Abstract
Background:
Ischemic postconditioning (IPostC) is an endogenous protective mechanism to reduce ischemia-reperfusion (I/R) injury. However, whether IPostC protects aged cardiomyocytes against I/R injury is not fully understood. Considering the protective function of microRNA 30a (miR-30a) against ischemia-induced injury in H9C2 cells, its role in the protective effects of IPostC on I/R injury of aged cardiomyocytes was investigated further.
Methods and Results:
To mimic I/R and IPostC in vitro, the aged cardiomyocyte model for hypoxia postconditioning (HPostC) treatment was established by 9 days of incubation with 8 mg/mL D-galactose and then followed by exposure to hypoxic environment. HPostC significantly alleviated hypoxia/reoxygenation (H/R) injury and reduced autophagy of aged cardiomyocytes, as evidenced by decreased LC3B-II expression and increased p62 by Western blot. Quantified by quantitative real-time polymerase chain reaction (qRT-PCR), miR-30a was increased in aged cardiomyocytes treated with HPostC compared with I/R injury group. Overexpression of miR-30a by LV3-rno-miR-30a mimic promoted cardioprotective effect of HPostC in aged cardiomyocytes by suppressing BECN1-mediated autophagy, all of which was abrogated by knockdown of miR-30a expression. Epigenetic analyses demonstrated that HPostC reduced DNA methyltransferase 3b-mediated DNA hypomethylation levels at miR-30a promoter, leading to upregulation of miR-30a.
Conclusions:
HPostC protected aged cardiomyocytes survival against H/R injury via DNMT3b-dependent activation of miR-30a. miR-30a could be a potential therapeutic target for ischemic myocardial infarction.
The incidence and mortality of cardiovascular diseases keep increasing due to aging population in the intensification, as old patients with aged myocardial are more prone to myocardial infarction (MI) and heart failure.1,2
After MI, ischemia/reperfusion (I/R) can cause additional injury such as cellular metabolic dysfunction and structural damage due to rapid reperfusion of the ischemic or hypoxia myocardium.3,4
It was reported that ischemic postconditioning (IPostC) can significantly protect cardiomyocytes against I/R injury through brief intermittent episodes of I/R at the onset of reperfusion after a prolonged period of ischemia or hypoxia.5
Evidence has indicated the urgent role of autophagy in the I/R injury of the heart by degrading long-lived proteins and organelles of cells.6
However, the exact relation and the related mechanism of autophagy in the cardioprotective effects of IPostC on aged myocardial I/R injury remains unclear.
MicroRNAs (miRNAs) are a class of endogenous, non-coding small ribonucleic acids containing 21–24 nucleotides.7
Evidence has indicated that miRNAs involved in a wide range of biologic processes, such as autophagy,8
proliferation,9
differentiation, and apoptosis, negatively modulate gene expression through binding of the target gene to the 3’UTR.10
For example, the regulation of miRNAs on autophagy was mediated by autophagy-related genes such as BECN1, light chain 3B (LC3B) and p62.11
Recently, it was reported that pathophysiological variation, including the process of cardiac remodeling and heart failure, can affect the abundance of many miRNAs.12,13
For example, miR-30a was found to abnormally expressed in the cardiac tissue of murine model with myocardial I/R.14
Although miR-30 has been demonstrated to have an important role in heart disease, whether it can mediate the protective effect of IPostC on I/R injury in aged myocardium has not been investigated.
Aberrant miRNA expression is often associated with epigenetic regulation such as DNA methylation and histone modifications.15
DNA methylation is recognized as the most important epigenetic regulation for gene expression,16
which is catalyzed by DNA methyltransferases (DNMTs). Among them, DNMT1 (maintenance DNMT), DNMT3a and DNMT3b (de novo DNMTs) are mainly responsible for modulating and maintaining DNA methylation levels of gene promoter.17
Alteration of DNA methylation level in miRNA promoter directly results in the change of its expression level.18
Vera et al19
reported that the increase of miR-7 methylation level could act as a potential clinical epigenetic biomarker for the identification of ovarian cancer patients with poor response to platinum-derived therapy. Undoubtedly, there is a crucial role for DNA methylation in many types of diseases, but its role in aged myocardial I/R injury is still poorly understood.
