapy for ANCA-associated vasculitis, GC+CY is proposed instead of GC+RTX.
Strength of recommendation: Weak
Reliability of evidence: Very low*/low**
For patients in whom the use of RTX is judged to be appropriate, GC+RTX may be used as an alternative of GC+CY by a physician with sufficient knowledge and experience in the treatment for ANCA-associated vasculitis.
*Comparison with IVCY, **Comparison with oral CY
(3) If neither CY nor RTX can be used, GC+MTX* is proposed as a remission induction therapy for patients with ANCA-associated vasculitis with no severe organ disorders and mild impairment of renal function.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Uncovered by insurance in Japan
(4) If neither CY nor RTX can be used, GC+MMF* is proposed when the above recommendation (3) cannot be applied.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Uncovered by insurance in Japan
CQ2
Is plasma exchange useful in remission induction therapy for ANCA-associated vasculitis with severe renal disorder?
(1)-(i) GC+oral CY+plasma exchange* is proposed instead of GC+oral CY+steroid pulse therapy for remission induction therapy for ANCA-associated vasculitis with severe renal disorder.
Strength of recommendation: Weak
Reliability of evidence: Very low
(1)-(ii) GC+oral CY+plasma exchange is proposed instead of GC+oral CY for remission induction therapy for ANCA-associated vasculitis with severe renal disorder.
Strength of recommendation: Weak
Reliability of evidence: Very low
*Covered by insurance in Japan from April 2018
Points of attention concerning the recommendations for CQ1 and CQ2 are as follows: Since the drugs/treatments indicated by * are uncovered by health insurance in Japan, they cannot be recommended in general. It is recommended to consult a nephrologist concerning patients with severe renal disorders including RPGN. Plasma exchange should be performed by a physician with sufficient experience.
7.2 Remission Maintenance Therapy
CQ3
What regimens are useful as remission maintenance therapy for ANCA-associated vasculitis?
The concomitant use of AZA in addition to GC is proposed in remission maintenance therapy for ANCA-associated vasculitis
Strength of recommendation: Weak
Reliability of evidence: Very low
RTX, MTX*, and MMF* can be alternative drugs to be used in maintenance remission therapy.
*Since the drugs/treatments indicated by * are uncovered by health insurance in Japan, they cannot be recommended in general.
8. Prognosis
In GPA, induction of remission has become possible in many patients by GC+CY therapy. However, relapse during maintenance therapy is often observed. In addition, severe infection is the most frequent cause of death. The prognosis of GPA is expected to be improved by measures to prevent complications including infection and the development of new maintenance therapies.
VIII. Eosinophilic Granulomatosis With Polyangiitis
1. Definition and Epidemiology
Eosinophilic granulomatosis with polyangiitis (EGPA) is a disease separated from polyarteritis nodosa (conventionally periarteritis nodosa) due to 3 characteristics: (1) Bronchial asthma, (2) peripheral blood eosinophilia, and (3) histological findings of eosinophil infiltration around middle/small-sized vessels (primarily arterioles) and necrotizing and/or necrotizing granulomatous vasculitis or the presence of extravascular granuloma. The disease used to be called Churg-Strauss syndrome,248
but was renamed to EGPA in 2012 and defined as “Eosinophil-rich and necrotizing granulomatous inflammation involving the respiratory tract, and necrotizing vasculitis predominantly affecting small to medium vessels, and clinically associated with asthma and eosinophilia.”1
The number of patients being treated is estimated to be about 1,900, the mean age at onset is about 55 years, and the male/female ratio is 1:1.7 slightly in favor of females.249
In Europe, 0.5–6.8 new patients/year/1 million people contract the disease, and the prevalence is reported to be 10.7–13/1 million people.250
2. Pathogenesis
The cause of the disease is unknown. Its relationship with the leukotriene receptor antagonist (LTRA) is dubious,251
and a reduction in GC after the use of LTRA has been suggested to be a possible cause.252
Genetically, relationships with HLA-DRB1*04, *07, and HLA-DRB4 have been reported.252
The pathological features include tissue damage due to eosinophil infiltration and immunological mechanisms such as IL-4, IL-5, and IL-13.252
Recently, a relationship with IgG4-related disease has also been suggested.253
3. Pathological Findings
Major histopathological findings of EGPA are infiltration of cells primarily eosinophils around middle/small vessels, fibrinoid necrosis, and the presence of extravascular granuloma.248,252
4. Signs and Symptoms
Typically, bronchial asthma occurs first, and after repeated relapses of asthma attack, eosinophilia, systemic signs and symptoms such as fever, and vasculitic lesions such as mononeuritis multiplex, ischemic enteritis, cutaneous vasculitis appear (Figure 37).252,254,255
Vasculitis often develops within 3 years after the onset of bronchial asthma. Mononeuritis multiplex is observed in more than 90% of the patients.249
5. Laboratory and Imaging Findings
Eosinophilia should be more than 10% of leukocytes or 1,500/μL. The percentage of patients positive for MPO-ANCA is about 40–50%,249
and PR3-ANCA (c-ANCA) is rare. Chest radiography often presents bilateral infiltrative shadows with non-segmental distribution, occasionally exhibits ground-glass appearance, and may show thickening of the bronchial wall or interlobular septa.256
6. Diagnosis and Diagnostic Criteria
Patients are classified as EGPA by Lanham’s criteria (Table 21)257
or ACR criteria (Table 22).258
However, in Japan, the diagnostic criteria by the Ministry of Health and Welfare of Japan proposed in 1998 are used. (Table 23).259
Table 21.
Lanham’s Criteria for Classification of Eosinophilic Granulomatosis With Polyangiitis
| 1. Asthma |
| 2. Peak peripheral blood eosinophil counts in excess of 1.5×109/L. |
| 3. Systemic vasculitis involving two or more extra-pulmonary organs. |
(From Lanham JG, et al. 1984257)
Table 22.
ACR Classification Criteria for Eosinophilic Granulomatosis With Polyangiitis
| Criterion |
Definition |
| 1. Asthma |
Wheezing or diffuse high pitched rales on expiration |
| 2. Eosinophilia |
Eosinophilia >10% on white blood cell differential count |
3. Mononeuropathy or
polyneuropathy |
Development of mononeuropathy, multiple mononeuropathies, or polyneuropathy (i.e., glove/socking
distribution) attributable to a systemic vasculitis |
4. Pulmonary infiltrates,
non-fixed |
Migratory or transitory pulmonary infiltrates on radiographs (not including fixed infiltrates) attributable to a
systemic vasculitis |
| 5. Paranasal sinus abnormality |
History of acute or chronic paranasal sinus pain or tenderness or radiographic opacification of the paranasal
sinuses |
| 6. Extravascular eosinophils |
Biopsy including artery, arteriole, or venule, showing accumulations of eosinophils in extravascular areas |
(From Masi AT, Hunder GG, Lie JT, et al., Arthritis Rheum 1990,258 John Wiley and Sons. (c) 1990, American College of Rheumatology.) The condition is classified as Churg-Strauss syndrome (presently EGPA) when 4 or more of the above criteria are met.
Table 23.
Criteria for Classification of Allergic Granulomatous Angiitis (Presently EGPA) by the Ministry of Health and Welfare of Japan, 1998
| 1. Major clinical findings |
| 1) Bronchial asthma or allergic rhinitis |
| 2) Eosinophilia |
| 3) Signs/symptoms of vasculitis: |
| Fever ≥38℃ for ≥2 weeks) |
| Weight loss (6 kg in ≤6 months) |
| Mononeuritis multiplex |
| Gastrointestinal hemorrhage |
| Purpura |
| Polyarthralgia (polyarthritis) |
| Myalgia, and muscular weakness |
| 2. Typical clinical course |
| Major clinical findings 1) and 2) develop first, followed by major clinical finding 3) |
| 3. Major histological findings |
1) Presence of granulomatous or fibrinoid necrotizing vasculitis in small vessels associated with significant infiltration of
eosinophils in the surrounding tissues |
| 2) Presence of extravascular granuloma |
| 4. Diagnosis |
| 1) Definite |
| (a) Patients with ≥1 major clinical findings of 1), 2), and 3) and with ≥1 major histological findings |
| (b) Patients with 3 major clinical findings and a typical clinical course |
| 2) Probable |
| (a) Patient with 1 major clinical finding and 1 major histological finding |
| (b) Patients with 3 major clinical findings without typical clinical course |
| 1. Supporting laboratory findings |
| 1) Leukocytosis (≥10,000/μL) |
| 2) Thrombocytosis (≥400,000/μL) |
| 3) Elevated serum IgE (≥600 U/mL) |
| 4) Positive MPO-ANCA |
| 5) Positive rheumatoid factor |
| 6) Pulmonary infiltration shadows on X-ray |
(From Sectional Meeting on Intractable Vasculitides, Specific Immune Disease Study Group, Ministry of Health and Welfare of Japan, 1999259)
Table 24
shows recommendations concerning treatments for EGPA. Treatments for ANCA-associated vasculitides vary with the severity and disease type.
Table 24.
Recommendations and Evidence Levels of Treatments for EGPA
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
I |
B |
| Steroid pulse therapy |
IIa |
C |
| Cyclophosphamide (CY) |
IIa |
B |
| Azathioprine (AZA) |
IIa |
C |
| Methotrexate (MTX)* |
IIa |
C |
| High-dose immunoglobulin therapy |
IIa |
B |
| Rituximab (RTX)* |
IIb |
B |
| Anti-IL5 therapy |
IIb |
B |
| Anti-IgE therapy* |
IIb |
B |
| Plasma exchange therapy* |
III |
B |
*Uncovered by health insurance in Japan.
7.1 Acute Phase
For patients with mild EGPA, oral PSL at 0.3–0.6 mg/kg/day (15–30 CY (1–2 mg/kg/day) or oral AZA (0.5–1.5 mg/kg/day) is used concomitantly as appropriate (recommendation class: IIa, evidence level: C). For patients with severe EGPA, oral PSL at 0.6–1.0 mg/kg/day (40–60 mg/day) or steroid pulse therapy is administered with concomitant administration of CY within 4 weeks260
(recommendation class: IIa, evidence level: B). CY is administered orally (2 mg/kg/day) or intravenously (0.5–0.75 g/m2/3–4 weeks, total of 3–6 times). For patients with severest EGPA showing conditions such as diffuse alveolar hemorrhage, plasma exchange (2.0–3.0 L for 3 days) should be performed simultaneously.254,260
Overseas, RTX, an anti-CD20 antibody preparation, is used for patients with ANCA-positive treatment-resistant cases260
(recommendation class: IIb, evidence level: B).
7.2 Chronic Phase
After induction of remission, PSL is reduced gradually to 10 mg/day or less in 6–18 months.261
High-dose intravenous γ-globulin (IVIG) therapy for residual peripheral neuropathy has been covered by health insurance in Japan from January 2010 (recommendation class: IIa, evidence level: B).
7.3 Remission Phase
PSL is maintained at 5–10 mg/day. CY should be discontinued after about 6 months and replaced by drugs such as MTX or AZA (recommendation class: IIa, evidence level: C).260
7.4 Exacerbation Phase (at the Time of Relapse)
In principle, treatments should be similar to those in the acute phase. A total dose of CY must be no more than 10–15 g by checking the past doses.250
Biological agents such as anti-IL-5 antibody mepolizumab and anti-IgE antibody omalizumab (omalizumab is uncovered by health insurance in Japan).252
8. Prognosis
By the above treatments, remission can be achieved within 6 months in 90% or more patients, and the 5-year survival rate is 97%.252
In rare instances, intestinal perforation, heart failure, renal dysfunction, cerebrovascular disorders may occur. Dysesthesia due to mononeuritis multiplex often persists and irreversible impairment remains in about two-thirds of the patients.
IX. Anti-Glomerular Basement Membrane Disease
1. Definitions and Epidemiology
Anti-glomerular basement membrane disease means poor-prognosis kidney/lung lesions caused by antibody to glomerular basement membrane (GBM). By the CHCC classification revised in 2012, it is classified as an immune complex small vessel vasculitis and is subclassified into 3 disease types: (1) renal-limited anti-GBM antibody nephritis, (2) Lung-limited anti-GBM antibody alveolar hemorrhage, and (3) the type with both kidney and lung involvements.1
Of these disease types, anti-GBM antibody glomerulonephritis is a disease with poor prognosis that clinically develops RPGN or, occasionally, acute kidney injury and histologically exhibits crescentic necrotizing glomerulonephritis and linear IgG staining (linear pattern) in the loop wall by immunofluorescence study. If complicated by lung hemorrhage, it is called Goodpasture’s disease.
It is a relatively rare disease with a reported incidence of 0.5–1.0 persons/year/1 million people. According to the survey by the MHLW of Japan, of the 1,772 patients with RPGN accumulated until 2006, 81 (4.6%) had anti-GBM antibody nephritis, 27 (1.5%) had Goodpasture’s disease with lung hemorrhage, and a total of 6.1% had anti-GBM disease.262
The mean ages of the patients with anti-GBM nephritis and Goodpasture’s disease, which used to be observed frequently in middle-aged individuals, are 61.6 (11–77) and 70.9 (57–93) years, respectively, indicating advanced aging of the patients.
2. Pathogenesis
The antigen for anti-GBM antibody is present in the noncollagenous domain 1 (NC1 domain) in the C-terminal of the α3 chain of type IV collagen distributed in the glomerular and pulmonary capillary basement membrane, and the amino acid residues at positions 17–31 on the N-terminal side (epitope A; EA) and those at positions 127–141 on the C-terminal side (epitope B; EB) have been identified.263,264
Usually, these epitopes are buried in the basement membrane, but, if the basement membrane of the lung or kidney is damaged due to infection such as influenza, smoking, or inhaled toxic substances, the α-chain hexamer is disassociated to expose the epitopes in the α3 and 5 chains, and anti-GBM antibody is produced.264
When it binds with the basement membrane antigen of the renal glomeruli or pulmonary capillaries, T-lymphocyte-mediated immune responses are induced, and disruption of the basement membrane and vasculitis occur due to local infiltration of inflammatory cells and subsequent inflammatory reactions.
3. Pathological Findings
By light microscopy, pathological features of severe crescentic necrotizing glomerulonephritis are observed. Immunofluorescence shows linear GBM staining for IgG is observed along the capillary walls.
4. Signs and Symptoms
In addition to symptoms due to nephritis and lung hemorrhage, systemic symptoms such as malaise, fever, body weight loss, and arthralgia are observed frequently.262
The symptoms observed at the onset are most frequently non-specific ones such as malaise and fever. Macroscopic hematuria is also a relatively frequent finding. It is also important to check a history of smoking and recent infection.
5. Laboratory Findings
In nearly all patients, hematuria, nephritic urinalysis findings, an elevated serum Cr level, and signs of marked inflammation are observed. Proteinuria of varying degrees is also observed, and it occasionally reaches nephrotic ranges. Anemia is also severe in many patients. Checking of anti-GBM antibody, which becomes positive, is essential for the definitive diagnosis. The anti-GBM antibody level is parallel to the disease activity and often becomes negative as the disease is treated.
Patients in whom both anti-GBM antibody and ANCA are positive (double positive) have been known, and ANCA (typically MPO-ANCA) is positive in about 20–30% of the patients with anti-GBM-antibody glomerulonephritis.265
Conversely, anti-GBM antibody is positive in about 5–10% of the patients with ANCA-associated vasculitis.266,267
6. Diagnosis and Diagnostic Criteria
Table 25
shows the diagnostic criteria for anti-GBM antibody glomerulonephritis as a designated intractable disease in Japan.
Table 25.
Diagnostic Criteria for Anti-GBM Antibody Glomerulonephritis
1. Nephritic urinalysis findings such as hematuria (often microscopic, rarely macroscopic), proteinuria, and urinary
cast are observed. |
| 2. Serum anti-GBM antibody is positive. |
3. Linear deposits of immunoglobulin along the glomerular loop wall and necrotizing crescentic glomerulonephritis are
demonstrated by renal biopsy. |
| “Anti-GBM nephritis” is definitively diagnosed when 1 and 2 or 1 and 3 of the above findings are positive. |
Table 26
shows recommendations about treatments for anti-GBM disease.
Table 26.
Recommendations and Evidence Levels Concerning Treatments for Anti-GBM Antibody Disease
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
I |
B |
| Steroid pulse therapy |
IIa |
B |
| Cyclophosphamide (CY) |
I |
B |
| Plasma exchange |
I |
B |
| Rituximab (RTX)* |
III |
C |
*Uncovered by health insurance in Japan.
7.1 Initial Treatment
The treatment for anti-GBM disease is determined in consideration of the presence or absence of organ lesions (kidney, lung lesions) and the disease severities.
7.1.1 When Non-Dialysis-Dependent RPGN or Lung Hemorrhage Is Noted
Usually, PSL is administered at 1 mg/kg/day (started by steroid pulse therapy using mPSL), and triplet combination therapy consisting of PSL, IVCY (500–750 mg/m2/month) or oral CY (1–2 mg/kg/day), and plasma exchange is carried out (recommendation class: I, evidence level: B). The dose of IVCY is adjusted according to the age and serum Cr level.
7.1.2 When Severe Renal Impairment Requiring Dialysis Is Present, and Recovery From Renal Failure Cannot Be Expected
The treatments are decided in consideration of the general conditions including the presence or absence of lung lesions.
7.2 Maintenance Therapy
After the initial treatment, PSL and CY are gradually reduced, and immunosuppressive therapy is usually continued for 6–12 months. Long-term maintenance therapy is usually unnecessary without relapse of the disease.
7.3 Comments About Treatments
The objective of treatment is to control the progression of nephritis and lung hemorrhage as soon as possible and to remove anti-GBM antibody that causes the disease immediately. Intensive anti-inflammatory therapy (GC including steroid pulse therapy), elimination of anti-GBM antibody (plasma exchange), and suppression of anti-GBM antibody production (GC and immunosuppressive therapy) are carried out simultaneously as soon as possible.268
Plasma exchange has recently been covered by health insurance in Japan, up to 2 courses of 7 sessions within two weeks each.
8. Prognosis
Once anti-GBM antibody becomes negative, and clinical remission is obtained, relapse of the disease is rare thereafter. Therefore, maintenance therapy is usually unnecessary. However, as cases of relapse after a long interval from the onset have also been reported, follow-up is essential.269
In patients who are also positive for ANCA, relapse tends to be observed more frequently than in those positive for anti-GBM antibody alone.