In this study, we explored the underlying mechanism of miR-30a in the cardioprotective effect of hypoxia postconditioning (HPostC) on aged cardiomyocyte hypoxia/reoxygenation (H/R) injury. The results indicated that HPostC upregulated the miR-30a level through DNMT3b-mediated hypomethylation of the miR-30a promoter in aged cardiomyocytes, thereby inhibiting autophagy.
Methods
Cell Culture, Lentiviral and Adenoviral Infection
Rat cardiomyocytes (H9C2 cells) purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) were cultured in the Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum and 100 U/mL, 100 μg/mL streptomycin in a normoxia incubator (5% CO2
and 37℃). Lentiviral vectors encoding Rattus miR-30a mimic (LV3-rno-miR-30a mimic: 5′-TGTAAACATCCTCGACTGGAAG3-3′), miR-30a inhibitor (LV3-rno-miR-30a inhibitor: 5′-CTTCCAGTCGAGGATGTTTACA-3′), and empty vector (LV3-rno-miR-neg: 5′- GTTCTCCGAACGTGTCACGT-3′) were obtained from GenePharma Co. Ltd (Shanghai, China). The adenovirus expressing Rattus BECN1, the adenovirus expressing Rattus shRNA targeting BECN1 and DNMT3b, and negative control adenovirus (Ad-NC) were provided by GenePharma Co. Ltd (Shanghai, China). Cardiomyocytes were infected with lentivirus or adenovirus following the reported protocols.20
Establishment of H/R and HPostC Models
Aged cardiomyocytes were induced by treatment of H9C2 cells with 8 mg/mL D-galactose (SigmaAldrich, G0750) for ∼9 days. To induce H/R injury, aged cardiomyocytes were cultured at 37℃ in a hypoxic incubator (Thermo Fisher Scientific, USA, 3131) filled with pre-mixed gas (5% CO2, 1% O2
and 94% N2) for 180 min followed by 120 min of reoxygenation. To establish HPostC model, aged cardiomyocytes were cultured under hypoxia for 180 min, followed by 3 cycles of 5 min of hypoxia and 5 min of reoxygenation before 120 min reoxygenation (Supplementary Figure A).
Senescence-Associated β-Galactosidase (SA-β-gal) Assay
A senescence-associated-β-galactosidase staining kit (Solarbio Science & Technology Co., Ltd., Beijing, China; G1580) was used according to the manufacturer’s instructions. Briefly, the cells were washed in phosphate buffered saline (PBS), fixed in fixative solution for 15 min at room temperature, and incubated with a staining solution mix overnight at 37℃. The percentage of senescent (SA-β-gal-positive) cells was calculated in five randomly chosen fields of view for each sample.
Immunofluorescence and Phalloidin Staining
Aged cardiomyocytes were fixed with 4% paraformaldehyde for immunofluorescence and phalloidin staining. The Rabbit anti-BECN1 antibody (Abcam; ab207612, IF 1:200) was used in this study. Phalloidin conjugated to phalloidin-iFluorTM
488 Conjugate (AAT Bioquest®, USA) was used to stain actin filaments (F-actin). After washing 3 times with PBS, samples were incubated with secondary antibody for 1 h at 37℃, followed by washing with PBS and nuclei staining with 4′,6-diamidino-2-phenylindole (DAPI) in the dark. The immunofluorescence images were captured under a Zeiss LSM800 Meta Laser Scanning confocal microscope (Carl Zeiss, Jena, Germany).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from aged cardiomyocytes and reverse-transcribed with a reverse transcriptase kit (Takara, Otsu, Japan). For qRT-PCR, the mRNA level of DNMT3b was analyzed using SYBR Green assay kit (Takara) on a FTC3000 qRT-PCR System (Funglyn Biotech Inc, Toronto, Canada). Both β-actin and U6 were used as internal controls for template normalization. Relative quantification of mRNA was calculated using the 2−∆∆CT
method.21
The primers for miR-30a-5p were synthesized by Ruibo Biotechnology (Guangzhou, China). The sequences of primers used in qRT-PCR were listed in the
Table.
Table.
Primer Sequences for Real-Time Polymerase Chain Reaction
| Gene |
GenBank |
Primer sequences |
| β-actin |
NM_031144.3 |
Forward: 5′-TGTCACCAACTGGGACGATA-3′ |
| Reverse: 5′-GGGGTGTTGAAGGTCTCAAA-3′ |
| DNMT3b |
NM_001003959.1 |
Forward: 5′-GATGATGGAGATGGCTCTGATA-3′ |
| Reverse: 5′-GGCTGGAGATACTGTTGCTGTT-3′ |
Other materials and methods information are available in the
Supplementary Materials
section (Supplementary File).
Statistical Analysis
All statistical analysis was performed using GraphPad Prism 5.0. Data were expressed as means±standard deviation (SD). More than 2 groups were compared using a one-way ANOVA, and a Student-Newman-Kaul’s test; differences between the 2 groups were evaluated using an unpaired Student’s t-test. P<0.05 was considered to be statistically different.
Results
HPostC Inhibited Autophagy and Prevented Aged Cardiomyocyte Death Induced by H/R Injury
Aged cardiomyocytes were established by 8 mg/mL D-galactose treatment of H9C2 cells for 9 days, as evidenced by increased SA-β-gal-positive cells (Figure 1A,B). Then, aged cardiomyocytes were exposed to hypoxia or a reoxygenation environment for several cycles to mimic I/R and IPostC as described before.22
After cell viability staining, the number of viable cardiomyocytes in the HPostC group is significantly higher than that of the H/R group (Figure 1C,D). Moreover, stress fiber disruption and loss of F-actin resulted from H/R were abrogated by HPostC treatment (Figure 1E). These results implied that HPostC promoted survival of aged cardiomyocytes.
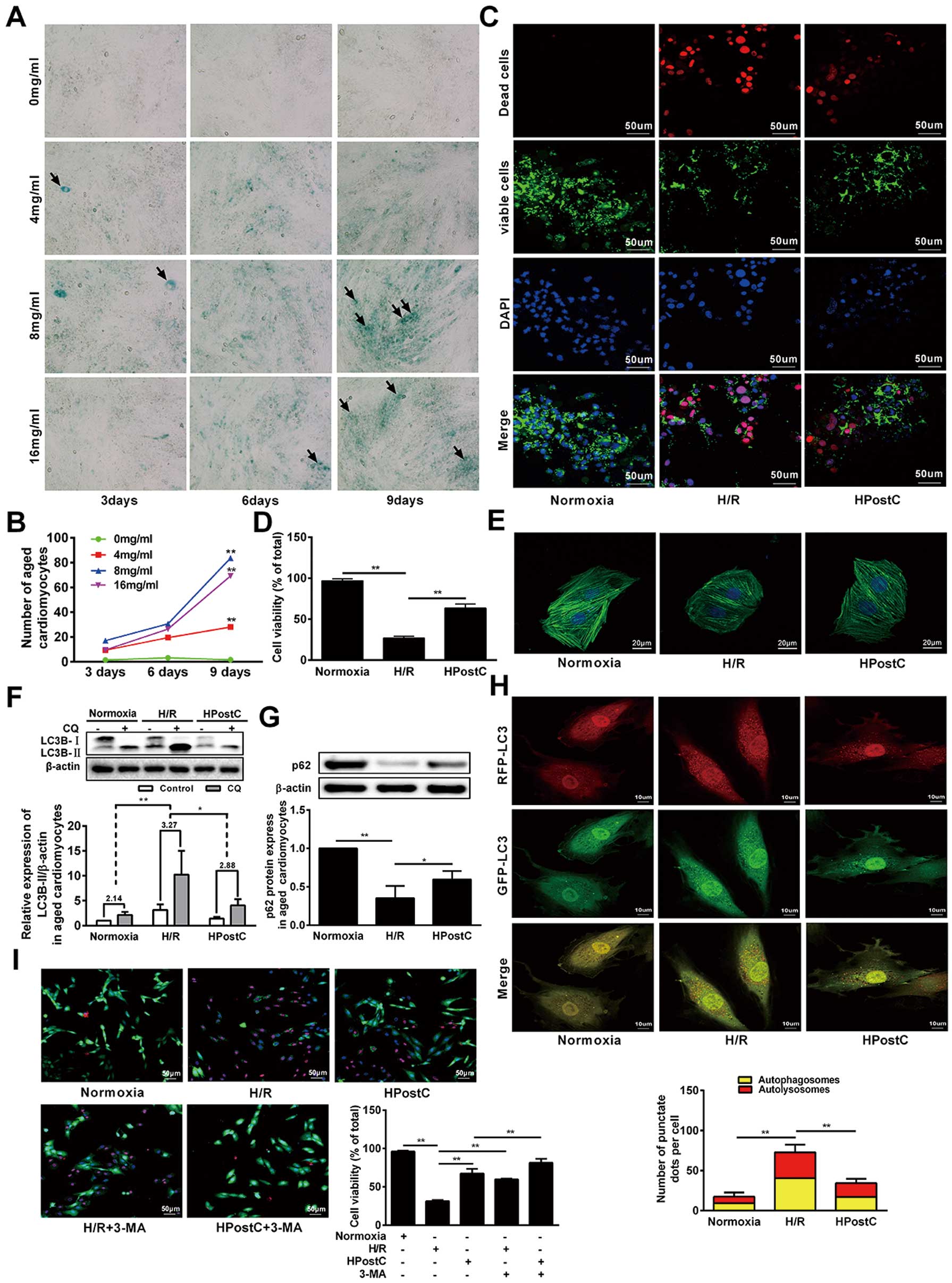
To testify whether HPostC can protect aged cardiomyocytes from H/R injury through autophagy, we assessed the expression of autophagy-related proteins, LC3B-II (a marker for autophagy activation) and p62 (a marker for autophagy inhibition), by Western blot. LC3B-II expression was significantly reduced, while p62 levels were significantly upregulated in aged cardiomyocytes under HPostC treatment compared to that of the H/R group (Figure 1F,G). It was reported that autophagosome–lysosome fusion can result in LC3B-II degradation;23
we suspect that the upregulation of LC3B-II in H/R-treated cells may be caused by the increase of autophagosomes synthesis or the blockage of autophagosome digestion in lysosomes. The effect of HPostC on autophagic flux was then investigated using chloroquine (CQ) to limit autophagosome–lysosome fusion. CQ administration increased the LC3B-II