X. Urticarial Vasculitis
1. Definition and Epidemiology
Long-persisting urticarial and cutaneous leukocytoclastic angiitis are observed. There is a report that about 20% of patients diagnosed with urticarial had urticarial vasculitis.270
The disease is classified into primary and secondary as well as hypocomplementemic and normocomplementemic types. Hypocomplementemic urticarial vasculitis was newly adopted by CHCC 2012 along with anti-C1q vasculitis as its another name.1
2. Pathogenesis
The disease, in which immunoglobulin and complement are deposited in the wall of the affected vessels, belongs to immune complex vasculitides. Viral infections (hepatitis B, hepatitis C) and collagen are known to be included as causative disorders of secondary urticarial vasculitis. The collagen-like region of C1q, which is the antigen for anti-C1q antibody, hepatitis virus, and drugs have been suggested as causes of the disease.271
3. Pathological Findings
Neutrophil-dominant perivascular infiltration, red blood cell extravasation, and fibrinoid deposits are observed in small arteries and veins in the upper and middle layers of the dermis. Deposits of immunoglobulin and complement are also observed. Mehregan et al. reported that immunoglobulin and complement deposits are observed in the cutaneous basement membrane in 69.6% and in the dermal capillary wall in 86.9% of the patients in hypocomplementemic cases but in 17.8% and 28.9%, respectively in those with normocomplementemic cases.272
Sanchez et al. showed that the positive rate increases even in the normocomplementemic type if systemic symptoms are present.273
4. Signs and Symptoms
Persisting (often for 24 hours or longer) urticarial is accompanied by signs and symptoms such as purpura, livedo reticularis, erythema, bullae, mixed presence of bloody bullae, burning sensation, and pain. Some patients exhibit ring-shaped or wood grain-like urticarial (Figure 38).
While 54% of the hypocomplementemic patients were diagnosed with SLE during the course of the disease,274
only 3% of the normocomplementemic patients were reported to have developed SLE.271
Hypocomplementemic urticarial vasculitis with characteristic systemic symptoms such as arthritis, renal symptoms, gastrointestinal symptoms, and ophthalmic symptoms is called hypotemplementemic urticarial vasculitis syndrome (HUVS). Kidney impairment is more likely to follow a serious course in children.275
Ophthalmic symptoms are observed in 15% of patients with SLE, but symptoms including episcleritis and uveitis are noted in 61% of patients with HUVS.274
5. Laboratory and Imaging Findings
5.1 Histopathological Examinations
Necrotizing vasculitis is detected, but fibrinoid degeneration is unlikely to occur, because the capillary wall is primarily affected (called leukocytoclastic vasculitis). Neutrophil infiltration is unclear because of edema due to urticarial. Deposits of immunoglobulin and complement are noted in the vascular wall.
5.2 Seroimmunological Examinations
Measurement of complements. Since there is the possibility of systemic vasculitis, ANCA and cryoglobulin are measured. With secondary pathogenesis in mind, anti-nuclear antibody (ANA), and-dsDNA antibody, anti-SSA antibody, and anti-SSB antibody are examined.
ANA is positive in 95% of SLE patients and 61–71% of HUVS patients274
and negative in 5% and 66%, respectively.276
There are reports that anti-dsDNA antibody is positive in ≥70% of the SLE patients and 17% of HUVS patients274
but negative in 5–30% and 83%, respectively,276
and that anti-C1q antibody is observed in 38–61% of SLE patients277,278
and 100% of HUVS patients.279
6. Diagnosis and Diagnostic Criteria
Urticarial vasculitis is definitively diagnosed based on persisting urticarial and necrotizing vasculitis demonstrated by skin biopsy (Evidence level: C). Schwartz et al. required fulfillment of the presence of urticarial and hypocomplementemia and at least 2 of cutaneous vasculitis, arthritis, glomerulonephritis, episcleritis or uveitis, recurrent abdominal pain, and anti-C1q antibody in blood for the diagnosis of HUVS.
7. Policies and Guidelines of Treatment
Table 27
shows recommendations concerning treatments for urticarial vasculitis. Treatment is initiated with anti-histamine and anti-allergic agents (recommendation class: IIb, evidence level: C). However, anti-histamine/anti-allergic drugs are reported to be ineffective.280,281
For patients with severe skin symptoms or organ disorders, GC is selected (recommendation class: IIa, evidence level: C). A dose of 20–30 mg/day is necessary,273,282
and steroid pulse therapy was reportedly necessary for conditions complicated by SLE.283,284
For patients who show serious systemic symptoms or GC resistance, the concomitant use of CyA, AZA or CY can be an option285–287
(recommendation class: IIb, evidence level: C). In addition, plasma exchange,288
IVIG,289
and RTX, an anti-CD20 antibody preparation,287
have also been reported (recommendation class: IIb, evidence level: C).
Table 27.
Recommendations and Evidence Levels Concerning Treatments for Urticarial Vasculitis
| |
Recommendations |
Levels of evidence |
| Anti-histamine/anti-allergic drugs |
IIb |
C |
| Glucocorticoid (GC) |
IIa |
C |
| Cyclosporine (CyA)* |
IIb |
C |
| Azathioprine (AZA)* |
IIb |
C |
| Cyclophosphamide (CY)* |
IIb |
C |
| Plasma exchange* |
IIb |
C |
| High-dose i.v. immunoglobulin therapy* |
IIb |
C |
| Rituximab (RTX)* |
IIb |
C |
| NSAIDs* |
IIb |
C |
| Colchicine* |
IIb |
C |
| Hydroxychloroquine* |
IIb |
C |
| Rectisol* |
IIb |
C |
*Uncovered by health insurance in Japan.
Non-steroidal anti-inflammatory drugs (NSAIDs) have been reported by Millns et al.290
and Mehregan272
to be effective (recommendation class: IIb, evidence level: C). Colchicine291
and hydroxychloroquine292
have been selected as first-line treatments based on the results of retrospective studies (recommendation level: IIb, evidence level: C). Rectisol is considered effective in patients who secondarily develop SLE293,294
(recommendation class: IIb, evidence level: C).
8. Prognosis
Systemic symptoms are absent or mild in normocomplementemic urticarial vasculitis but severer in the hypocomplementemic type.
XI. IgA Vasculitis (Henoch-Schönlein Purpura)
1. Definition and Epidemiology
Henoch- Schönlein purpura (HSP) was renamed as IgA vasculitis.1,295
The disease is classified as a small vessel vasculitis based on the size of the affected vessels. It may occur in people of all ages but most frequently affects those aged 3–10 years and appears to have a slight predilection for boys. It is a common vasculitis in children.296
It is reportedly complicated by glomerulonephritis in about 50–80% of adults and 20–50% of children.297
2. Pathogenesis
The pathogenesis remains unclear.
3. Pathological Findings
Histologically, the disease is characterized by infiltration of inflammatory cells, which are primarily multinuclear leukocytes, around small vessels and IgA deposits in vessel walls. In the skin, histological features of leukocytoclastic necrotizing small-vessel vasculitis accompanied by dermal hemorrhage are observed. In the renal glomeruli, deposits of IgA and C3 are observed primarily in the mesangial region by immunofluorescent staining similarly to IgA nephropathy.
4. Signs and Symptoms
In children, 50% of the patients have a history of upper respiratory infection as an antecedent infection. The symptoms are purpura (100%), arthritis (80%), abdominal pain (60%), and nephritis (50%).297
4.1 Skin Signs and Symptoms
Symmetric palpable purpura with punctate distribution are observed in the lower limbs and back in nearly all patients (Figure 39).
4.2 Joint Symptoms
Pain and swelling of joints are observed in 70–80% of the patients.
4.3 Gastrointestinal Symptoms
Symptoms including abdominal pain, vomiting, bloody stools, and melena are observed in 50–70% of the patients.
4.4 Renal Symptoms
Abnormal urinalysis findings often appear within a few days to 1 month after the onset of vasculitis. The onset is with hematuria alone in 15%, hematuria+proteinuria in 38%, acute nephritic syndrome in 15%, nephritis+nephrotic syndrome in 23%, and nephrotic syndrome in 8%.
5. Laboratory and Imaging Findings
The platelet count is normal, and indices of the clotting system such as the prothrombin time and partial thromboplastin time are also normal. The plasma factor XIII activity is reduced in about 3/4 of the patients and decreases with exacerbation of the clinical condition. The serum IgA level increases in about 40–60% of the patients in an early stage of the disease. Rumpel-Leede test is positive, which indicates weakening of the capillary resistance, in about 30% of the patients.
Patients with IgA vasculitis complicated by glomerulonephritis show hematuria (microscopic, macroscopic), erythrocyte deformation in urine sediment, granular cast and red blood cell cast, and proteinuria of varying degrees depending on the severity of nephritis.
6. Diagnosis and Diagnostic Criteria
In children, IgA vasculitis can be diagnosed if 2 or more of the 4 classification criteria of the American College of Rheumatology298
are met: [(1) palpable purpura, (2) acute abdominal colic, (3) presence of granulocytes in the walls of small arteries and veins on biopsy, and (4) age of ≤20 years]. Histopathological examinations by biopsy show features of leukocytoclastic vasculitis with infiltration of multinuclear leukocytes and monocytes around small vessels and deposits of IgA, C3, and IgG in vascular walls. Adult purpura nephritis was designated as an intractable disease in July 2015 and is classified according to the criteria by the American College of Rheumatism.298,299
The renal tissue classification of purpura nephritis by the International Study of Kidney Disease in Children (ISKDC)300
is shown below.
Grade I Minimal changes
Grade II Mesangial proliferation alone
Grade III a) focal, b) diffuse mesangial proliferation; crescents<50% of glomeruli
Grade IV a) focal, b) diffuse mesangial proliferation; crescents 50–75% of glomeruli
Grade V a) focal, b) diffuse mesangial proliferation; crescents >75% of glomeruli
Grade VI Membranoproliferative glomerulonephritis
7. Policies and Guidelines of Treatment
Table 28
shows recommendations about treatment for IgA vasculitis.
Table 28.
IgA Recommendations and Evidence Levels About Treatments for IgA Vasculitis
| |
Recommendations |
Levels of evidence |
| Extrarenal manifestations (skin symptoms, arthralgia, abdominal pain) |
| Anti-histamine drugs |
IIb |
C |
| Non-steroidal anti-inflammatory drugs (NSAIDs) |
IIa |
C |
| Glucocorticoid (GC) |
IIa |
C |
| Dried human factor XIII concentrate |
IIa |
B |
| Renal symptoms |
| Antiplatelet drugs |
IIb |
C |
| Glucocorticoid (GC) |
IIa |
C |
| Steroid pulse therapy+Urokinase*+cyclophosphamide (CY) |
IIa |
C |
| GC+azathioprine (AZA) |
IIa |
C |
| GC+mycophenolate mofetil (MMF)* |
IIb |
C |
| GC+cyclosporine (CyA)* |
IIb |
C |
| Plasma exchange therapy* |
IIb |
C |
| Cyclophosphamide (CY) |
III |
B |
| Tonsillectomy*+steroid pulse therapy |
IIa |
C |
*Uncovered by health insurance in Japan.
7.1 Treatment for Extrarenal Manifestations
Rest is maintained, and symptomatic treatments are performed depending on the symptoms.
1) Anti-histamine drugs are administered for painful edema and itching of the skin (recommendation class: IIb, evidence level: C)
2) Non-steroidal anti-inflammatory drugs (NSAIDs) are administered for joint symptoms (recommendation class: IIa, evidence level: C)
3) If severe abdominal symptoms cannot be controlled even by the administration of analgesics, glucocorticoid (GC) is administered, tapered, and discontinued after 1–2 weeks.301,302
(recommendation class: IIa, evidence level: C)
4) If a decrease in the plasma factor XIII level to 90% or less is accompanied by abdominal symptoms and joint symptoms, the administration of dried human clotting factor XIII concentrate should be considered (recommendation class: IIa, evidence level: B)
5) In children, the administration of GC at the onset does not contribute to a decrease in the rate of subsequent occurrence of nephritis or abdominal symptoms.303–305
The KDIGO guidelines recommend no use of GC to prevent the occurrence of nephritis306
(recommendation class: III, evidence level: B)
7.2 When Complicated by Nephritis
ISKDC class I–II: Antiplatelets are often administered (recommendation class: IIb, evidence level: C).
ISKDC class III–VI: The following treatments are considered.
1) GC (PSL 1 mg/kg/day)301
(recommendation class: IIa, evidence level: C)
2) Steroid pulse therapy+iv urokinase*+oral CY307
(recommendation class: IIa, evidence level: C)
3) AZA+GC combination therapy308,309
(recommendation class: IIa, evidence level: C)
4) MMF alone*310
or MMF*+GC combination therapy311
adults) (recommendation class: IIb, evidence level: C)
5) CyA* alone312
or CyA*+GC combination therapy313
(recommendation class: IIb, evidence level: C)
6) Plasma exchange therapy* (recommendation class: IIb, evidence level: C): There are reports that plasma exchange was effective in children.314,315
7) CY alone316
or IVCY+GC combination therapy317
is not effective (recommendation class: III, evidence level: B)
8) There is a report suggesting the effectiveness of tonsillectomy*+steroid pulse therapy also for purpura nephritis318
(recommendation class: IIa, evidence level: C)
*Not covered by health insurance in Japan
The above recommendations are based primarily on reports concerning children, but the KDIGO guidelines suggest treating adults with nephritis complicating adult IgA vasculitis similarly to children.306
8. Prognosis
The prognosis of IgA vasculitis is basically favorable, and the disease often follows a monophasic course and is remitted spontaneously in several weeks. Relapse is observed in 10–20% of the patients and the episode occasionally persists for more than a year.
In children, nephritis is observed in about half the patients, but exacerbation to end-stage renal disease is rare. In adults, 85% of the patients suffer nephritis, which often develops into end-stage renal disease, and the prognosis is poorer than in children.
XII. Cryoglobulinemic Vasculitis
1. Definition and Epidemiology
Cryoglobulin (CG) is protein that precipitates in serum or plasma at a low temperature and dissolves when heated to 37℃. Since cryoglobulinemia is asymptomatic in some patients, a condition that exhibits clinical features of immune complex-mediated vasculitis is called cryoglobulinemic vasculitis (CV).245,319
CV may be a primary disorder without an underlying disorder, or a secondary disorder. Cryoglobulinemia occurs secondarily to HCV infection in 40–65% and collagen disease in 15–25% of the patients.320–323
2. Pathogenesis
Cryoglobulinemia is classified into 3 types (Table 29).324,325
Recently, essential CV has decreased to about 10% or less with accumulation of anti-HCV antibody-positive cases.326,327
Table 29.
Cryoglobulin Classification
| |
Type I |
Type II |
Type III |
| Components |
Simple |
Mixed |
Mixed |
Monoclonal immunoglobulins (IgG,
IgM, or IgA) or Bence Jones protein/
monoclonal free light chains |
Monoclonal immunoglobulins (IgM,
IgG, or IgA) and polyclonal
immunoglobulin (mostly IgG) |
Polyclonal immunoglobulins of all
isotypes |
Pathological
features |
Microthrombosis |
Immune complex-mediated vasculitis |
Immune complex-mediated vasculitis |
Disease
association |
• Waldenstrom’s macroglobulinemia
• Multiple myeloma
• Monoclonal gammopathy associated
with lymphoproliferative disorder
• Light chain disease |
• Hepatitis C
• Essential cryoglobulinemia
• Sjogren’s syndrome
• Rheumatoid arthritis
• Chronic lymphocytic leukemia |
• Essential cryoglobulinemia
• Sjogren’s syndrome
• Systemic lupus erythematosus
• Viral infections (Hepatitis B and C,
CMV, EBV, HIV)
• Endocarditis, other bacterial
infections
• Biliary cirrhosis |
(Reproduced from Brouet JC, et al. 1974324 and Motyckova G, et al. 2011.325 Motyckova G, Murali M., Am J Hematol 2011325 with modification, John Wiley and Sons. (c) 2011, Wiley-Liss, Inc.)
Type I shows microthrombi due to non-inflammatory eosinophilic amorphous substances can be detected in the kidney and skin-biopsy specimens. In Types II and III, necrotizing vasculitis in small vessels of the upper and middle layers of the dermis is observed. Histological findings of membranoproliferative glomerulonephritis are characteristic in the kidney.
4. Clinical Features
See
Table 30.325
Table 30.
Clinical and Laboratory Features of Cryoglobulinemia
| Clinical manifestations |
Type I |
Type II |
Type III |
| Arthralgia > arthritis |
+ |
++ |
+++ |
| Purpura |
+ |
+++ |
+++ |
| Gangrene/acrocyanosis |
+++ |
+ |
± |
| Hyperviscosity |
+++ |
± |
− |
| Hematologic |
++ |
± |
± |
| Renal |
+ |
++ |
+ |
| Neurologic |
+ |
++ |
++ |
| Liver |
± |
++ |
+++ |
| Lung |
− |
+ |
+ |
| C3 |
Normal |
Decreased |
Decreased |
| C4 |
Normal |
Decreased |
Decreased |
| CH50 |
Normal |
Decreased |
Decreased |
| RF |
+ |
++ |
++ |
| Autoantibodies (ANA, etc.) |
− |
++ |
++ |
| Hepatitis B |
− |
+ |
+ |
| Hepatitis C |
− |
+++ |
+++ |
ANA, antinuclear antibodies, RF, rheumatoid factor; Hematologic features include thrombosis and bleeding and spurious/artifact thrombocytosis. (Reproduced from Motyckova G, Murali M., Am J Hematol 2011325 with modification, John Wiley and Sons. (c) 2011, Wiley-Liss, Inc.)
If vasculitic lesions associated with cold exposure are noted, examinations for serum CG and causative disorders (lymphoproliferative disease, hepatitis virus infection, collagen disease, etc.) should be performed.
6. Diagnosis and Diagnostic Criteria
As a result of a multicenter collaborative study in Europe, preliminary classification criteria for CV have been proposed (Table 31).328
Table 31.
Preliminary Criteria for Classification of Cryoglobulinemic Vasculitis
Satisfied if at least two of the three items (questionnaire, clinical, laboratory) are positive the patient must be positive for serum
cryos in at least 2 determinations at ≥12 week interval |
| (i) Questionnaire item: at least two out of the following |
| • Do you remember one or more episodes of small red spots on your skin particularly involving the lower limbs? |
| • Have you ever had red spots on your lower extremities which leave a brownish color after their disappearance? |
| • Has a doctor ever told you that you have viral hepatitis? |
| (ii) Clinical item: at least three out of the following four (present or past) |
| • Constitutional symptoms |
Fatigue
Low grade fever (37–37.9℃, >10 days, no cause)
Fever (>38℃, no cause)
Fibromyalgia |
| • Articular involvement |
Arthralgias
Arthritis |
| • Vascular involvement |
Purpura
Skin ulcers
Necrotizing vasculitis
Hyperviscosity syndrome
Raynaud’s phenomenon |
| • Neurological involvement |
Peripheral neuropathy
Cranial nerve involvement
Vasculitic CNS involvement |
| (iii) Laboratory item: At least 2 out of the 3 questions. (present) |
| • Reduced serum C4 |
| • Positive serum rheumatoid factor |
| • Positive serum M component |
CG, cryoglobulin; CV, cryoglobulinemic vasculitis. (Cited from Ann Rheum Dis De Vita S, Soldano F, Isola M, et al. 2011,328 with permission from BMJ Publishing Group Ltd.)