level under all investigated conditions, while the variability of LC3B-II in the HPostC and Normoxia groups was significantly lower than that of the H/R group (Figure 1F). These data demonstrated that HPostC can inhibit autophagosome synthesis and, accordingly, reduce the autophagy flux. To confirm that, mRFP-GFP-LC3 adenovirus infection was used to monitor the autophagy flux in aged cardiomyocytes. In this experiment, only red puncta implied autolysosome, whereas yellow puncta, the overlaying of red puncta with green ones, represented autophagosome.24
Both the number of autolysosomes and autophagosomes remarkably decreased in the HPostC group (Figure 1H). In contrary, both the red and yellow puncta increased in the H/R group. Moreover, aged cardiomyocytes were treated with the autophagy inhibitor, 3-MA; H/R-induced cell viability was similar with HPostC while HPostC-induced cell viability increased further, indicating that 3-MA can act in the same way with HPostC to enhance the cell viability and the protective effects through inhibiting autophagic activity (Figure 1I). Collectively, these results indicated that HPostC could protect aged cardiomyocytes from H/R injury via inhibiting autophagy.
miR-30a Was Involved in HPostC-Mediated Inhibition of Autophagy in Aged Cardiomyocytes
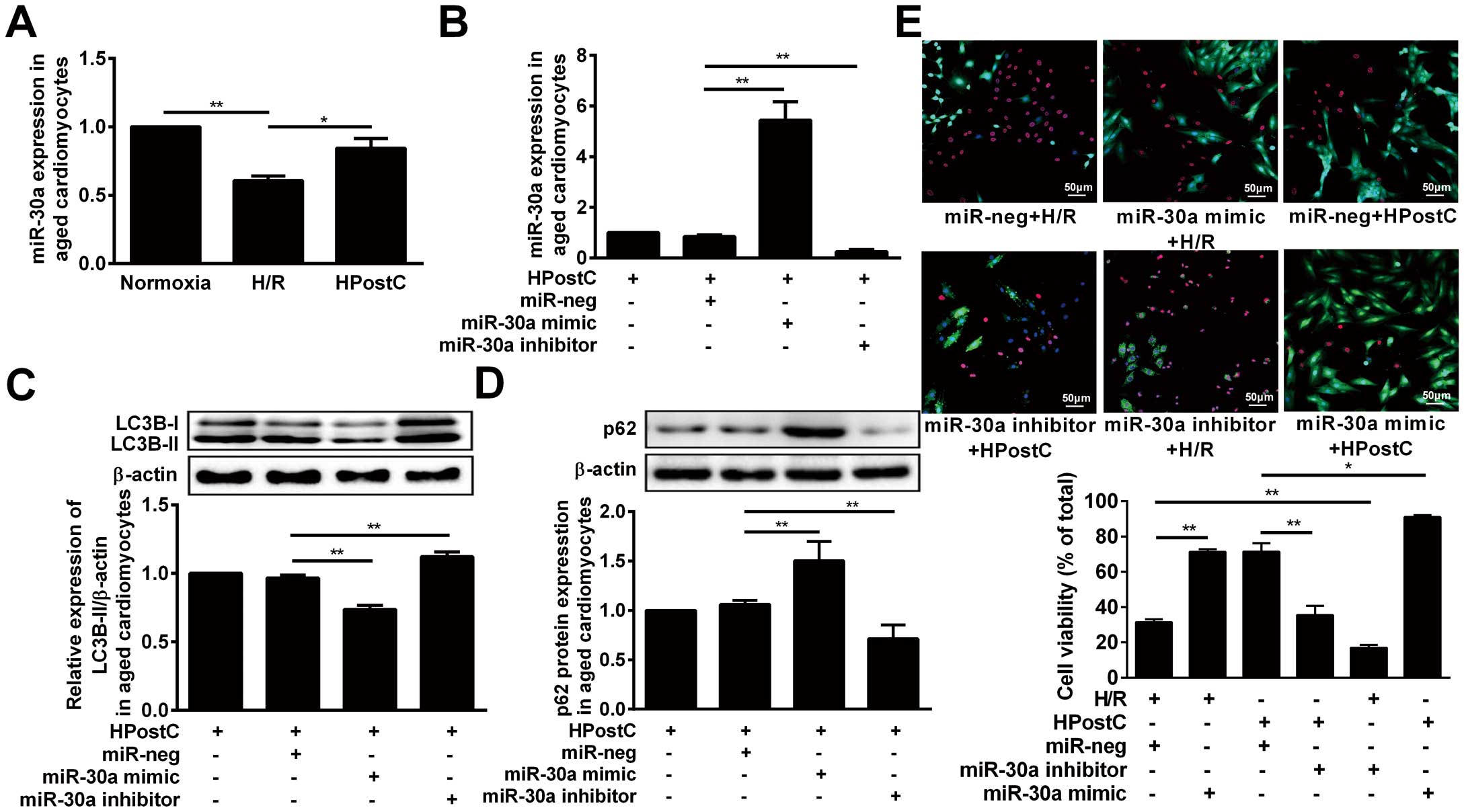
A previous study reported specific miR-30a expression in mouse cardiac tissue.25
The qRT-PCR assay showed a dramatic increase of miR-30a expression in aged cardiomyocytes exposed to HPostC compared to that of H/R-treated cells (Figure 2A
and
Supplementary Figure B). In order to further investigate the effect of miR-30a on autophagy, we established cell lines with miR-30a knock-down or over-expression by transfecting LV3-rno-miR-30a inhibitors or LV3-rno-miR-30a mimic, respectively (Figure 2B). As depicted in
Figure 2C, over-expression of miR-30a could decrease the LC3B-II level, whereas it is the opposite for miR-30a knockdown. In contrast, p62 protein levels were the opposite to the LC3B-II levels (Figure 2D). And cell viability staining elucidated that over-expression of miR-30a induced a significant increase of cell viability under H/R conditions compared with miR-neg, while miR-30a knock-down abolished protection afforded by HPostC (Figure 2E). In summary, these data suggested that miR-30a mediated the protection of HPostC against aged cardiomyocyte H/R injury through inhibiting autophagy.
TargetScan (http://www.targetscan.org/vert_71/) predicted a putative miR-30a complementary site in the 3′-UTR of BECN1, meaning that
BECN1
is a potential target gene of miR-30a (Figure 3A). A luciferase-reporter assay also confirmed the reliability of this prediction as miR-30a mimic significantly inhibited the luciferase activity of wild-type (WT) 3′UTR of
BECN1, but not for the mutated type (Mut) (Figure 3B), suggesting that miR-30a can specifically target the
BECN1
3′-UTR region by binding to their putative sequences. Meanwhile, a Western blot assay indicated a negative relation between miR-30a and
BECN1
under HPostC treatments (Figure 3C). In addition, the BECN1 protein level markedly decreased in aged cardiomyocytes subjected to HPostC as compared with H/R (Figure 3D
and
Supplementary Figure C). Consistent with the Western blot analysis, immunofluorescence staining also showed that HPostC dramatically reduced the number of
BECN1-positive cells compared with H/R (Figure 3E). A mRFP-GFP-LC3 adenovirus reporter was further used to investigate the role of
BECN1
in autophagy flux of aged cardiomyocytes under HPostC. As expected, knock-down of
BECN1
reduced the number of red and yellow puncta in the aged cardiomyocytes. In contrast, ectopic expression of
BECN1
promoted the total autophagic structures under HPostC treatment (Figure 3F,G). Moreover, upregulating autophagy by restoration of BECN1 expression prevents HPostC to protect aged cardiomyocytes from H/R-induced cell death (Figure 3H). Therefore, these data verified that miR-30a was involved in the autophagy inhibition under HPostC by targeting
BECN1.