Table 32
shows recommendations about treatments for CV.
Table 32.
Recommendations and Evidence Levels About Treatments for CV
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
IIa |
B |
| Cyclophosphamide (CY) |
IIa |
B |
| Rituximab (RTX)* |
IIa |
B |
| Plasma exchange therapy* |
IIb |
C |
*Uncovered by health insurance in Japan.
In addition to avoiding cold exposure and keeping warm, analgesics such as non-steroidal anti-inflammatory drugs (NSAIDs) are used. If there are rapidly progressive organ disorders, the use of glucocorticoid (GC) (including pulse therapy)+RTX (often 375 mg/m2/week×4 times) or CY (2 mg/kg/day) (recommendation class: IIa, evidence level: B) is recommended. If RTX cannot be used, treatment using CY is recommended.329–331
In secondary cryoglobulinemia, treatments for the primary disorder are performed in addition to immunosuppressants (recommendation class: IIa, evidence level: B).331–333
For essential cryoglobulinemia, combination therapy of GC and RTX is recommended (recommendation class: IIa, evidence level: B).
Plasma exchange therapy should be considered in cases with (1) hyperviscosity syndrome, (2) acute respiratory insufficiency, alveolar hemorrhage, and acute mesenteric vasculitis,334
and (3) RPGN requiring dialysis330
(recommendation class: IIb, evidence level: C). RTX and plasma exchange therapy are not covered by health insurance in Japan.
8. Prognosis
Regarding the prognosis of mixed cryoglobulinemia without the involvement of HCV infection, the 5-year survival rate is 79%. Age (≥65 years or older), sex (male), complication by respiratory and gastrointestinal lesions, and renal failure are important prognostic factors.327
XIII. Behçet’s Disease
1. Definition and Epidemiology
1.1 Definition
Behçet’s disease is an inflammatory disorder characterized by oral aphtha, skin symptoms, ophthalmic lesions, and genital ulcers.335
Vascular involvement is one of serious manifestations which appear in some patients.336,337
1.2 Epidemiology
There are about 20,000 patients with Behçet’s disease in Japan. The frequency of vascular involvement has been reported 6.3–15.3%.336
2. Signs and Symptoms
2.1 Deep-Vein Thrombosis
Deep-vein thrombosis is the most common vascular lesion and prevalent in the lower extremities.336,337
More serious venous lesions such as superior vena cava syndrome, Budd-Chiari syndrome, and cerebral venous sinus thrombosis can occur.336,338
2.2 Aneurysms and Arterial Occlusion
Aneurysms and arterial occlusions develop in the large and medium-sized arteries. Aortic insufficiency cause by aneurysm is associated with poor prognosis.336,337,339
Peripheral aneurysms, which are often at risk of rupture and bleeding, are multiple in the one-third of the patients.336,340
An arterial occlusion causes the local ischemic symptoms.
2.3 Pulmonary Artery Involvement
Rupture of pulmonary artery aneurysm causes fatal hemoptysis.336,341
The pulmonary lesions are commonly complicated with pre-existing deep-vein thrombosis. Frequency of the lesions is low in Japanese patients compared with the other regions.
3. Laboratory and Imaging Findings
3.1 Laboratory Findings
There is no specific data for Behçet’s disease. A pathergy test, HLA-B*51, A*26, and inflammatory responses on blood tests are listed as suggestive findings in the diagnostic criteria of the MHLW of Japan.
3.2 Imaging Findings
3.2.1 Venous Lesions
Venous thrombotic lesions are illustrated by ultrasonography and contrast-enhanced CT.336,337
3.2.2 Arterial Lesions
Contrast-enhanced CT is a standard modality to detect arterial lesions.336,337
PET-CT (not approved),336
which simultaneously assesses the morphological changes and localization of inflammation, is considered promising.
3.2.3 Pulmonary Lesions
Pulmonary artery aneurysms are examined by chest CT scan after screening of chest radiography. Lung perfusion scintigraphy is helpful to detect occlusive lesions in the pulmonary vessels.336,341
4. Diagnosis and Diagnostic Criteria
4.1 Diagnosis
The diagnosis of Behçet’s disease is made based on combinations of symptoms, according to the diagnostic criteria of the MHLW, Japan.
Table 33
compares the MHLW criteria with other sets of representative international criteria.342,343
In addition, local assessment of the vascular lesions is essential.337
Table 33.
Comparison of Diagnostic Criteria for Behçet’s Disease
| |
MHLW337 |
ISG342 |
ITR-ICBD343 |
| Oral aphtha |
Major symptom |
Essential |
2 |
| Skin symptoms |
Major symptom |
✓ |
1 |
| Ophthalmic lesions |
Major symptom |
✓ |
2 |
| Ulcer of the vulva |
Major symptom |
✓ |
2 |
| Arthritis |
Minor symptom |
|
|
| Epididymitis |
Minor symptom |
|
|
| Intestinal lesions |
Minor symptom |
|
|
| Vascular lesions |
Minor symptom |
|
1 |
| Nerve lesions |
Minor symptom |
|
1 |
| Pathergy test |
Suggestive finding |
✓ |
(1) |
| HLA-B51 A26 |
Suggestive finding |
|
|
Diagnoses according to the MHLW criteria.
(1) Complete form: Appearance of the 4 Major symptoms during the course.
(2) Incomplete type: 3 Major symptoms, 2 Major symptoms+2 Minor symptoms, typical ophthalmic symptoms+1 other Major symptom or 2 Minor symptoms.
(3) Suspected: Appearance of some Major symptoms, typical Minor symptoms recurring or exacerbated.
(4) Variant types: (a) Intestinal type, (b) Vascular type, (c) Neurological type.
Behçet’s disease can be diagnosed or classified by the ISG criteria, which require oral aphtha plus 2 or more symptoms out of four symptoms indicated by (✓), or by the ITR-ICBD criteria in which the total score is more than 4.
ISG: International Study Group for Behçet’s Disease; ITR-ICBD: International Team for the Revision of the International Criteria for Behçet’s Disease (from Japan Intractable Diseases Information Center, Behçet’s disease,337 International Study Group for Behçet’s Disease. 1990,342 International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD). 2014343)
4.2 Differential Diagnosis
4.2.1 Venous Lesions
Major differential diagnoses include idiopathic venous thrombosis, congenital thrombotic tendency (protein C deficiency, protein S deficiency, antithrombin III deficiency, etc.), antiphospholipid antibody syndrome, surgical invasion, malignant neoplasm, and local compression.
4.2.2 Arterial Lesions
Arterial lesions that should be differentiated from Behçet’s disease include Takayasu arteritis, GCA, PAN, infectious aneurysm, and Buerger disease and arteriosclerosis obliterans.
4.2.3 Pulmonary Lesions
Hughes-Stovin syndrome is characterized by pulmonary artery aneurysms and venous thrombosis without the mucocutaneous and ocular symptoms.
5. Policies and Guidelines of Treatment
Table 34
summarizes recommendations for treatments of Behçet’s disease, according to the guidelines from Behçet’s Disease Research Committee (BDRC), MHLW, Japan.
Table 34.
Recommendations and Evidence Levels About Treatments for Behçet’s Disease
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
IIa |
B |
| Azathioprine (AZA) |
IIa |
B |
| Cyclophosphamide (CY) |
IIa |
B |
| Cyclosporine (CyA) |
IIb |
C |
| Methotrexate (MTX)* |
IIb |
C |
| TNF inhibitors |
IIb |
B |
| Anticoagulants |
IIa |
C |
| Vascular reconstruction |
IIb |
B |
| Endovascular treatment |
IIb |
B |
| Perioperative immunosuppressive therapy |
IIa |
B |
*Uncovered by health insurance in Japan.
5.1 Pharmacological Therapy
5.1.1 Glucocorticoid/Immunosuppressants/Biologics
a. Combination of GC and AZA (recommendation class: IIa, evidence level: B)
For acute deep venous thrombosis, 0.5–1 mg/kg of PSL is given as the initial therapy followed by gradually tapering while concomitantly using AZA.344–346
b. Combination of GC and CY (recommendation class: IIa, evidence level: B)
For severe venous lesions and aneurysms, high-dose GC including steroid pulse therapy and IVCY (1,000 mg/month) are conducted as the remission induction therapy followed by maintenance therapy using oral PSL and AZA.
c. TNF Inhibitors (recommendation class: IIb, evidence level: B)
Sporadic case reports have shown efficacy of anti-TNF monoclonal antibody for serious forms of vascular involvement in Behçet’s disease. IFX has been approved since 2015 in Japan.
5.1.2 Anticoagulant Therapy (recommendation class: IIa, evidence level: C)
Anticoagulant therapy is not recommended by EULAR, though retrospective studies have shown warfarin is used frequently in Japan and other countries.337,345–347
The guidelines by BDRC, MHLW, Japan recommends the use of anticoagulants or antiplatelet drugs in patients having risk of bleeding.337
5.2 Surgery and Endovascular Treatment
5.2.1 Operative Indication (recommendation class: IIb, evidence level: B)
Rupture or impending rupture of aneurysm and arterial occlusion are indications for surgery. Postoperative complications such as suture failure, occlusion, and anastomotic aneurysm are frequent.336,348
5.2.2 Perioperative Immunosuppressive Therapy (recommendation class: IIa, evidence level: B)
The perioperative concomitant use of immunosuppressive therapy has been shown to prevent post-operative complications and relapses.336,349
5.2.3 Catheterization and Endovascular Treatment (recommendation class: IIb, evidence level: B)
Endovascular treatment is optional for some patients.350–352
6. Prognosis
Several studies have shown that vascular lesions, particularly arterial lesions, are critical prognostic factors of Behçet’s disease.353
The vascular involvement such as pulmonary artery aneurysm, aortic aneurysm, and Budd-Chiari syndrome can be fatal.
XIV. Malignant Rheumatoid Arthritis
1. Definition and Epidemiology
Vasculitis of small and medium-sized vessels associated with rheumatoid arthritis (RA) is called rheumatoid vasculitis (RV). Malignant RA (MRA) is Japan’s original concept defined as “RA that has a severe clinical condition due to extra-articular symptoms including vasculitis”,354
and is not synonymous with RV. A clinical profile of MRA is shown in
Figure 40.
MRA is observed in 0.6–1.0% of the patients with RA, the peak age at the diagnosis is 60–69 years, and the male/female ratio is 1:2 (Figure 25).11
The incidence of RV has been decreasing recently.355
Risk factors for RV include HLA-C*03,356
male sex, a long duration of illness, severe RA, and the presence of peripheral/cerebral vascular lesions.
2. Pathogenesis
The pathogenesis of MRA is unclear, but mechanisms including an involvement of auto-antibody targeted to blood vessels, induction of inflammation due to deposition of immune complexes, and local activation of cellular immunity have been suggested.
3. Pathological Findings
Infiltration of mononuclear cells and neutrophils into the vascular wall are observed, and it is accompanied by necrosis, leukocytoclast, and features of destruction of the vascular wall including rupture of the elastic lamina. MRA is classified into the systemic vasculitis type characterized by necrotizing vasculitis of muscular arteries and peripheral arteritis type characterized by fibrous proliferation of the intima and endarteritis obliterans. Generally, the pathological findings are primarily those of leukocytoclastic vasculitis accompanied by deposits of immune complex in lesions of small arteries, capillaries, and small vessels, and pauci-immune type in lesions of medium-sized arteries and glomeruli.
4. Signs and Symptoms
MRA is more frequent in long-standing RA with advanced joint destruction,357
and rheumatoid nodules are often observed. Systemic type of MRA follows an acute course and causes symptoms such as a fever of ≥38℃, body weight loss, purpura, episcleritis, myalgia, interstitial pneumonia, pleurisy, mononeuritis multiplex, and gastrointestinal bleeding. Interstitial lesions of the lung often progress chronically but occasionally show an acute or subacute course.
5. Laboratory and Imaging Findings
Findings including inflammatory reaction, hypergammaglobulinemia, anti-citrullinated peptide antibody (ACPA), immune complexes, and a decrease in complement are observed. The rheumatoid factor (RF) level is high.
6. Diagnosis and Diagnostic Criteria
The diagnostic criteria shown in
Table 35358
are used.
Table 35.
Diagnostic Criteria for Malignant Rheumatoid Arthritis (Rheumatoid Vasculitis)
| 1. Clinical features |
| (1) Mononeuritis multiplex: Sensory and/or motor disturbance may present. |
| (2) Skin ulcer or infarction or finger gangrene: Not including lesions due to infection or injury. |
| (3) Subcutaneous nodules: Subcutaneous nodules may be present in the apophyseal area, extensor surfaces, and tissues surrounding joints. |
| (4) Episcleritis or iritis: Conditions confirmed by ophthalmologists, not including those due to other causes. |
(5) Exudative pleuritis or pericarditis: Not including those due to other causes such as infection. The presence of pleurodesis alone should
not be considered a positive finding. |
(6) Myocarditis: Diagnosis should be made on the basis of clinical findings, inflammatory reactions, muscle enzyme levels, ECG, and
echocardiography, among other parameters. |
(7) Interstitial pneumonia or pulmonary fibrosis: Diagnosis should be made on the basis of physical findings, chest X-ray, and pulmonary
function testing, regardless of the extent of lesions. |
| (8) Organ infarction: Infarction of intestine, myocardium, lungs, and other organs caused by ischemia or necrosis due to vasculitis. |
(9) Elevated RF: RF should be positive at a titer of 1:2,560 or higher in ≥2 sessions of the RAHA or RAPA test (or ≥960 IU/mL on
quantification of RF). |
(10) Decreased serum complement level or the presence of immune complexes in blood: A decrease in serum complement level should be
demonstrated by a decrease in serum concentration of a complement component such as C3 and C4 or a decrease of CH50-induced
complement activation in ≥2 sessions. The presence of immune complexes in blood should be demonstrated on the basis of C1q
binding activity in >2 sessions. |
| 2. Histological findings |
Biopsy of skin, muscle, nerve, or other affected organs should reveal the presence of necrotic vasculitis, granulomatous vasculitis, or
occlusive endoangiitis in small- or medium-sized vessels. |
| 3. Diagnosis |
Diagnosed as malignant rheumatoid arthritis (MRA) when the 2010 ACR/EULAR criteria for classification of rheumatoid arthritis are fulfilled,
and when |
| (1) 3 or more of the clinical signs and symptoms (1)∼(10) are observed, or |
| (2) at least 1 of the clinical signs and symptoms (1)∼(10) and histological findings in 2. are present. |
| 4. Differential diagnosis |
MRA should be differentiated from infection, secondary amyloidosis, and adverse reactions to drugs (drug-induced interstitial pneumonia,
drug-induced vasculitis, etc.). In amyloidosis, amyloid deposits are demonstrated in the stomach, rectum, skin, kidney, or liver by biopsy.
Attention to overlap syndrome with connective tissue diseases other than RA (e.g., SLE, scleroderma, and polymyositis) is also necessary.
Sjögren’s syndrome most frequently complicates RA and is also observed in about 10% of the patients with MRA. Felty’s syndrome should
also be differentiated from MRA, but it shows leukocytopenia, splenomegaly, and increased susceptibility to infection. |
RAHA, rheumatoid arthritis hemagglutination; RAPA, rheumatoid arthritis particle agglutination; RF, rheumatoid factor; ACR, American College of Rheumatology; EULAR, European League Against Rheumatism (from Malignant Rheumatoid Arthritis, MHLW of Japan358).
Table 36
shows recommendations about treatments for MRA. The dosage/regimen of MTX and bDMARDs are the same as those for the treatment of usual RA.
Table 36.
Recommendations and Evidence Levels About Treatments for Malignant Rheumatoid Arthritis
| |
Recommendations |
Levels of evidence |
| Glucocorticoid (GC) |
IIa |
C |
| Steroid pulse therapy |
IIb |
C |
| Methotrexate (MTX) |
IIa |
C |
| Azathioprine (AZA) |
IIa |
C |
| Cyclophosphamide (CY) |
IIa |
B |
| TNF inhibitors |
IIa |
B |
| Tocilizumab (TCZ) |
IIa |
C |
| Rituximab (RTX)* |
IIb |
B |
| Abatacept |
IIb |
C |
*Uncovered by health insurance in Japan. Note: MTX and biological drugs are covered by health insurance for rheumatoid arthritis.
MTX is administered for active RA without contraindications (recommendation class: IIa, evidence level: C) in patients with MRA. Glucocorticoid (GC) is administered at a medium dose (0.5 mg/kg/day as PSL) or less if there are only skin lesions and serositis but at a high dose (1 mg/kg/day as PSL) including steroid pulse therapy for acute/subacute interstitial pneumonia, mononeuritis multiplex, and multiple organ involvement.
For severe systemic vasculitis, IVCY (500–700 mg/m2/month) or oral CY (1–2 mg/kg/day) may be used (recommendation class: IIa, evidence level: B). AZA (1–2 mg/kg/day) is used for mild cases and as maintenance therapy359
(recommendation class: IIa, evidence level: C). bDMARDs such as TNF inhibitors and IL-6 inhibitors are used for RV that occurs during the administration of MTX,360
but the occurrence of vasculitis associated with the administration of these drugs has also been reported.361–365
There is also a report that plasma exchange therapy, leukocytapheresis, and IVIG366
(not covered by health insurance in Japan) were effective in treatment-resistant and severe cases.
8. Prognosis
The 1-, 5-, and 10-year survival rates of patients with systemic RV are 89.6, 47.7, and 26%, respectively,355
and no improvement is observed compared with the past. According to a domestic epidemiological survey, the outcome was alleviation in 21%, no change in 26%, exacerbation in 31%, death in 14%, and unknown/others in 8%, and the most frequent cause of death was respiratory failure.367
Acknowledgments
The drafting of the present guideline in Japanese was started in December 2015. The manuscript was evaluated by the Independent Assessment Committee in September 2017. The completed Japanese version was published online in March 2018. The English translation of the digest version was started by Medical English Service, Ltd. in July 2018. Collection, revision, and management were conducted by H. Yoshifuji and supervised by M. Isobe. The COI of group members are shown in
Appendix 2.
Appendix 1. JCS Joint Working Group
Chair:
• Mitsuaki Isobe, Sakakibara Heart Institute
Members:
• Koichi Amano, Department of Rheumatology and Clinical Immunology, Saitama Medical Center, Saitama Medical University
• Yoshihiro Arimura, Department of Rheumatology and Nephrology, Kyorin University School of Medicine / Internal Medicine, Kichijoji Asahi Hospital
• Shouichi Fujimoto, Department of Hemovascular Medicine and Artificial Organs, Faculty of Medicine, University of Miyazaki
• Masayoshi Harigai, Department of Rheumatology, School of Medicine, Tokyo Women’s Medical University
• Hitoshi Hasegawa, Department of Hematology, Clinical Immunology, and Infectious Diseases, Ehime University Graduate School of Medicine
• Akihiro Ishizu, Department of Medical Laboratory Science, Faculty of Health Sciences, Hokkaido University
• Shuichi Ito, Department of Pediatrics, Graduate School of Medicine, Yokohama City University
• Shinya Kaname, Department of Nephrology and Rheumatology, Kyorin University School of Medicine
• Shigeto Kobayashi, Department of Internal Medicine, Juntendo Koshigaya Hospital
• Yoshinori Komagata, Department of Nephrology and Rheumatology, Kyorin University School of Medicine
• Kimihiro Komori, Division of Vascular Surgery, Department of Surgery, Nagoya University Graduate School of Medicine
• Issei Komuro, Department of Cardiovascular Medicine, The University of Tokyo Graduate School of Medicine
• Tetsuro Miyata, Vascular Center, Sanno Hospital and Sanno Medical Center
• Tatsuhiko Miyazaki, Department of Pathology, Gifu University Hospital
• Kei Takahashi, Department of Pathology, Toho University Ohashi Medical Center
• Kazuo Tanemoto, Department of Cardiovascular Surgery, Kawasaki Medical School
• Hidehiro Yamada, Medical Center for Rheumatic Diseases, Seirei Yokohama Hospital
• Akitoshi Yoshida, Asahikawa Medical University
• Takashi Wada, Department of Nephrology and Laboratory Medicine, Graduate School of Medical Sciences, Kanazawa University
Collaborators:
• Hiroaki Dobashi, Division of Hematology, Rheumatology and Respiratory Medicine Department of Internal Medicine, Faculty of Medicine, Kagawa University
• Yoshinori Inoue, Ambulatory Vascular Surgical Clinic Tokyo
• Tamihiro Kawakami, Division of Dermatology, Tohoku Medical and Pharmaceutical University
• Reiko Kinouchi, (1) Medicine and Engineering Combined Research Institute, Asahikawa Medical University, (2) Department of Ophthalmology, Asahikawa Medical University
• Hisanori Kosuge, Department of Cardiology, Tokyo Medical University
• Atsushi Kurata, Department of Molecular Pathology, Tokyo Medical University
• Yasuhiro Maejima, Department of Cardiovascular Medicine, Tokyo Medical and Dental University
• Kenji Nagasaka, Department of Rheumatology, Ome Municipal General Hospital
• Yoshikazu Nakaoka, Department of Vascular Physiology, National Cerebral and Cardiovascular Center Research Institute
• Takahiro Okazaki, Vice-Director, Shizuoka Medical Center, National Hospital Organization
• Mitsuho Onimaru, Division of Pathophysiological and Experimental Pathology, Department of Pathology, Graduate School of Medical Sciences, Kyushu University
• Hideki Ota, Department of Advanced MRI Collaboration Research, Tohoku University Graduate School of Medicine
• Ken-ei Sada, Department of Nephrology, Rheumatology, Endocrinology and Metabolism, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences
• Kunihiro Shigematsu, Department of Vascular Surgery, International University of Health and Welfare Mita Hospital
• Eiichi Suematsu, Division of Internal Medicine and Rheumatology, National Hospital Organization, Kyushu Medical Center
• Eijun Sueyoshi, Department of Radiological Science, Nagasaki University Graduate School of Biomedical Sciences
• Takahiko Sugihara, Department of Lifetime Clinical Immunology, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University
• Hitoshi Sugiyama, Department of Human Resource Development of Dialysis Therapy for Kidney Disease, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences
• Mitsuhiro Takeno, Department of Allergy and Rheumatology, Nippon Medical School Graduate School of Medicine
• Naoto Tamura, Department of Internal Medicine and Rheumatology, Juntendo University Faculty of Medicine
• Michi Tsutsumino, Institute of Rheumatology, Tokyo Women’s Medical University
• Haruhito A. Uchida, Department of Chronic Kidney Disease and Cardiovascular Disease, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences
• Hajime Yoshifuji, Department of Rheumatology and Clinical Immunology, Graduate School of Medicine, Kyoto University
• Yoshiko Watanabe, Frist Department of Physiology, Kawasaki Medical School
Independent Assessment Committee:
• Takeshi Kimura, Department of Cardiovascular Medicine, Kyoto University Graduate School of Medicine
• Shin-ichi Momomura, Saitama Medical Center Jichi Medical University
• Toyoaki Murohara, Department of Cardiology, Nagoya University Graduate School of Medicine
• Shoichi Ozaki, Division of Rheumatology and Allergology, Department of Internal Medicine, St. Marianna University School of Medicine
• Hiroshi Shigematsu, Clinical Research Center for Medicine, International University of Health and Welfare
• Keiko Yamauchi-Takihara, Health and Counseling Center, Osaka University
(*The affiliations of the authors and independent assessment committee members are as of April 2019)
Appendix 2. Disclosure of Potential Conflicts of Interest (COI): JCS 2017 Guideline on Management of Vasculitis Syndrome
| Author |
Employment
or supervisory
position
(private firm) |
Shareholder |
Patent
royalty |
Reward |
Manuscript
fee |
Research fund offered |
Scholarship donation/endowed
chair |
Other
rewards |
Declaration
concerning
spouse/relatives
within the first
degree or those
who share the
income/property |
Chair:
Mitsuaki Isobe |
|
|
|
Daiichi Sankyo Co., Ltd. |
|
Chugai Pharmaceutical
Co., Ltd.
Ono Pharmaceutical Co.,
Ltd. |
Teijin Pharma Ltd.
Otsuka Pharmaceutical Co., Ltd.
Daiichi Sankyo Co., Ltd.
Ono Pharmaceutical Co., Ltd.
Mitsubishi Tanabe Pharma
Corporation. |
|
|
Member:
Koichi Amano |
|
|
|
Pfizer Japan Inc.
Chugai Pharmaceutical Co.,
Ltd.
Mitsubishi Tanabe Pharma
Corporation.
AbbVie
Eli Lilly Japan K.K. |
|
|
Chugai Pharmaceutical Co., Ltd.
Asahi Kasei Pharma |
|
|
Member:
Shuichi Ito |
|
|
|
Sanofi K.K. |
|
|
Astellas Pharma Inc.
Pfizer Japan Inc. |
|
|
Member:
Shinya Kaname |
|
|
|
|
|
|
Chugai Pharmaceutical Co., Ltd.
Astellas Pharma Inc. |
|
|
Member:
Issei Komuro |
|
|
|
Actelion Pharmaceuticals
Japan Ltd.
MSD
Daiichi Sankyo Co., Ltd.
Takeda Pharmaceutical Co.,
Ltd.
Mitsubishi Tanabe Pharma
Corporation.
Boehringer Ingelheim
Ono Pharmaceutical Co.,
Ltd.
Toa Eiyo Ltd.
Amgen Astellas BioPharma
K.K. |
|
Ono Pharmaceutical Co.,
Ltd. |
Astellas Pharma Inc.
Edwards Lifesciences
Corporation.
Otsuka Pharmaceutical Co., Ltd.
Kowa Pharmaceutical Co., Ltd.
Daiichi Sankyo Co., Ltd.
Sumitomo Dainippon Pharma
Co., Ltd.
Takeda Pharmaceutical Co.,
Ltd.
Mitsubishi Tanabe Pharma
Corporation.
Teijin Pharma Ltd.
Nipro
Terumo Corporation. |
|
|
Member:
Kimihiro Komori |
|
|
|
Daiichi Sankyo Co., Ltd. |
|
|
Otsuka Pharmaceutical Co., Ltd.
Sanofi-aventis K.K.
Daiichi Sankyo Co., Ltd. |
|
|
Member:
Masayoshi
Harigai |
|
|
|
Chugai Pharmaceutical Co.,
Ltd.
Teijin Pharma Ltd. |
|
Eisai Co., Ltd.
Teijin Pharma Ltd. |
Mitsubishi Tanabe Pharma
Corporation.
Ayumi Pharmaceutical
Corporation.
Nippon Kayaku Co., Ltd.
Takeda Pharmaceutical Co.,
Ltd.
Chugai Pharmaceutical Co., Ltd.
Taisho Toyama Pharmaceutical
Co., Ltd.
Abbvie |
|
|
Member:
Tetsuro Miyata |
|
|
|
Daiichi Sankyo Co., Ltd.
Bayer Yakuhin, Ltd |
|
|
Daiichi Sankyo Co., Ltd. |
|
|
Member:
Hidehiro Yamada |
|
|
|
Chugai Pharmaceutical Co.,
Ltd. |
|
|
|
|
|
Member:
Akitoshi Yoshida |
|
|
|
|
|
Topcon Corporation
Bayer Yakuhin, Ltd |
Abbott Medical Optics Inc.
Otsuka Pharmaceutical Co., Ltd.
HOYA
Alcon Japan Ltd.
Santen Pharmaceutical Co., Ltd.
Senju Pharmaceutical Co., Ltd.
Hokkaido Welfare Federation
of Agricultural Cooperatives ·
HOYA Corporation. |
|
|
Member:
Takashi Wada |
|
|
|
Kyowa Hakko Kirin Co.,
Ltd. |
|
Shiseido Japan Co., Ltd. |
Kyowa Hakko Kirin Co., Ltd.
Chugai Pharmaceutical Co., Ltd. |
|
|
Collaborator:
Haruhito A.
Uchida |
|
|
|
|
|
|
Kawanishi Holdings, Inc.
MSD
Chugai Pharmaceutical Co., Ltd.
Boehringer Ingelheim |
|
|
Collaborator:
Hideki Ota |
|
|
|
|
|
|
Canon Medical Systems
Corporation |
|
|
Collaborator:
Takahiro Okazaki |
|
|
|
|
|
|
Actelion Pharmaceuticals Japan
Ltd. |
|
|
Collaborator:
Reiko Kinouchi |
|
|
|
|
|
|
Ain Pharmaciez Inc. |
|
|
Collaborator:
Ken-ei Sada |
|
|
|
Chugai Pharmaceutical Co.,
Ltd. |
|
|
|
|
|
Collaborator:
Eiichi Suematsu |
|
|
|
|
|
|
AstraZeneca |
|
|
Collaborator:
Takahiko
Sugihara |
|
|
|
Mitsubishi Tanabe Pharma
Corporation.
Chugai Pharmaceutical Co.,
Ltd. |
|
|
|
|
|
Collaborator:
Mitsuhiro Takeno |
|
|
|
Celgene |
|
|
Novartis
Eisai Co., Ltd. |
|
|
Collaborator:
Naoto Tamura |
|
|
|
AbbVie
Mitsubishi Tanabe Pharma
Corporation.
Janssen Pharmaceutical
K.K.
Bristol-Myers Squibb Co. |
|
|
Chugai Pharmaceutical Co., Ltd.
Asahi Kasei Pharma
Corporation.
Bayer Yakuhin, Ltd
MSD
Astellas Pharma Inc.
Mitsubishi Tanabe Pharma
Corporation.
Takeda Pharmaceutical Co.,
Ltd.
Actelion Pharmaceuticals Japan
Ltd.
Eisai Co., Ltd. |
|
|
Collaborator:
Yoshikazu
Nakaoka |
|
|
|
Chugai Pharmaceutical Co.,
Ltd. |
|
|
Chugai Pharmaceutical Co., Ltd.
Bayer Yakuhin, Ltd |
|
|
Collaborator:
Hajime Yoshifuji |
|
|
|
Chugai Pharmaceutical Co.,
Ltd. |
|
|
Astellas Pharma Inc. |
|
|
Member: Yoshihiro Arimura: None.
Member: Akihiro Ishizu: None.
Member: Shigeto Kobayashi: None.
Member: Yoshinori Komagata: None.
Member: Kei Takahashi: None.
Member: Kazuo Tanemoto: None.
Member: Hitoshi Hasegawa: None.
Member: Shouichi Fujimoto: None.
Member: Tatsuhiko Miyazaki: None.
Collaborator: Yoshinori Inoue: None.
Collaborator: Mitsuho Onimaru: None.
Collaborator: Tamihiro Kawakami: None.
Collaborator: Atsushi Kurata: None.
Collaborator: Hisanori Kosuge: None.
Collaborator: Kunihiro Shigematsu: None.
Collaborator: Eijun Sueyoshi: None.
Collaborator: Hitoshi Sugiyama: None.
Collaborator: Michi Tsutsumino: None.
Collaborator: Hiroaki Dobashi: None.
Collaborator: Kenji Nagasaka: None.
Collaborator: Yasuhiro Maejima: None.
Collaborator: Yoshiko Watanabe: None.
References
- 1.
Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 2013; 65: 1–11. PMID: 23045170
- 2.
Guideline for management of vasculitis syndrome (JCS 2008). Circ J 2008; 72 Suppl IV: 1253–1318. http://www.j-circ.or.jp/guideline/pdf/JCS2008_ozaki_h.pdf
- 3.
World Health Organization. WHO Handbook for Guideline Development – 2nd Edition. 2014. http://apps.who.int/medicinedocs/documents/s22083en/s22083en.pdf
- 4.
Fukui T, Yamaguchi N (ed.) Minds Handbook for the Preparation of Clinical Practice Guidelines 2014. Igaku-shoin Ltd. 2014. http://minds4.jcqhc.or.jp/minds/guideline/handbook2014.html
- 5.
Isobe M. Takayasu arteritis revisited: Current diagnosis and treatment. Int J Cardiol 2013; 168: 3–10. PMID: 23415176
- 6.
Ohigashi H, Haraguchi G, Konishi M, et al. Improved prognosis of Takayasu arteritis over the past decade: Comprehensive analysis of 106 patients. Circ J 2012; 76: 1004–1011. PMID: 22301847
- 7.
Takayasu M. Peculiar changes of the central retinal vessels. Juzenkai Journal 1908; 50: 1–4.
- 8.
Shimizu K, Sano K. Pulseless disease. J Neuropathol Clin Neurol 1951; 1: 37–47. PMID: 24538949
- 9.
Ueda H, Saito Y, Ito I, et al. Immunological studies of aortitis syndrome. Jpn Heart J 1967; 8: 4–18.
- 10.
Ito I, Saito Y, Nonaka Y. Immunological aspects of aortitis syndrome. Jpn Circ J 1975; 39: 459–462. PMID: 1121058
- 11.
Japan Intractable Diseases Information Center. Number of holders of certificates as recipients of medical benefit for specified diseases. http://www.nanbyou.or.jp/entry/1356
- 12.
Watanabe Y, Miyata T, Tanemoto K. Current clinical Features of new patients with Takayasu arteritis observed from cross-country research in Japan: Age and sex specificity. Circulation 2015; 132: 1701–1709. PMID: 26354799
- 13.
Hata A, Noda M, Moriwaki R, et al. Angiographic findings of Takayasu arteritis: New classification. Int J Cardiol 1996; 54 Suppl: S155–S163. PMID: 9119518
- 14.
Isobe M. Current diagnosis and therapy of Takayasu arteritis. J Jpn Soc Intern Med 2014; 103: 2131–2136.
- 15.
Kato Y, Terashima M, Ohigashi H, et al. Vessel wall inflammation of Takayasu arteritis detected by contrast-enhanced magnetic resonance imaging: Association with disease distribution and activity. PLoS One 2015; 10: e0145855. PMID: 26720837
- 16.
Isohisa I, Numano F, Maezawa H, et al. HLA-Bw52 in Takayasu disease. Tissue Antigens 1978; 12: 246–248. PMID: 31708
- 17.
Kimura A, Kitamura H, Date Y, et al. Comprehensive analysis of HLA genes in Takayasu arteritis in Japan. Int J Cardiol 1996; 54 Suppl: S61–S69. PMID: 9119528
- 18.
Terao C, Yoshifuji H, Kimura A, et al. Two susceptibility loci to Takayasu arteritis reveal a synergistic role of the IL12B and HLA-B regions in a Japanese population. Am J Hum Genet 2013; 93: 289–297. PMID: 23830516
- 19.
Hotchi M. Pathological studies on Takayasu arteritis. Heart Vessels Suppl 1992; 7: 11–17. PMID: 1360954
- 20.
Yutani C. Takayasu arteritis (aortitis syndrome), Volume of pathology. MHLW research project for conquering intractable diseases Study group on intractable vasculitides. Vasculitis Atlas
2005: 7–8.
- 21.
Nasu T. Takayasu’s truncoarteritis: Pulseless disease or aortitis syndrome. Acta Pathol Jpn 1982; 32 Suppl: 117–131. PMID: 6139930
- 22.
Jain D, Dietz HC, Oswald GL, et al. Causes and histopathology of ascending aortic disease in children and young adults. Cardiovasc Pathol 2011; 20: 15–25. PMID: 19926309
- 23.
Takamura C, Ohhigashi H, Ebana Y, et al. New human leukocyte antigen risk allele in Japanese patients with Takayasu arteritis. Circ J 2012; 76: 1697–1702. PMID: 22664757
- 24.
Terao C, Yoshifuji H, Ohmura K, et al. Association of Takayasu arteritis with HLA-B 67:01 and two amino acids in HLA-B protein. Rheumatology (Oxford) 2013; 52: 1769–1774. PMID: 23873822
- 25.
Hoffman GS, Ahmed AE. Surrogate markers of disease activity in patients with Takayasu arteritis: A preliminary report from The International Network for the Study of the Systemic Vasculitides (INSSYS). Int J Cardiol 1998; 66 Suppl: S191–S194; discussion S195. PMID: 9951819
- 26.
Kerr GS. Takayasu’s arteritis. Rheum Dis Clin North Am 1995; 21: 1041–1058. PMID: 8592736
- 27.
Koga T, Nishino Y, Makiyama J, et al. Serum amyloid A is a useful marker to evaluate the disease activity of Takayasu’s arteritis. Rheumatol Int 2010; 30: 561–563. PMID: 20020137
- 28.
Ishihara T, Haraguchi G, Kamiishi T, et al. Sensitive assessment of activity of Takayasu’s arteritis by pentraxin3, a new biomarker. J Am Coll Cardiol 2011; 57: 1712–1713. PMID: 21492771
- 29.
Dagna L, Salvo F, Tiraboschi M, et al. Pentraxin-3 as a marker of disease activity in Takayasu arteritis. Ann Intern Med 2011; 155: 425–433. PMID: 21969341
- 30.
Tombetti E, Di Chio MC, Sartorelli S, et al. Systemic pentraxin-3 levels reflect vascular enhancement and progression in Takayasu arteritis. Arthritis Res Ther 2014; 16: 479. PMID: 25394473
- 31.
Noris M, Daina E, Gamba S, et al. Interleukin-6 and RANTES in Takayasu arteritis: A guide for therapeutic decisions? Circulation 1999; 100: 55–60. PMID: 10393681
- 32.
Park MC, Lee SW, Park YB, et al. Serum cytokine profiles and their correlations with disease activity in Takayasu’s arteritis. Rheumatology (Oxford) 2006; 45: 545–548. PMID: 16352633
- 33.
Mahajan N, Dhawan V, Malik S, et al. Serum levels of soluble receptor for advanced glycation end products (sRAGE) in Takayasu’s arteritis. Int J Cardiol 2010; 145: 589–591. PMID: 20579752
- 34.
Noguchi S, Numano F, Gravanis MB, et al. Increased levels of soluble forms of adhesion molecules in Takayasu arteritis. Int J Cardiol 1998; 66 Suppl: S23–S33; discussion S35–S36. PMID: 9951800
- 35.
Matsuyama A, Sakai N, Ishigami M, et al. Matrix metalloproteinases as novel disease markers in Takayasu arteritis. Circulation 2003; 108: 1469–1473. PMID: 12952836
- 36.
Matsunaga N, Hayashi K, Sakamoto I, et al. Takayasu arteritis: Protean radiologic manifestations and diagnosis. Radiographics 1997; 17: 579–594. PMID: 9153698
- 37.
Zhu FP, Luo S, Wang ZJ, et al. Takayasu arteritis: Imaging spectrum at multidetector CT angiography. Br J Radiol 2012; 85: e1282–e1292. PMID: 23175494
- 38.
Yamada I, Nakagawa T, Himeno Y, et al. Takayasu arteritis: Evaluation of the thoracic aorta with CT angiography. Radiology 1998; 209: 103–109. PMID: 9769819
- 39.
Yamada I, Nakagawa T, Himeno Y, et al. Takayasu arteritis: Diagnosis with breath-hold contrast-enhanced three-dimensional MR angiography. J Magn Reson Imaging 2000; 11: 481–487. PMID: 10813857
- 40.
Li D, Lin J, Yan F. Detecting disease extent and activity of Takayasu arteritis using whole-body magnetic resonance angiography and vessel wall imaging as a 1-stop solution. J Comput Assist Tomogr 2011; 35: 468–474. PMID: 21765303
- 41.
Japan Radiological Society, Japanese College of Radiology. Guidelines for Imaging Diagnosis, 2013 edition. KANEHARA & Co., LTD. 2013.
- 42.
Mason JC. Takayasu arteritis: Advances in diagnosis and management. Nat Rev Rheumatol 2010; 6: 406–415. PMID: 20596053
- 43.
Isobe M. The Asia Pacific meeting on vasculitis and ANCA 2012 workshop on Takayasu arteritis: Advances in diagnosis and medical treatment. Clin Exp Nephrol 2013; 17: 686–689. PMID: 24068578
- 44.
Maeda H, Handa N, Matsumoto M, et al. Carotid lesions detected by B-mode ultrasonography in Takayasu’s arteritis: “Macaroni sign” as an indicator of the disease. Ultrasound Med Biol 1991; 17: 695–701. PMID: 1685816
- 45.
Soussan M, Nicolas P, Schramm C, et al. Management of large-vessel vasculitis with FDG-PET: A systematic literature review and metaanalysis. Medicine (Baltimore) 2015; 94: e622. PMID: 25860208
- 46.
Cong XL, Dai SM, Feng X, et al. Takayasu’s arteritis: Clinical features and outcomes of 125 patients in China. Clin Rheumatol 2010; 29: 973–981. PMID: 20589520
- 47.
Peter J, David S, Danda D, et al. Ocular manifestations of Takayasu arteritis: A cross-sectional study. Retina 2011; 31: 1170–1178. PMID: 21317836
- 48.
Shelhamer JH, Volkman DJ, Parrillo JE, et al. Takayasu’s arteritis and its therapy. Ann Intern Med 1985; 103: 121–126. PMID: 2860834
- 49.
Bicakcigil M, Aksu K, Kamali S, et al. Takayasu’s arteritis in Turkeyclinical and angiographic features of 248 patients. Clin Exp Rheumatol 2009; 27: S59–S64. PMID: 19646348
- 50.
Freitas DS, Camargo CZ, Mariz HA, et al. Takayasu arteritis: Assessment of response to medical therapy based on clinical activity criteria and imaging techniques. Rheumatol Int 2012; 32: 703–709. PMID: 21152919
- 50a.
Kerr GS, Hallahan CW, Giordano J, et al. Takayasu arteritis. Ann Intern Med 1994; 120: 919–929. PMID: 7909656
- 51.
Maksimowicz-McKinnon K, Clark TM, Hoffman GS. Limitations of therapy and a guarded prognosis in an American cohort of Takayasu arteritis patients. Arthritis Rheum 2007; 56: 1000–1009. PMID: 17328078
- 52.
Ozen S, Duzova A, Bakkaloglu A, et al. Takayasu arteritis in children: Preliminary experience with cyclophosphamide induction and corticosteroids followed by methotrexate. J Pediatr 2007; 150: 72–76. PMID: 17188618
- 53.
Ito I. Medical treatment of Takayasu arteritis. Heart Vessels Suppl 1992; 7: 133–137. PMID: 1360959
- 54.
Shimizu M, Ueno K, Ishikawa S, et al. Successful multitarget therapy using mizoribine and tacrolimus for refractory Takayasu arteritis. Rheumatology (Oxford) 2014; 53: 1530–1532. PMID: 24609060
- 55.
Goel R, Danda D, Mathew J, et al. Mycophenolate mofetil in Takayasu’s arteritis. Clin Rheumatol 2010; 29: 329–332. PMID: 20054700
- 56.
Guideline for Management of Vasculitis Syndrome (JCS 2008). Circ J 2008; 72 Suppl IV: 1260–1275. http://www.j-circ.or.jp/guideline/pdf/JCS2008_ozaki_h.pdf
- 57.
Valsakumar AK, Valappil UC, Jorapur V, et al. Role of immunosuppressive therapy on clinical, immunological, and angiographic outcome in active Takayasu’s arteritis. J Rheumatol 2003; 30: 1793–1798. PMID: 12913937
- 58.
Hoffman GS, Leavitt RY, Kerr GS, et al. Treatment of glucocorticoidresistant or relapsing Takayasu arteritis with methotrexate. Arthritis Rheum 1994; 37: 578–582. PMID: 7908520
- 59.
Ohigashi H, Tamura N, Ebana Y, et al. Effects of immunosuppressive and biological agents on refractory Takayasu arteritis patients unresponsive to glucocorticoid treatment. J Cardiol 2017; 69: 774–778. PMID: 27567177
- 60.
Shinjo SK, Pereira RM, Tizziani VA, et al. Mycophenolate mofetil reduces disease activity and steroid dosage in Takayasu arteritis. Clin Rheumatol 2007; 26: 1871–1875. PMID: 17332971
- 61.
Yokoe I, Haraoka H, Harashima H. A patient with Takayasu’s arteritis and rheumatoid arthritis who responded to tacrolimus hydrate. Intern Med 2007; 46: 1873–1877. PMID: 18025771
- 62.
Yamazaki H, Nanki T, Harigai M, et al. Successful treatment of refractory Takayasu arteritis with tacrolimus. J Rheumatol 2012; 39: 1487–1488. PMID: 22753804
- 63.
Kusunoki R, Ishihara S, Sato M, et al. Rare case of Takayasu’s arteritis associated with Crohn’s disease. Intern Med 2011; 50: 1581–1585. PMID: 21804285
- 64.
Horigome H, Kamoda T, Matsui A. Treatment of glucocorticoiddependent Takayasu’s arteritis with cyclosporin. Med J Aust 1999; 170: 566. PMID: 10397054
- 65.
Nishimoto N, Nakahara H, Yoshio-Hoshino N, et al. Successful treatment of a patient with Takayasu arteritis using a humanized antiinterleukin-6 receptor antibody. Arthritis Rheum 2008; 58: 1197–1200. PMID: 18383395
- 66.
Abisror N, Mekinian A, Lavigne C, et al. Club Rhumatismes et Inflammation, and SNFMI. Tocilizumab in refractory Takayasu arteritis: A case series and updated literature review. Autoimmun Rev 2013; 12: 1143–1149. PMID: 23820042
- 67.
Salvarani C, Magnani L, Catanoso M, et al. Tocilizumab: A novel therapy for patients with large-vessel vasculitis. Rheumatology (Oxford) 2012; 51: 151–156. PMID: 22075063
- 68.
Seitz M, Reichenbach S, Bonel HM, et al. Rapid induction of remission in large vessel vasculitis by IL-6 blockade: A case series. Swiss Med Wkly 2011; 141: w13156. PMID: 21243547
- 69.
Unizony S, Arias-Urdaneta L, Miloslavsky E, et al. Tocilizumab for the treatment of large-vessel vasculitis (giant cell arteritis, Takayasu arteritis) and polymyalgia rheumatica. Arthritis Care Res (Hoboken) 2012; 64: 1720–1729. PMID: 22674883
- 70.
Nakaoka Y, Higuchi K, Arita Y, et al. Tocilizumab for the treatment of patients with refractory Takayasu arteritis. Int Heart J 2013; 54: 405–411. PMID: 24309452
- 71.
Mekinian A, Comarmond C, Resche-Rigon M, et al. French Takayasu Network. Efficacy of biological-targeted treatments in Takayasu arteritis: Multicenter, retrospective study of 49 patients. Circulation 2015; 132: 1693–1700. PMID: 26354797
- 72.
Nakaoka Y, Isobe M, Takei S, et al. Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: Results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study). Ann Rheum Dis 2018; 77: 348–354. PMID: 29191819
- 73.
Hoffman GS, Merkel PA, Brasington RD, et al. Anti-tumor necrosis factor therapy in patients with difficult to treat Takayasu arteritis. Arthritis Rheum 2004; 50: 2296–2304. PMID: 15248230
- 74.
Schmidt J, Kermani TA, Bacani AK, et al. Tumor necrosis factor inhibitors in patients with Takayasu arteritis: Experience from a referral center with long-term followup. Arthritis Care Res (Hoboken) 2012; 64: 1079–1083. PMID: 22328491
- 75.
de Souza AW, Machado NP, Pereira VM, et al. Antiplatelet therapy for the prevention of arterial ischemic events in takayasu arteritis. Circ J 2010; 74: 1236–1241. PMID: 20467149
- 76.
Fields CE, Bower TC, Cooper LT, et al. Takayasu’s arteritis: Operative results and influence of disease activity. J Vasc Surg 2006; 43: 64–71. PMID: 16414389
- 77.
Min PK, Park S, Jung JH, et al. Endovascular therapy combined with immunosuppressive treatment for occlusive arterial disease in patients with Takayasu’s arteritis. J Endovasc Ther 2005; 12: 28–34. PMID: 15683269
- 78.
Park MC, Lee SW, Park YB, et al. Post-interventional immunosuppressive treatment and vascular restenosis in Takayasu’s arteritis. Rheumatology (Oxford) 2006; 45: 600–605. PMID: 16352637
- 79.
Perera AH, Youngstein T, Gibbs RG, et al. Optimizing the outcome of vascular intervention for Takayasu arteritis. Br J Surg 2014; 101: 43–50. PMID: 24375298
- 80.
Saadoun D, Lambert M, Mirault T, et al. Retrospective analysis of surgery versus endovascular intervention in Takayasu arteritis: A multicenter experience. Circulation 2012; 125: 813–819. PMID: 22230484
- 81.
Ueno A, Awane Y, Wakabayashi A, et al. Successfully operated obliterative brachiocephalic arteritis (Takayasu) associated with the elongated coarctation. Jpn Heart J 1967; 8: 538–544. PMID: 5299711
- 82.
Uyama M, Asayama K. Retinal Vascular Changes in Takayasu’s Disease (Pulseless Disease), Occurrence and Evolution of the Lesion.
In: Doc Ophthalmol Proc Ser 9. 1976: 549–554.
- 83.
Tada Y, Sato O, Ohshima A, et al. Surgical treatment of Takayasu arteritis. Heart Vessels Suppl 1992; 7: 159–167. PMID: 1360963
- 84.
Pajari R, Hekali P, Harjola PT. Treatment of Takayasu’s arteritis: An analysis of 29 operated patients. Thorac Cardiovasc Surg 1986; 34: 176–181. PMID: 2426832
- 85.
Wu X, Duan HY, Gu YQ, et al. Surgical treatment of brachiocephalic vessel involvement in Takayasu’s arteritis. Chin Med J (Engl) 2010; 123: 1122–1126. PMID: 20529549
- 86.
Kumar A, Dubey D, Bansal P, et al. Surgical and radiological management of renovascular hypertension in a developing country. J Urol 2003; 170: 727–730. PMID: 12913683
- 87.
Miyata T, Sato O, Koyama H, et al. Long-term survival after surgical treatment of patients with Takayasu’s arteritis. Circulation 2003; 108: 1474–1480. PMID: 12952846
- 88.
Kim YW, Kim DI, Park YJ, et al. Surgical bypass vs endovascular treatment for patients with supra-aortic arterial occlusive disease due to Takayasu arteritis. J Vasc Surg 2012; 55: 693–700. PMID: 22119246
- 89.
Mason JC. Takayasu arteritis: Surgical interventions. Curr Opin Rheumatol 2015; 27: 45–52. PMID: 25405823
- 90.
Lee GY, Jeon P, Do YS, et al. Comparison of outcomes between endovascular treatment and bypass surgery in Takayasu arteritis. Scand J Rheumatol 2014; 43: 153–161. PMID: 24134435
- 91.
Vahanian A, Alfieri O, Andreotti F, et al. Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC): Guidelines on the management of valvular heart disease (version 2012). Eur Heart J 2012; 33: 2451–2496. PMID: 22922415
- 92.
Nishimura RA, Otto CM, Bonow RO, et al. 2014 AHA/ACC guideline for the management of patients with valvular heart disease: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Thorac Cardiovasc Surg 2014; 148: e1–e132. PMID: 24939033
- 93.
Enomoto S, Miyamoto T, Shimada I, et al. Surgical therapy for aortic regurgitation due to aortitis syndrome: Effectiveness and extended indication of Bentall’s operation. Jpn J Thorac and Cardiovasc Surg 1992; 40: 500–505.
- 94.
Ando M, Kosakai Y, Okita Y, et al. Surgical treatment for aortic regurgitation caused by Takayasu’s arteritis. J Card Surg 1998; 13: 202–207. PMID: 10193991
- 95.
Ando M, Okita Y, Sasako Y, et al. Evaluation of the results of surgical treatment for dilative lesions associated with Takayasu’s arteritis. Int J Angiol 2000; 9: 194–197.
- 96.
Ogino H, Matsuda H, Minatoya K, et al. Overview of late outcome of medical and surgical treatment for Takayasu arteritis. Circulation 2008; 118: 2738–2747. PMID: 19106398
- 97.
Matsuura K, Ogino H, Kobayashi J, et al. Surgical treatment of aortic regurgitation due to Takayasu arteritis: Long-term morbidity and mortality. Circulation 2005; 112: 3707–3712. PMID: 16330687
- 98.
Fujiwara T, Masaki H, Yamane H, et al. Coronary ostial endarterectomy in Takayasu’s aortitis: Confirmation of patency nine years postsurgically. Jpn Circ J 1992; 56: 556–559. PMID: 1352558
- 99.
Dong H, Jiang X, Peng M, et al. Percutaneous transluminal angioplasty for symptomatic pulmonary stenosis in Takayasu arteritis. J Rheumatol 2014; 41: 1856–1862. PMID: 25128508
- 100.
Ishikawa K, Maetani S. Long-term outcome for 120 Japanese patients with Takayasu’s disease: Clinical and statistical analyses of related prognostic factors. Circulation 1994; 90: 1855–1860. PMID: 7923672
- 101.
Schmidt J, Kermani TA, Bacani AK, et al. Diagnostic features, treatment, and outcomes of Takayasu arteritis in a US cohort of 126 patients. Mayo Clin Proc 2013; 88: 822–830. PMID: 23849994
- 102.
Maeda M, Kobayashi M, Okamoto S, et al. Aortitis syndrome in children: Clinical observation of 35 cases in Japan. Acta Paediatr Jpn 1997; 39: 280–284. PMID: 9141273
- 103.
Nakano N. Study of intractable pathology and treatment of vasculitis syndrome in children (Takayasu arteritis). Health and Labour Sciences Research Grant (Research Project for the Prevention and Treatment of Immunological and Allergic Diseases) Study of the Diagnosis and Treatment of Intractable Pathology of Rheumatism/Collagen Disease in Childhood. Report in 2010.
- 104.
Information Center for Chronic Diseases, Japan. https://www.shouman.jp/
- 105.
Batu ED, Sönmez HE, Hazırolan T, et al. Tocilizumab treatment in childhood Takayasu arteritis: Case series of four patients and systematic review of the literature. Semin Arthritis Rheum 2017; 46: 529–535. PMID: 27515155
- 106.
Kobayashi S, Yano T, Matsumoto Y, et al. Clinical and epidemiologic analysis of giant cell (temporal) arteritis from a nationwide survey in 1998 in Japan: The first government-supported nationwide survey. Arthritis Rheum 2003; 49: 594–598. PMID: 12910568
- 107.
Carmona FD, González-Gay MA, Martín J. Genetic component of giant cell arteritis. Rheumatology (Oxford) 2014; 53: 6–18. PMID: 23843109
- 108.
Carmona FD, Mackie SL, Martín JE, et al. Spanish GCA Group. A large-scale genetic analysis reveals a strong contribution of the HLA class II region to giant cell arteritis susceptibility. Am J Hum Genet 2015; 96: 565–580. PMID: 25817017
- 109.
Elling P, Olsson AT, Elling H. Synchronous variations of the incidence of temporal arteritis and polymyalgia rheumatica in different regions of Denmark; association with epidemics of Mycoplasma pneumoniae infection. J Rheumatol 1996; 23: 112–119. PMID: 8838518
- 110.
Health and Labour Sciences Research Grant – Policy Research Project on Intractable Diseases Study Group on Intractable Vasculitis. Vasculitis Pathology Atlas, web edition. http://www.vas-mhlw.org/html/pathology/index.html
- 111.
Singh AG, Kermani TA, Crowson CS, et al. Visual manifestations in giant cell arteritis: Trend over 5 decades in a population-based cohort. J Rheumatol 2015; 42: 309–315. PMID: 25512481
- 112.
Muratore F, Kermani TA, Crowson CS, et al. Large-vessel giant cell arteritis: A cohort study. Rheumatology (Oxford) 2015; 54: 463–470. PMID: 25193809
- 113.
Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant-cell arteritis. Lancet 2008; 372: 234–245. PMID: 18640460
- 114.
Mazlumzadeh M, Hunder GG, Easley KA, et al. Treatment of giant cell arteritis using induction therapy with high-dose glucocorticoids: A double-blind, placebo-controlled, randomized prospective clinical trial. Arthritis Rheum 2006; 54: 3310–3318. PMID: 17009270
- 115.
Seror R, Baron G, Hachulla E, et al. Adalimumab for steroid sparing in patients with giant-cell arteritis: Results of a multicentre randomized controlled trial. Ann Rheum Dis 2014; 73: 2074–2081. PMID: 23897775
- 116.
Mahr AD, Jover JA, Spiera RF, et al. Adjunctive methotrexate for treatment of giant cell arteritis: An individual patient data meta-analysis. Arthritis Rheum 2007; 56: 2789–2797. PMID: 17665429
- 117.
Weyand CM, Fulbright JW, Hunder GG, et al. Treatment of giant cell arteritis: Interleukin-6 as a biologic marker of disease activity. Arthritis Rheum 2000; 43: 1041–1048. PMID: 10817557
- 118.
Roche NE, Fulbright JW, Wagner AD, et al. Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis. Arthritis Rheum 1993; 36: 1286–1294. PMID: 8216422
- 119.
Blockmans D. Diagnosis and extension of giant cell arteritis. Contribution of imaging techniques. Presse Med 2012; 41: 948–954. PMID: 22795837
- 120.
Schmidt WA, Kraft HE, Vorpahl K, et al. Color duplex ultrasonography in the diagnosis of temporal arteritis. N Engl J Med 1997; 337: 1336–1342. PMID: 9358127
- 121.
Salvarani C, Silingardi M, Ghirarduzzi A, et al. Is duplex ultrasonography useful for the diagnosis of giant-cell arteritis? Ann Intern Med 2002; 137: 232–238. PMID: 12186513
- 122.
Nordborg E, Nordborg C. Giant cell arteritis: Strategies in diagnosis and treatment. Curr Opin Rheumatol 2004; 16: 25–30. PMID: 14673385
- 123.
Liu GT, Glaser JS, Schatz NJ, et al. Visual morbidity in giant cell arteritis: Clinical characteristics and prognosis for vision. Ophthalmology 1994; 101: 1779–1785. PMID: 7800356
- 124.
Liozon E, Boutros-Toni F, Ly K, et al. Silent, or masked, giant cell arteritis is associated with a strong inflammatory response and a benign short term course. J Rheumatol 2003; 30: 1272–1276. PMID: 12784402
- 125.
Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol 1998; 125: 509–520. PMID: 9559737
- 126.
Tanano I, Kinouchi R, Ohya S, et al. Gradual visual loss after visual recovery by pulsed corticosteroid therapy in a patient with biopsy-proven giant cell arteritis. Ophthalmol 2011; 53: 1769–1773.
- 127.
Hamidou MA, Batard E, Trewick D, et al. Silent versus cranial giant cell arteritis. Initial presentation and outcome of 50 biopsy-proven cases. Eur J Intern Med 2005; 16: 183–186. PMID: 15967333
- 128.
Cid MC, Font C, Oristrell J, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum 1998; 41: 26–32. PMID: 9433866
- 129.
Hunder GG, Bloch DA, Michel BA, et al. The American College of Rheumatology 1990 criteria for the classification of giant cell arteritis. Arthritis Rheum 1990; 33: 1122–1128. PMID: 2202311
- 130.
Karassa FB, Matsagas MI, Schmidt WA, et al. Meta-analysis: Test performance of ultrasonography for giant-cell arteritis. Ann Intern Med 2005; 142: 359–369. PMID: 15738455
- 131.
Nesher G, Shemesh D, Mates M, et al. The predictive value of the halo sign in color Doppler ultrasonography of the temporal arteries for diagnosing giant cell arteritis. J Rheumatol 2002; 29: 1224–1226. PMID: 12064840
- 132.
Smetana GW, Shmerling RH. Does this patient have temporal arteritis? JAMA 2002; 287: 92–101. PMID: 11754714
- 133.
Muratore F, Cavazza A, Boiardi L, et al. Histopathologic findings of patients with biopsy-negative giant cell arteritis compared to those without arteritis: A population-based study. Arthritis Care Res (Hoboken) 2016; 68: 865–870. PMID: 26415044
- 134.
Breuer GS, Nesher R, Nesher G. Negative temporal artery biopsies: Eventual diagnoses and features of patients with biopsy-negative giant cell arteritis compared to patients without arteritis. Clin Exp Rheumatol 2008; 26: 1103–1106. PMID: 19210879
- 135.
Esteban MJ, Font C, Hernández-Rodríguez J, et al. Small-vessel vasculitis surrounding a spared temporal artery: Clinical and pathological findings in a series of twenty-eight patients. Arthritis Rheum 2001; 44: 1387–1395. PMID: 11407699
- 136.
Généreau T, Lortholary O, Pottier MA, et al. French Vasculitis Study Group. Temporal artery biopsy: A diagnostic tool for systemic necrotizing vasculitis. Arthritis Rheum 1999; 42: 2674–2681. PMID: 10616017
- 137.
Salvarani C, Gabriel SE, Gertz MA, et al. Primary systemic amyloidosis presenting as giant cell arteritis and polymyalgia rheumatica. Arthritis Rheum 1994; 37: 1621–1626. PMID: 7980674
- 138.
Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: Frequency of occurence in a population-based study. Arthritis Rheum 2001; 45: 140–145. PMID: 11324777
- 139.
Salvarani C, Cantini F, Boiardi L, et al. Laboratory investigations useful in giant cell arteritis and Takayasu’s arteritis. Clin Exp Rheumatol 2003; 21 Suppl: S23–S28. PMID: 14740424
- 140.
Mukhtyar C, Guillevin L, Cid MC, et al. European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis. Ann Rheum Dis 2009; 68: 318–323. PMID: 18413441
- 141.
Achkar AA, Lie JT, Hunder GG, et al. How does previous corticosteroid treatment affect the biopsy findings in giant cell (temporal) arteritis? Ann Intern Med 1994; 120: 987–992. PMID: 8185147
- 142.
Brack A, Martinez-Taboada V, Stanson A, et al. Disease pattern in cranial and large-vessel giant cell arteritis. Arthritis Rheum 1999; 42: 311–317. PMID: 10025926
- 143.
Kermani TA, Warrington KJ, Crowson CS, et al. Large-vessel involvement in giant cell arteritis: A population-based cohort study of the incidence-trends and prognosis. Ann Rheum Dis 2013; 72: 1989–1994. PMID: 23253927
- 144.
Nuenninghoff DM, Hunder GG, Christianson TJ, et al. Incidence and predictors of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: A population-based study over 50 years. Arthritis Rheum 2003; 48: 3522–3531. PMID: 14674004
- 145.
Robson JC, Kiran A, Maskell J, et al. The relative risk of aortic aneurysm in patients with giant cell arteritis compared with the general population of the UK. Ann Rheum Dis 2015; 74: 129–135. PMID: 24095936
- 146.
Nuenninghoff DM, Hunder GG, Christianson TJ, et al. Mortality of large-artery complication (aortic aneurysm, aortic dissection, and/or large-artery stenosis) in patients with giant cell arteritis: A population-based study over 50 years. Arthritis Rheum 2003; 48: 3532–3537. PMID: 14674005
- 147.
Andersson R, Malmvall BE, Bengtsson BA. Long-term corticosteroid treatment in giant cell arteritis. Acta Med Scand 1986; 220: 465–469. PMID: 3812030
- 148.
Proven A, Gabriel SE, Orces C, et al. Glucocorticoid therapy in giant cell arteritis: Duration and adverse outcomes. Arthritis Rheum 2003; 49: 703–708. PMID: 14558057
- 149.
Yates M, Loke YK, Watts RA, et al. Prednisolone combined with adjunctive immunosuppression is not superior to prednisolone alone in terms of efficacy and safety in giant cell arteritis: Meta-analysis. Clin Rheumatol 2014; 33: 227–236. PMID: 24026674
- 150.
Henes JC, Mueller M, Pfannenberg C, et al. Cyclophosphamide for large vessel vasculitis: Assessment of response by PET/CT. Clin Exp Rheumatol 2011; 29 Suppl: S43–S48. PMID: 21385544
- 151.
Loock J, Henes J, Kötter I, et al. Treatment of refractory giant cell arteritis with cyclophosphamide: A retrospective analysis of 35 patients from three centres. Clin Exp Rheumatol 2012; 30 Suppl: S70–S76. PMID: 22640650
- 152.
de Boysson H, Boutemy J, Creveuil C, et al. Is there a place for cyclophosphamide in the treatment of giant-cell arteritis? A case series and systematic review. Semin Arthritis Rheum 2013; 43: 105–112. PMID: 23453684
- 153.
Quartuccio L, Maset M, De Maglio G, et al. Role of oral cyclophosphamide in the treatment of giant cell arteritis. Rheumatology (Oxford) 2012; 51: 1677–1686. PMID: 22627726
- 154.
De Silva M, Hazleman BL. Azathioprine in giant cell arteritis/polymyalgia rheumatica: A double-blind study. Ann Rheum Dis 1986; 45: 136–138. PMID: 3511861
- 155.
Boureau AS, de Faucal P, Espitia O, et al. Place of azathioprine in the treatment of giant cell arteritis. [Article in French] Rev Med Interne 2016; 37: 723–729. PMID: 27260788
- 156.
Hoffman GS, Cid MC, Rendt-Zagar KE, et al. Infliximab-GCA Study Group. Infliximab for maintenance of glucocorticosteroid-induced remission of giant cell arteritis: A randomized trial. Ann Intern Med 2007; 146: 621–630. PMID: 17470830
- 157.
Villiger PM, Adler S, Kuchen S, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387: 1921–1927. PMID: 26952547
- 158.
Unizony SH, Dasgupta B, Fisheleva E, et al. Design of the tocilizumab in giant cell arteritis trial. Int J Rheumatol 2013; 2013: 912562. PMID: 23653652
- 159.
Pazzola G, Padovano I, Boiardi L, et al. Tocilizumab in glucocorticoid-naïve large-vessel vasculitis. Clin Exp Rheumatol 2013; 31 Suppl: S59–S61. PMID: 23306184
- 160.
Loricera J, Blanco R, Hernández JL, et al. Tocilizumab in giant cell arteritis: Multicenter open-label study of 22 patients. Semin Arthritis Rheum 2015; 44: 717–723. PMID: 25697557
- 161.
Salvarani C, Giannini C, Miller DV, et al. Giant cell arteritis: Involvement of intracranial arteries. Arthritis Rheum 2006; 55: 985–989. PMID: 17139651
- 162.
Lee MS, Smith SD, Galor A, et al. Antiplatelet and anticoagulant therapy in patients with giant cell arteritis. Arthritis Rheum 2006; 54: 3306–3309. PMID: 17009265
- 163.
Nesher G, Berkun Y, Mates M, et al. Low-dose aspirin and prevention of cranial ischemic complications in giant cell arteritis. Arthritis Rheum 2004; 50: 1332–1337. PMID: 15077317
- 164.
Nesher G, Berkun Y, Mates M, et al. Risk factors for cranial ischemic complications in giant cell arteritis. Medicine (Baltimore) 2004; 83: 114–122. PMID: 15028965
- 165.
Gonzalez-Gay MA, Garcia-Porrua C, Piñeiro A, et al. Aortic aneurysm and dissection in patients with biopsy-proven giant cell arteritis from northwestern Spain: A population-based study. Medicine (Baltimore) 2004; 83: 335–341. PMID: 15525845
- 166.
Dehaine-Bamberger N, Amar R, Touboul C, et al. Buerger disease, clinical and prognostic aspects: 83 cases. [Article in French] Presse Med 1993; 22: 945–948. PMID: 8367418
- 167.
Mills JL, Taylor LM, Porter JM. Buerger’s disease in the modern era. Am J Surg 1987; 154: 123–129. PMID: 3605510
- 168.
Lie JT. Thromboangiitis obliterans (Buerger’s disease) in women. Medicine (Baltimore) 1987; 66: 65–72. PMID: 3492659
- 169.
Olin JW, Young JR, Graor RA, et al. The changing clinical spectrum of thromboangiitis obliterans (Buerger’s disease). Circulation 1990; 82 Suppl: IV3–IV8. PMID: 2225420
- 170.
Shionoya S, Ban I, Nakata Y, et al. Diagnosis, pathology, and treatment of Buerger’s disease. Surgery 1974; 75: 695–700. PMID: 4824428
- 171.
Adar R, Papa MZ, Halpern Z, et al. Cellular sensitivity to collagen in thromboangiitis obliterans. N Engl J Med 1983; 308: 1113–1116. PMID: 6835334
- 172.
Buerger L. Thromboangiitis obliterans: A study of the vascular lesions leading to presenile spontaneous gangrene. Am J Med Sci 1908; 136: 567–580.
- 173.
Gulati SM, Madhra K, Thusoo TK, et al. Autoantibodies in thromboangiitis obliterans (Buerger’s disease). Angiology 1982; 33: 642–651. PMID: 6751163
- 174.
Roncon de Albuquerque R, Delgado L, Correia P, et al. Circulating immune complexes in Buerger’s disease. Endarteritis obliterans in young men. J Cardiovasc Surg (Torino) 1989; 30: 821–825. PMID: 2808505
- 175.
Majewski W, Marszalek A, Staniszewiski R, et al. Clinical and morphological aspects of Buerger’s disease. Int Angiol 1997; 16: 239–244. PMID: 9543220
- 176.
Eichhorn J, Sima D, Lindschau C, et al. Antiendothelial cell antibodies in thromboangiitis obliterans. Am J Med Sci 1998; 315: 17–23. PMID: 9427570
- 177.
Shionoya S, Leu HJ, Lie JT. Buerger’s disease (thromboangiitis obliterans).
In: Stehbens WE, Lie JT, editors. Vascular pathology. Chapman & Hall Medical, 1995: 657–678.
- 178.
Juergens JL. Thromboangiitis obliterans (Buerger’s disease, TAO).
In: Juergens JL, Spittel JA, Fairbairn JF, editors. Peripheral vascular diseases. WB Saunders, 1980: 469–491.
- 179.
Allen EV, Brown GE. Thromboangiitis obliterans. A clinical study of 200 cases. Ann Intern Med 1928; 1: 535–549.
- 180.
Iwai T, Inoue Y, Umeda M, et al. Oral bacteria in the occluded arteries of patients with Buerger disease. J Vasc Surg 2005; 42: 107–115. PMID: 16012459
- 181.
Iwase S, Okamoto T, Mano T, et al. Skin sympathetic outflow in Buerger’s disease. Auton Neurosci 2001; 87: 286–292. PMID: 11476291
- 182.
Roncon-Albuquerque R, Serrão P, Vale-Pereira R, et al. Plasma catecholamines in Buerger’s disease: Effects of cigarette smoking and surgical sympathectomy. Eur J Vasc Endovasc Surg 2002; 24: 338–343. PMID: 12323177
- 183.
Halacheva K, Gulubova MV, Manolova I, et al. Expression of ICAM-1, VCAM-1, E-selectin and TNF-alpha on the endothelium of femoral and iliac arteries in thromboangiitis obliterans. Acta Histochem 2002; 104: 177–184. PMID: 12086338
- 184.
Pietraszek MH, Choudhury NA, Baba S, et al. Serotonin as a factor involved in pathophysiology of thromboangiitis obliterans. Int Angiol 1993; 12: 9–12. PMID: 8376917
- 185.
Shionoya S. What is Buerger’s disease? World J Surg 1983; 7: 544–551. PMID: 6624130
- 186.
Kim EJ, Cho BS, Lee TS, et al. Morphologic change of the internal elastic lamina in Buerger’s disease. J Korean Med Sci 2000; 15: 44–48. PMID: 10719807
- 187.
Kurata A, Franke FE, Machinami R, et al. Thromboangiitis obliterans: Classic and new morphological features. Virchows Arch 2000; 436: 59–67. PMID: 10664163
- 188.
Kurata A, Machinami R, Schulz A, et al. Different immunophenotypes in Buerger’s disease. Pathol Int 2003; 53: 608–615. PMID: 14507318
- 189.
Shionoya S. Clinical manifestation in Buerger’s disease. Shionoya S. Buerger’s disease-Pathology, Diagnosis and Treatment. The University of Nagoya Press 1990: 101–117.
- 190.
Kobayashi M, Nishikimi N, Komori K. Current pathological and clinical aspects of Buerger’s disease in Japan. Ann Vasc Surg 2006; 20: 148–156. PMID: 16378138
- 191.
No YJ, Lee EM, Lee DH, et al. Cerebral angiographic findings in thromboangiitis obliterans. Neuroradiology 2005; 47: 912–915. PMID: 16133479
- 192.
Becit N, Unlü Y, Koçak H, et al. Involvement of the coronary artery in a patient with thromboangiitis obliterans. A case report. Heart Vessels 2002; 16: 201–203. PMID: 12181595
- 193.
Donatelli F, Triggiani M, Nascimbene S, et al. Thromboangiitis obliterans of coronary and internal thoracic arteries in a young woman. J Thorac Cardiovasc Surg 1997; 113: 800–802. PMID: 9104994
- 194.
Goktas S, Bedir S, Bozlar U, et al. Intrarenal arterial stenosis in a patient with thromboangiitis obliterans. Int J Urol 2006; 13: 1243–1244. PMID: 16984562
- 195.
Kurata A, Nonaka T, Arimura Y, et al. Multiple ulcers with perforation of the small intestine in buerger’s disease: A case report. Gastroenterology 2003; 125: 911–916. PMID: 12949735
- 196.
Lee KS, Paik CN, Chung WC, et al. Colon ischemia associated with Buerger’s disease: Case report and review of the literature. Gut Liver 2010; 4: 287–291. PMID: 20559539
- 197.
Sasaki S, Sakuma M, Kunihara T, et al. Distribution of arterial involvement in thromboangiitis obliterans (Buerger’s disease): Results of a study conducted by the Intractable Vasculitis Syndromes Research Group in Japan. Surg Today 2000; 30: 600–605. PMID: 10930225
- 198.
Sakaguchi S, Mishima Y. Report of Angiographic Findings. Buerger Disease. Ministry of Health and Welfare Specific Diseases Systematic Study Group on Vascular Lesions Subcommittee Report 1976. 1977: 1–38.
- 199.
Shionoya S, Hirai M, Kawai S, et al. Pattern of arterial occlusion in Buerger’s disease. Angiology 1982; 33: 375–384. PMID: 7091768
- 200.
Shionoya S. Buerger’s disease-Pathology, Diagnosis and Treatment. The University of Nagoya Press 1990: 118–150.
- 201.
Sakaguchi S, Baba S, Kameda T, et al. Peripheral Circulation Disorders in Collagen Disease — Arteriography Findings. Ministry of Health and Welfare Specific Diseases Systematic Study Group on Vascular Lesions Subcommittee Report 1976. 1977: 295–302.
- 202.
Mishima Y, Kobayashi H, Ohashi S, et al. Disorders of Major Arteries of the Limbs Associated with Collagen Disease — Arteriography Findings. Ministry of Health and Welfare Specific Diseases Systematic Study Group on Vascular Lesions Subcommittee Report 1976. 1977: 133–136.
- 203.
Olin JW, Shih A. Thromboangiitis obliterans (Buerger’s disease). Curr Opin Rheumatol 2006; 18: 18–24. PMID: 16344615
- 204.
Mills JL Sr. Buerger’s disease in the 21st century: Diagnosis, clinical features, and therapy. Semin Vasc Surg 2003; 16: 179–189. PMID: 12975757
- 205.
Papa MZ, Rabi I, Adar R. A point scoring system for the clinical diagnosis of Buerger’s disease. Eur J Vasc Endovasc Surg 1996; 11: 335–339. PMID: 8601245
- 206.
Shionoya S. Buerger’s disease: Diagnosis and management. Cardiovasc Surg 1993; 1: 207–214. PMID: 8076031
- 207.
Nakajima N. Buerger’s disease. Health science research project for the control of intractable diseases Study group on intractable vasculitis: Clinical practice manual for intractable vasculitis. 2002: 9–12.
- 208.
Shigematsu K, Shigematsu H, Yasuda K. Study on intractable vasculitis. Long term prognosis of Buerger disease. Research with (nationwide questionnaire survey results). 2003 Summary and Report of Assigned Studies. 2004: 115–119.
- 209.
Japanese College of Angiology (ed.) Inter-society consensus for the management of PAD (TASC II). Medical Tribune, Inc. 2007: 44–45.
- 210.
Saito Y, Sasaki K, Katsuda Y, et al. Effect of autologous bone-marrow cell transplantation on ischemic ulcer in patients with Buerger’s disease. Circ J 2007; 71: 1187–1192. PMID: 17652879
- 211.
Matoba S, Tatsumi T, Murohara T, et al. TACT Follow-up Study Investigators. Long-term clinical outcome after intramuscular implantation of bone marrow mononuclear cells (Therapeutic Angiogenesis by Cell Transplantation [TACT] trial) in patients with chronic limb ischemia. Am Heart J 2008; 156: 1010–1018. PMID: 19061721
- 212.
Inaba S, Egashira K, Komori K. Peripheral-blood or bone-marrow mononuclear cells for therapeutic angiogenesis? Lancet 2002; 360: 2083. PMID: 12504445
- 213.
Shigematsu H, Shigematsu K. Factors affecting the long-term outcome of Buerger’s disease (thromboangiitis obliterans). Int Angiol 1999; 18: 58–64. PMID: 10392482
- 214.
Sugimoto M, Miyachi H, Morimae H, et al. Fate of ischemic limbs in patients with Buerger’s disease based on our 30-year experience: Does smoking have a definitive impact on the late loss of limbs? Surg Today 2015; 45: 466–470. PMID: 24845736
- 215.
Nakabayashi K. A subcommittee report on epidemiology, prognosis and QOL of medium and small angiitis. Ministry of Health and Welfare Specific Diseases Immunological Diseases Research Group Intractable Vasculitis Subcommittee. 1998 Research Report 1999: 38–48.
- 216.
Arkin A. A clinical and pathological study of periarteritis nodosa: A report of five cases, one histologically healed. Am J Pathol 1930; 6: 401–426.5. PMID: 19969916
- 217.
Pagnoux C, Seror R, Henegar C, et al. Clinical features and outcomes in 348 patients with polyarteritis nodosa: A systematic retrospective study of patients diagnosed between 1963 and 2005 and entered into the French Vasculitis Study Group Database. Arthritis Rheum 2010; 62: 616–626. PMID: 20112401
- 218.
Lindberg K. Ein Beitrag zur Kenntnis der periarteriitis nodosa. Acta Med Scand 1931; 76: 183–225.
- 219.
MHLW health and labour sciences research grant Research project for conquering intractable diseases Study on intractable vasculitides. 2005 Summary and report of assigned studies.
- 220.
Guillevin L, Cohen P, Mahr A, et al. Treatment of polyarteritis nodosa and microscopic polyangiitis with poor prognosis factors: A prospective trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in sixty-five patients. Arthritis Rheum 2003; 49: 93–100. PMID: 12579599
- 221.
Guillevin L, Lhote F, Cohen P, et al. Corticosteroids plus pulse cyclophosphamide and plasma exchanges versus corticosteroids plus pulse cyclophosphamide alone in the treatment of polyarteritis nodosa and Churg-Strauss syndrome patients with factors predicting poor prognosis. A prospective, randomized trial in sixty-two patients. Arthritis Rheum 1995; 38: 1638–1645. PMID: 7488285
- 222.
Ribi C, Cohen P, Pagnoux C, et al. French Vasculitis Study Group. Treatment of polyarteritis nodosa and microscopic polyangiitis without poor-prognosis factors: A prospective randomized study of one hundred twenty-four patients. Arthritis Rheum 2010; 62: 1186–1197. PMID: 20131268
- 223.
Phillip R, Luqmani R. Mortality in systemic vasculitis: A systematic review. Clin Exp Rheumatol 2008; 26 Suppl: S94–S104. PMID: 19026150
- 224.
Samson M, Puéchal X, Devilliers H, et al. French Vasculitis Study Group (FVSG). Long-term follow-up of a randomized trial on 118 patients with polyarteritis nodosa or microscopic polyangiitis without poor-prognosis factors. Autoimmun Rev 2014; 13: 197–205. PMID: 24161361
- 225.
Guillevin L, Lhote F, Gayraud M, et al. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine (Baltimore) 1996; 75: 17–28. PMID: 8569467
- 226.
Guillevin L, Pagnoux C, Seror R, et al. The Five-Factor Score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine (Baltimore) 2011; 90: 19–27. PMID: 21200183
- 227.
Sada KE, Yamamura M, Harigai M, et al. Research Committee on Intractable Vasculitides, the MHLW of Japan. Classification and characteristics of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis in a nationwide, prospective, inception cohort study. Arthritis Res Ther 2014; 16: R101. PMID: 24758294
- 228.
Fujimoto S, Watts RA, Kobayashi S, et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology (Oxford) 2011; 50: 1916–1920. PMID: 21798892
- 229.
Reinhold-Keller E, Herlyn K, Wagner-Bastmeyer R, et al. Stable incidence of primary systemic vasculitides over five years: Results from the German vasculitis register. Arthritis Rheum 2005; 53: 93–99. PMID: 15696553
- 230.
Tsuchiya N, Kobayashi S, Kawasaki A, et al. Genetic background of Japanese patients with antineutrophil cytoplasmic antibody-associated vasculitis: Association of HLA-DRB1*0901 with microscopic polyangiitis. J Rheumatol 2003; 30: 1534–1540. PMID: 12858454
- 231.
Lyons PA, Rayner TF, Trivedi S, et al. Genetically distinct subsets within ANCA-associated vasculitis. N Engl J Med 2012; 367: 214–223. PMID: 22808956
- 232.
Kessenbrock K, Krumbholz M, Schönermarck U, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 2009; 15: 623–625. PMID: 19448636
- 233.
Kawashima S, Arimura Y, Sano K, et al. Immunopathologic co-localization of MPO, IgG, and C3 in glomeruli in human MPO-ANCAassociated glomerulonephritis. Clin Nephrol 2013; 79: 292–301. PMID: 23537682
- 234.
Nakazawa D, Tomaru U, Suzuki A, et al. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: Implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2012; 64: 3779–3787. PMID: 22777766
- 235.
Supervising editor: Matsuno S, editor: Arimura Y. Guidelines for management of rapidly progressive nephritic syndrome Q&A Revised 2nd edition. Shindan To Chiryo Sha, Inc. 2015: 7.
- 236.
Yoshida M, Kobayashi S, Sueishi K, et al. A subcommittee report on the clinics of medium and small vasculitis. Intractable Vasculitis Study Group of the MHLW. Report in 1998. 1999: 239–246.
- 237.
Watts R, Lane S, Hanslik T, et al. Development and validation of a consensus methodology for the classification of the ANCA-associated vasculitides and polyarteritis nodosa for epidemiological studies. Ann Rheum Dis 2007; 66: 222–227. PMID: 16901958
- 238.
Policy Research Project for Intractable Diseases funded by Health and Labour Sciences Research Grants (Intractable Diseases Policy Research Project) Study Group on Intractable Vasculitis Arimura Y, Study Group on Intractable Kidney Diseases Maruyama S, Study Group on Diffuse Lung Diseases Honma S. Guidelines for the Management of ANCA-associated Vasculitides 2017. Shindan To Chiryo Sha, Inc. 2017.
- 239.
Matsuo S, Kimura K, Arimura Y, et al. Health and labour scientific research “Study on progressive renal disorders” Subcommittee for the preparation of evidence-based guidelines for management of rapidly progressive glomerulonephritis (RPGN). Evidence-based guidelines for management of rapidly progressive glomerulonephritis (RPGN) 2014. Jpn J Nephrol 2015; 57: 139–232.
- 240.
Mukhtyar C, Flossmann O, Hellmich B, et al. European Vasculitis Study Group (EUVAS). Outcomes from studies of antineutrophil cytoplasm antibody associated vasculitis: A systematic review by the European League Against Rheumatism systemic vasculitis task force. Ann Rheum Dis 2008; 67: 1004–1010. PMID: 17911225
- 241.
Lauque D, Cadranel J, Lazor R, et al. Microscopic polyangiitis with alveolar hemorrhage. A study of 29 cases and review of the literature. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore) 2000; 79: 222–233. PMID: 10941351
- 242.
Ozaki S, Atsumi T, Hayashi T, et al. Severity-based treatment for Japanese patients with MPO-ANCA-associated vasculitis: The JMAAV study. Mod Rheumatol 2012; 22: 394–404. PMID: 21928092
- 243.
Wegener F. Über eine eigenartige rhinogene granulomotose mit besonderer: Beteiligung des arteriensystems und der nieren. Beitr Pathol Anat 1939; 102: 36–68.
- 244.
Japan Rheumatism Foundation Education and Training Committee, Japan College of Rheumatology Lifelong Education Committee. Textbook of Rheumatology revised 2nd edition. Shindan To Chiryo Sha, Inc. 2016.
- 245.
Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 1994; 37: 187–192. PMID: 8129773
- 246.
Csernok E. Anti-neutrophil cytoplasmic antibodies and pathogenesis of small vessel vasculitides. Autoimmun Rev 2003; 2: 158–164. PMID: 12848957
- 247.
Ministry of Health and Welfare Specific Diseases Systematic Study Group on Vascular Lesions. Report in 1998. (Immunological Disease Study Group, Clinical Study Group, funded by a research grant for specific diseases, Ministry of Health and Welfare. Summary Concerning Intractable Vasculitides 1998)
- 248.
Churg J, Strauss L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 1951; 27: 277–301. PMID: 14819261
- 249.
Sada KE, Amano K, Uehara R, et al. Research Committee on Intractable Vasculitides, the Ministry of Health, Labour, Welfare of Japan. A nationwide survey on the epidemiology and clinical features of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) in Japan. Mod Rheumatol 2014; 24: 640–644. PMID: 24289197
- 250.
Gioffredi A, Maritati F, Oliva E, et al. Eosinophilic granulomatosis with polyangiitis: An overview. Front Immunol 2014; 5: 549. PMID: 25404930
- 251.
Keogh KA. Leukotriene receptor antagonists and Churg-Strauss syndrome: Cause, trigger or merely an association? Drug Saf 2007; 30: 837–843. PMID: 17867722
- 252.
Greco A, Rizzo MI, De Virgilio A, et al. Churg-Strauss syndrome. Autoimmun Rev 2015; 14: 341–348. PMID: 25500434
- 253.
Vaglio A, Strehl JD, Manger B, et al. IgG4 immune response in Churg-Strauss syndrome. Ann Rheum Dis 2012; 71: 390–393. PMID: 22121132
- 254.
Nagasawa T, Yoshida M. Clinical features of allergic granulomatous angiitis in Japan and the propose of guideline for clinical diagnosis of this disease. J Jpn Soc Intern Med 1989; 78: 352–356.
- 255.
Ministry of Health and Welfare Specific Disease Systematic Study Group on Vascular Lesions. 1987 Research Report. 1988.
- 256.
Feragalli B, Mantini C, Sperandeo M, et al. The lung in systemic vasculitis: Radiological patterns and differential diagnosis. Br J Radiol 2016; 89: 20150992. PMID: 26876879
- 257.
Lanham JG, Elkon KB, Pusey CD, et al. Systemic vasculitis with asthma and eosinophilia: A clinical approach to the Churg-Strauss syndrome. Medicine (Baltimore) 1984; 63: 65–81. PMID: 6366453
- 258.
Masi AT, Hunder GG, Lie JT, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis). Arthritis Rheum 1990; 33: 1094–1100. PMID: 2202307
- 259.
Ministry of Health and Welfare Specific Diseases Immunological Diseases Study Group Subcommittee on Intractable Vasculitides. 1998 Research Report. 1999.
- 260.
Groh M, Pagnoux C, Baldini C, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur J Intern Med 2015; 26: 545–553. PMID: 25971154
- 261.
Mukhtyar C, Guillevin L, Cid MC, et al. European Vasculitis Study Group. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis 2009; 68: 310–317. PMID: 18413444
- 262.
Koyama A, Yamagata K, Makino H, et al. Japan RPGN Registry Group. A nationwide survey of rapidly progressive glomerulonephritis in Japan: Etiology, prognosis and treatment diversity. Clin Exp Nephrol 2009; 13: 633–650. PMID: 19533266
- 263.
Netzer KO, Leinonen A, Boutaud A, et al. The goodpasture autoantigen. Mapping the major conformational epitope(s) of alpha3(IV) collagen to residues 17–31 and 127–141 of the NC1 domain. J Biol Chem 1999; 274: 11267–11274. PMID: 10196215
- 264.
Pedchenko V, Bondar O, Fogo AB, et al. Molecular architecture of the Goodpasture autoantigen in anti-GBM nephritis. N Engl J Med 2010; 363: 343–354. PMID: 20660402
- 265.
Hellmark T, Burkhardt H, Wieslander J. Goodpasture disease. Characterization of a single conformational epitope as the target of pathogenic autoantibodies. J Biol Chem 1999; 274: 25862–25868. PMID: 10464328
- 266.
Levy JB, Hammad T, Coulthart A, et al. Clinical features and outcome of patients with both ANCA and anti-GBM antibodies. Kidney Int 2004; 66: 1535–1540. PMID: 15458448
- 267.
Jayne DR, Marshall PD, Jones SJ, et al. Autoantibodies to GBM and neutrophil cytoplasm in rapidly progressive glomerulonephritis. Kidney Int 1990; 37: 965–970. PMID: 2179617
- 268.
Cui Z, Zhao J, Jia XY, et al. Anti-glomerular basement membrane disease: Outcomes of different therapeutic regimens in a large singlecenter Chinese cohort study. Medicine (Baltimore) 2011; 90: 303–311. PMID: 21862934
- 269.
Coombes JS, Fassett RG. Antioxidant therapy in hemodialysis patients: A systematic review. Kidney Int 2012; 81: 233–246. PMID: 21975860
- 270.
Davis MD, Brewer JD. Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol Allergy Clin North Am 2004; 24: 183–213, vi. PMID: 15120147
- 271.
Davis MD, Daoud MS, Kirby B, et al. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol 1998; 38: 899–905. PMID: 9631995
- 272.
Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: A histopathologic and clinical review of 72 cases. J Am Acad Dermatol 1992; 26: 441–448. PMID: 1564151
- 273.
Sanchez NP, Winkelmann RK, Schroeter AL, et al. The clinical and histopathologic spectrums of urticarial vasculitis: Study of forty cases. J Am Acad Dermatol 1982; 7: 599–605. PMID: 6754776
- 274.
Aydogan K, Karadogan SK, Adim SB, et al. Hypocomplementemic urticarial vasculitis: A rare presentation of systemic lupus erythematosus. Int J Dermatol 2006; 45: 1057–1061. PMID: 16961508
- 275.
Pasini A, Bracaglia C, Aceti A, et al. Renal involvement in hypocomplementaemic urticarial vasculitis syndrome: A report of three paediatric cases. Rheumatology (Oxford) 2014; 53: 1409–1413. PMID: 24625505
- 276.
Werder M, Truniger B. Hypocomplementaemic urticarial vasculitis. Nephrol Dial Transplant 1997; 12: 1278–1279. PMID: 9198071
- 277.
Saadoun D, Sadallah S, Trendelenburg M, et al. Anti-C1q antibodies in hepatitis C virus infection. Clin Exp Immunol 2006; 145: 308–312. PMID: 16879251
- 278.
Lienesch DW, Sherman KE, Metzger A, et al. Anti-Clq antibodies in patients with chronic hepatitis C infection. Clin Exp Rheumatol 2006; 24: 183–185. PMID: 16762156
- 279.
Wisnieski JJ, Jones SM. IgG autoantibody to the collagen-like region of Clq in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus, and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992; 19: 884–888. PMID: 1404125
- 280.
Black AK, Greaves MW. Antihistamines in urticaria and angioedema. Clin Allergy Immunol 2002; 17: 249–286. PMID: 12113219
- 281.
Black AK. Unusual urticarias. J Dermatol 2001; 28: 632–634. PMID: 11770721
- 282.
Berg RE, Kantor GR, Bergfeld WF. Urticarial vasculitis. Int J Dermatol 1988; 27: 468–472. PMID: 3065257
- 283.
Her MY, Song JY, Kim DY. Hypocomplementemic urticarial vasculitis in systemic lupus erythematosus. J Korean Med Sci 2009; 24: 184–186. PMID: 19270838
- 284.
Macêdo PA, Garcia CB, Schmitz MK, et al. Juvenile systemic lupus erythematosus and dermatomyositis associated with urticarial vasculitis syndrome: A unique presentation. Rheumatol Int 2012; 32: 3643–3646. PMID: 20429007
- 285.
Wisnieski JJ. Urticarial vasculitis. Curr Opin Rheumatol 2000; 12: 24–31. PMID: 10647951
- 286.
Soma J, Sato H, Ito S, et al. Nephrotic syndrome associated with hypocomplementaemic urticarial vasculitis syndrome: Successful treatment with cyclosporin A. Nephrol Dial Transplant 1999; 14: 1753–1757. PMID: 10435889
- 287.
Jachiet M, Flageul B, Deroux A, et al. French Vasculitis Study Group. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: Data from a French nationwide study of fifty-seven patients. Arthritis Rheumatol 2015; 67: 527–534. PMID: 25385679
- 288.
Kartal O, Gulec M, Caliskaner Z, et al. Plasmapheresis in a patient with “refractory” urticarial vasculitis. Allergy Asthma Immunol Res 2012; 4: 245–247. PMID: 22754719
- 289.
Yamazaki-Nakashimada MA, Duran-McKinster C, Ramírez-Vargas N, et al. Intravenous immunoglobulin therapy for hypocomplementemic urticarial vasculitis associated with systemic lupus erythematosus in a child. Pediatr Dermatol 2009; 26: 445–447. PMID: 19689522
- 290.
Millns JL, Randle HW, Solley GO, et al. The therapeutic response of urticarial vasculitis to indomethacin. J Am Acad Dermatol 1980; 3: 349–355. PMID: 7430455
- 291.
Asherson RA, Buchanan N, Kenwright S, et al. The normocomplementemic urticarial vasculitis syndrome--report of a case and response to colchicine. Clin Exp Dermatol 1991; 16: 424–427. PMID: 1806316
- 292.
Lopez LR, Davis KC, Kohler PF, et al. The hypocomplementemic urticarial-vasculitis syndrome: Therapeutic response to hydroxychloroquine. J Allergy Clin Immunol 1984; 73: 600–603. PMID: 6371104
- 293.
Ruzicka T, Goerz G. Systemic lupus erythematosus and vasculitic urticaria. Effect of dapsone and complement levels. Dermatologica 1981; 162: 203–205. PMID: 7250465
- 294.
Holtman JH, Neustadt DH, Klein J, et al. Dapsone is an effective therapy for the skin lesions of subacute cutaneous lupus erythematosus and urticarial vasculitis in a patient with C2 deficiency. J Rheumatol 1990; 17: 1222–1225. PMID: 2290166
- 295.
Takahashi K, Oharaseki T, Yokouchi Y, et al. New classification of vasculitides: 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides (CHCC2012). Jpn J Nephrol 2014; 56: 70–79.
- 296.
Saulsbury FT. Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999; 78: 395–409. PMID: 10575422
- 297.
Guidelines for the diagnosis and treatment of cardiovascular diseases. Guidelines for management of vasculitis syndrome. Circ J 2008; 72 Suppl IV: 1303–1305. http://www.j-circ.or.jp/guideline/pdf/JCS2008_ozaki_h.pdf
- 298.
Mills JA, Michel BA, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheum 1990; 33: 1114–1121. PMID: 2202310
- 299.
Japan Intractable Diseases Information Center. Purpura nephritis (designated intractable disease 224). http://www.nanbyou.or.jp/entry/4658
- 300.
Counahan R, Winterborn MH, White RH, et al. Prognosis of Henoch-Schönlein nephritis in children. Br Med J 1977; 2: 11–14. PMID: 871734
- 301.
Ronkainen J, Koskimies O, Ala-Houhala M, et al. Early prednisone therapy in Henoch-Schönlein purpura: A randomized, double-blind, placebo-controlled trial. J Pediatr 2006; 149: 241–247. PMID: 16887443
- 302.
Shin JI, Park JM, Shin YH, et al. Predictive factors for nephritis, relapse, and significant proteinuria in childhood Henoch-Schönlein purpura. Scand J Rheumatol 2006; 35: 56–60. PMID: 16467044
- 303.
Dudley J, Smith G, Llewelyn-Edwards A, et al. Randomised, double-blind, placebo-controlled trial to determine whether steroids reduce the incidence and severity of nephropathy in Henoch-Schonlein Purpura (HSP). Arch Dis Child 2013; 98: 756–763. PMID: 23845696
- 304.
Hahn D, Hodson EM, Willis NS, et al. Interventions for preventing and treating kidney disease in Henoch-Schönlein Purpura (HSP). Cochrane Database Syst Rev
2015: CD005128. PMID: 26258874
- 305.
Hong S, Ahn SM, Lim DH, et al. Late-onset IgA vasculitis in adult patients exhibits distinct clinical characteristics and outcomes. Clin Exp Rheumatol 2016; 34 Suppl: S77–S83. PMID: 26842218
- 306.
Chapter 11: Henoch-Schönlein purpura nephritis. Kidney Int Suppl 2012; 2: 218–220. PMID: 25018936
- 307.
Kawasaki Y, Suzuki J, Suzuki H. Efficacy of methylprednisolone and urokinase pulse therapy combined with or without cyclophosphamide in severe Henoch-Schoenlein nephritis: A clinical and histopathological study. Nephrol Dial Transplant 2004; 19: 858–864. PMID: 15031341
- 308.
Bergstein J, Leiser J, Andreoli SP. Response of crescentic Henoch-Schoenlein purpura nephritis to corticosteroid and azathioprine therapy. Clin Nephrol 1998; 49: 9–14. PMID: 9491279
- 309.
Foster BJ, Bernard C, Drummond KN, et al. Effective therapy for severe Henoch-Schonlein purpura nephritis with prednisone and azathioprine: A clinical and histopathologic study. J Pediatr 2000; 136: 370–375. PMID: 10700695
- 310.
Du Y, Hou L, Zhao C, et al. Treatment of children with Henoch-Schönlein purpura nephritis with mycophenolate mofetil. Pediatr Nephrol 2012; 27: 765–771. PMID: 22081165
- 311.
Ren P, Han F, Chen L, et al. The combination of mycophenolate mofetil with corticosteroids induces remission of Henoch-Schönlein purpura nephritis. Am J Nephrol 2012; 36: 271–277. PMID: 22965140
- 312.
Jauhola O, Ronkainen J, Autio-Harmainen H, et al. Cyclosporine A vs. methylprednisolone for Henoch-Schönlein nephritis: A randomized trial. Pediatr Nephrol 2011; 26: 2159–2166. PMID: 21626222
- 313.
Park JM, Won SC, Shin JI, et al. Cyclosporin A therapy for Henoch-Schönlein nephritis with nephrotic-range proteinuria. Pediatr Nephrol 2011; 26: 411–417. PMID: 21184240
- 314.
Hattori M, Ito K, Konomoto T, et al. Plasmapheresis as the sole therapy for rapidly progressive Henoch-Schönlein purpura nephritis in children. Am J Kidney Dis 1999; 33: 427–433. PMID: 10070905
- 315.
Kawasaki Y, Suzuki J, Murai M, et al. Plasmapheresis therapy for rapidly progressive Henoch-Schönlein nephritis. Pediatr Nephrol 2004; 19: 920–923. PMID: 15197640
- 316.
Tarshish P, Bernstein J, Edelmann CM Jr. Henoch-Schönlein purpura nephritis: Course of disease and efficacy of cyclophosphamide. Pediatr Nephrol 2004; 19: 51–56. PMID: 14634864
- 317.
Pillebout E, Alberti C, Guillevin L, et al. CESAR study group. Addition of cyclophosphamide to steroids provides no benefit compared with steroids alone in treating adult patients with severe Henoch Schönlein Purpura. Kidney Int 2010; 78: 495–502. PMID: 20505654
- 318.
Kanai H, Sawanobori E, Kobayashi A, et al. Early treatment with methylprednisolone pulse therapy combined with tonsillectomy for heavy proteinuric henoch-schönlein purpura nephritis in children. Nephron Extra 2011; 1: 101–111. PMID: 22470384
- 319.
Lamprecht P, Gause A, Gross WL. Cryoglobulinemic vasculitis. Arthritis Rheum 1999; 42: 2507–2516. PMID: 10615995
- 320.
García-Carrasco M, Ramos-Casals M, Cervera R, et al. Cryoglobulinemia in systemic lupus erythematosus: Prevalence and clinical characteristics in a series of 122 patients. Semin Arthritis Rheum 2001; 30: 366–373. PMID: 11303309
- 321.
Ramos-Casals M, Cervera R, Yagüe J, et al. Cryoglobulinemia in primary Sjögren’s syndrome: Prevalence and clinical characteristics in a series of 115 patients. Semin Arthritis Rheum 1998; 28: 200–205. PMID: 9872481
- 322.
Cicardi M, Cesana B, Del Ninno E, et al. Prevalence and risk factors for the presence of serum cryoglobulins in patients with chronic hepatitis C. J Viral Hepat 2000; 7: 138–143. PMID: 10760044
- 323.
Ramos-Casals M, Muñoz S, Medina F, et al. HISPAMEC Study Group. Systemic autoimmune diseases in patients with hepatitis C virus infection: Characterization of 1020 cases (The HISPAMEC Registry). J Rheumatol 2009; 36: 1442–1448. PMID: 19369460
- 324.
Brouet JC, Clauvel JP, Danon F, et al. Biologic and clinical significance of cryoglobulins: A report of 86 cases. Am J Med 1974; 57: 775–788. PMID: 4216269
- 325.
Motyckova G, Murali M. Laboratory testing for cryoglobulins. Am J Hematol 2011; 86: 500–502. PMID: 21594887
- 326.
Trejo O, Ramos-Casals M, García-Carrasco M, et al. Cryoglobulinemia: Study of etiologic factors and clinical and immunologic features in 443 patients from a single center. Medicine (Baltimore) 2001; 80: 252–262. PMID: 11470986
- 327.
Terrier B, Carrat F, Krastinova E, et al. Prognostic factors of survival in patients with non-infectious mixed cryoglobulinaemia vasculitis: Data from 242 cases included in the CryoVas survey. Ann Rheum Dis 2013; 72: 374–380. PMID: 22586172
- 328.
De Vita S, Soldano F, Isola M, et al. Preliminary classification criteria for the cryoglobulinaemic vasculitis. Ann Rheum Dis 2011; 70: 1183–1190. PMID: 21571735
- 329.
Dammacco F, Sansonno D. Therapy for hepatitis C virus-related cryoglobulinemic vasculitis. N Engl J Med 2013; 369: 1035–1045. PMID: 24024840
- 330.
Pietrogrande M, De Vita S, Zignego AL, et al. Recommendations for the management of mixed cryoglobulinemia syndrome in hepatitis C virus-infected patients. Autoimmun Rev 2011; 10: 444–454. PMID: 21303705
- 331.
De Vita S, Quartuccio L, Isola M, et al. A randomized controlled trial of rituximab for the treatment of severe cryoglobulinemic vasculitis. Arthritis Rheum 2012; 64: 843–853. PMID: 22147661
- 332.
Saadoun D, Resche Rigon M, Sene D, et al. Rituximab plus Peginterferon-alpha/ribavirin compared with Peg-interferon-alpha/ribavirin in hepatitis C-related mixed cryoglobulinemia. Blood 2010; 116: 326–334. PMID: 20439619
- 333.
Ferri C, Cacoub P, Mazzaro C, et al. Treatment with rituximab in patients with mixed cryoglobulinemia syndrome: Results of multicenter cohort study and review of the literature. Autoimmun Rev 2011; 11: 48–55. PMID: 21821153
- 334.
Ramos-Casals M, Robles A, Brito-Zerón P, et al. Life-threatening cryoglobulinemia: Clinical and immunological characterization of 29 cases. Semin Arthritis Rheum 2006; 36: 189–196. PMID: 16996578
- 335.
Sakane T, Takeno M, Suzuki N, et al. Behçet’s disease. N Engl J Med 1999; 341: 1284–1291. PMID: 10528040
- 336.
Takeno M, Ideguchi H, Suda A, et al. Vascular involvement of Behçet’s disease.
In
: Ishigatsubo Y, editor. Behçet’s disease. Springer, 2015: 79–100.
- 337.
Japan Intractable Diseases Information Center. Behçet’s disease (designated intractable disease 56). http://www.nanbyou.or.jp/entry/330
- 338.
Seyahi E, Caglar E, Ugurlu S, et al. An outcome survey of 43 patients with Budd-Chiari syndrome due to Behçet’s syndrome followed up at a single, dedicated center. Semin Arthritis Rheum 2015; 44: 602–609. PMID: 25476470
- 339.
Saadoun D, Asli B, Wechsler B, et al. Long-term outcome of arterial lesions in Behçet disease: A series of 101 patients. Medicine (Baltimore) 2012; 91: 18–24. PMID: 22198498
- 340.
Tüzün H, Beşirli K, Sayin A, et al. Management of aneurysms in Behçet’s syndrome: An analysis of 24 patients. Surgery 1997; 121: 150–156. PMID: 9037226
- 341.
Seyahi E, Melikoglu M, Akman C, et al. Pulmonary artery involvement and associated lung disease in Behçet disease: A series of 47 patients. Medicine (Baltimore) 2012; 91: 35–48. PMID: 22210555
- 342.
International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990; 335: 1078–1080. PMID: 1970380
- 343.
Davatchi F. International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD). The International Criteria for Behçet’s Disease (ICBD): A collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J Eur Acad Dermatol Venereol 2014; 28: 338–347. PMID: 23441863
- 344.
Ahn JK, Lee YS, Jeon CH, et al. Treatment of venous thrombosis associated with Behcet’s disease: Immunosuppressive therapy alone versus immunosuppressive therapy plus anticoagulation. Clin Rheumatol 2008; 27: 201–205. PMID: 17636362
- 345.
Desbois AC, Wechsler B, Resche-Rigon M, et al. Immunosuppressants reduce venous thrombosis relapse in Behçet’s disease. Arthritis Rheum 2012; 64: 2753–2760. PMID: 22354392
- 346.
Alibaz-Oner F, Karadeniz A, Ylmaz S, et al. Behçet disease with vascular involvement: Effects of different therapeutic regimens on the incidence of new relapses. Medicine (Baltimore) 2015; 94: e494. PMID: 25674739
- 347.
Mehta P, Laffan M, Haskard DO. Thrombosis and Behçet’s syndrome in non-endemic regions. Rheumatology (Oxford) 2010; 49: 2003–2004. PMID: 20421214
- 348.
Ha YJ, Jung SY, Lee KH, et al. Long-term clinical outcomes and risk factors for the occurrence of post-operative complications after cardiovascular surgery in patients with Behçet’s disease. Clin Exp Rheumatol 2012; 30 Suppl: S18–S26. PMID: 22776346
- 349.
Tuzun H, Seyahi E, Arslan C, et al. Management and prognosis of nonpulmonary large arterial disease in patients with Behçet disease. J Vasc Surg 2012; 55: 157–163. PMID: 21944910
- 350.
Kim WH, Choi D, Kim JS, et al. Effectiveness and safety of endovascular aneurysm treatment in patients with vasculo-Behçet disease. J Endovasc Ther 2009; 16: 631–636. PMID: 19842735
- 351.
Yang SS, Park KM, Park YJ, et al. Peripheral arterial involvement in Behcet’s disease: An analysis of the results from a Korean referral center. Rheumatol Int 2013; 33: 2101–2108. PMID: 23417141
- 352.
Liu Q, Ye W, Liu C, et al. Outcomes of vascular intervention and use of perioperative medications for nonpulmonary aneurysms in Behçet disease. Surgery 2016; 159: 1422–1429. PMID: 26765098
- 353.
Saadoun D, Wechsler B, Desseaux K, et al. Mortality in Behçet’s disease. Arthritis Rheum 2010; 62: 2806–2812. PMID: 20496419
- 354.
Hashimoto H, et al. Revised diagnostic criteria and guidelines for treatment of malignant rheumatoid arthritis (draft). MRA subcommittee, MHLW Specified Diseases Systematic Study Group on Vascular Lesions. 1987 Research Report. 1988: 23–25.
- 355.
Ntatsaki E, Mooney J, Scott DG, et al. Systemic rheumatoid vasculitis in the era of modern immunosuppressive therapy. Rheumatology (Oxford) 2014; 53: 145–152. PMID: 24108586
- 356.
Turesson C, Schaid DJ, Weyand CM, et al. Association of HLA-C3 and smoking with vasculitis in patients with rheumatoid arthritis. Arthritis Rheum 2006; 54: 2776–2783. PMID: 16947780
- 357.
Scott DG, Bacon PA, Tribe CR. Systemic rheumatoid vasculitis: A clinical and laboratory study of 50 cases. Medicine (Baltimore) 1981; 60: 288–297. PMID: 7242322
- 358.
MHLW of Japan. Malignant Rheumatoid Arthritis. Summary, Diagnostic Criteria, etc. Intractable diseases designated on January 1, 2015 (newly designated, updated). http://www.mhlw.go.jp/stf/seisakunitsuite/bunya/0000062437.html
- 359.
Heurkens AH, Westedt ML, Breedveld FC. Prednisone plus azathioprine treatment in patients with rheumatoid arthritis complicated by vasculitis. Arch Intern Med 1991; 151: 2249–2254. PMID: 1953230
- 360.
Bartels CM, Bridges AJ. Rheumatoid vasculitis: Vanishing menace or target for new treatments? Curr Rheumatol Rep 2010; 12: 414–419. PMID: 20842467
- 361.
Saint Marcoux B, De Bandt M. CRI (Club Rhumatismes et Inflammation). Vasculitides induced by TNFalpha antagonists: A study in 39 patients in France. Joint Bone Spine 2006; 73: 710–713. PMID: 17127088
- 362.
Sokumbi O, Wetter DA, Makol A, et al. Vasculitis associated with tumor necrosis factor-α inhibitors. Mayo Clin Proc 2012; 87: 739–745. PMID: 22795634
- 363.
Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol 2004; 31: 1955–1958. PMID: 15468359
- 364.
Kim MJ, Kim HO, Kim HY, et al. Rituximab-induced vasculitis: A case report and review of the medical published work. J Dermatol 2009; 36: 284–287. PMID: 19382999
- 365.
Carvajal Alegria G, Uguen A, Genestet S, et al. New onset of rheumatoid vasculitis during abatacept therapy and subsequent improvement after rituximab. Joint Bone Spine 2016; 83: 605–606. PMID: 27052429
- 366.
Katz-Agranov N, Khattri S, Zandman-Goddard G. The role of intravenous immunoglobulins in the treatment of rheumatoid arthritis. Autoimmun Rev 2015; 14: 651–658. PMID: 25870941
- 367.
MHLW Health Service Bureau, Study group on the management of intractable diseases. Summary of measures against intractable diseases. 2007 edition. Taiyou Bijutsu Co., Ltd. 2007: 318.