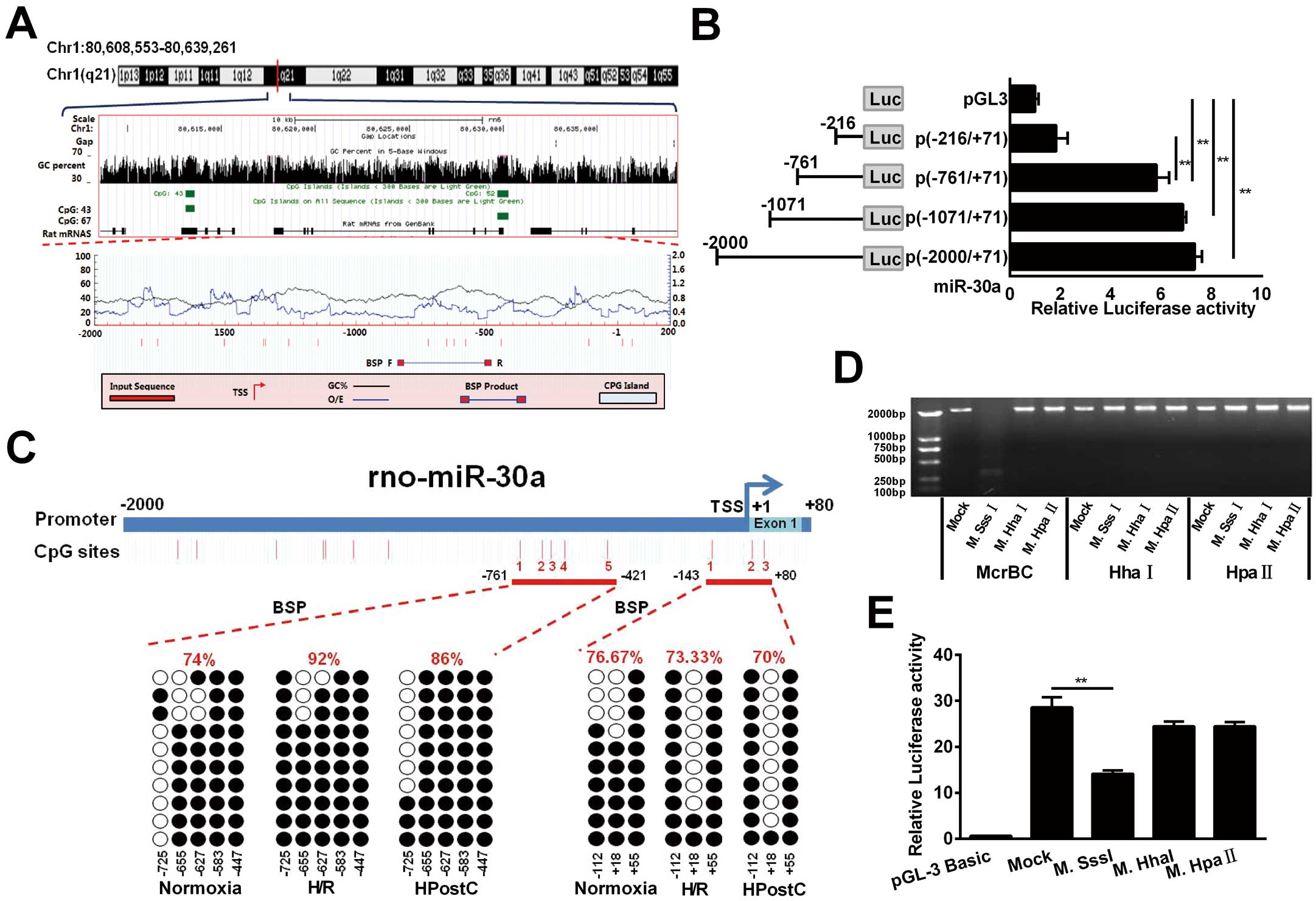
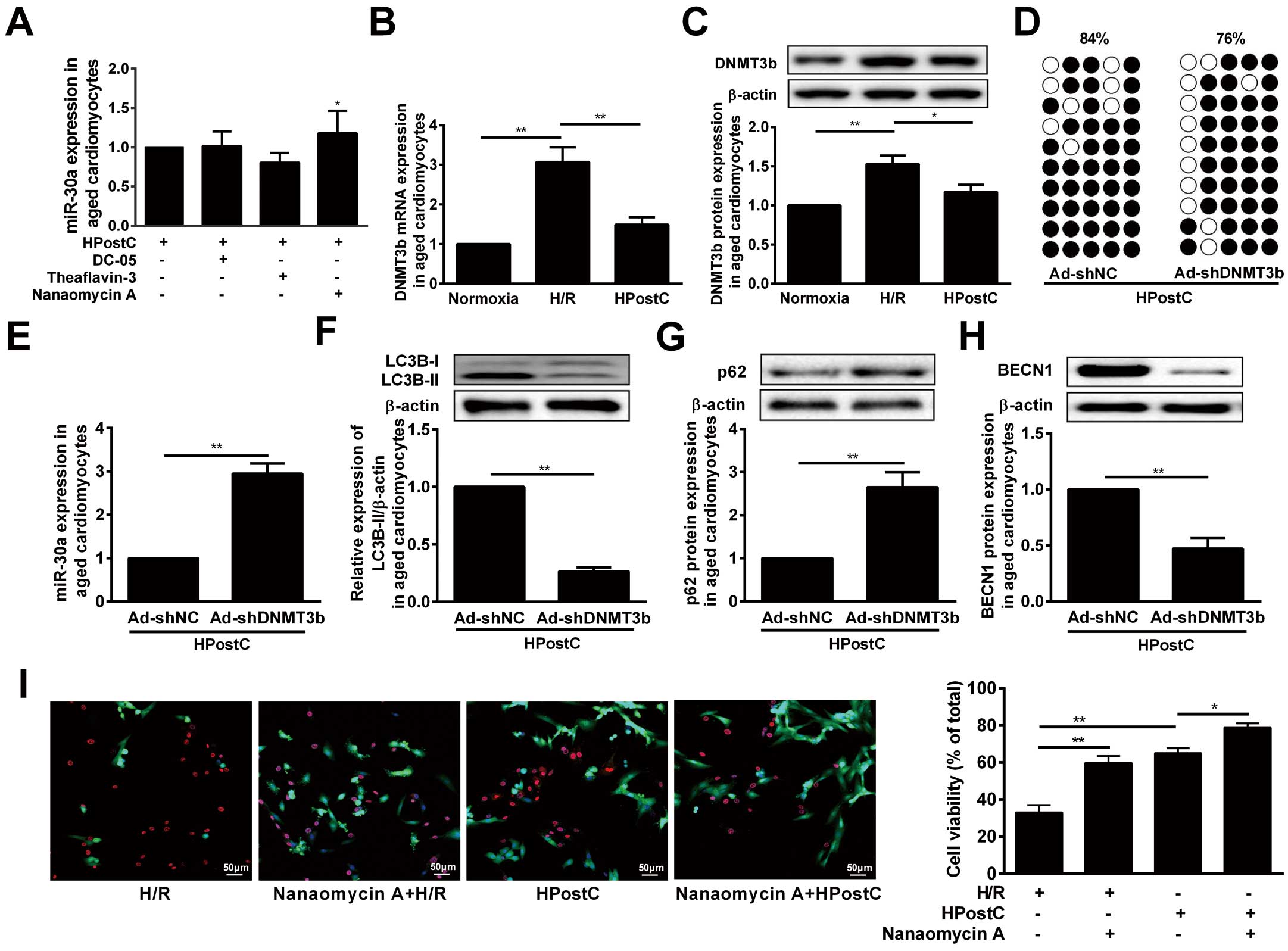
Next, we explored the mechanism responsible for miR-30a upregulation in aged cardiomyocytes under HPostC. Analysis of the genomic DNA sequence by an UCSC genome browser indicated 2 CpG island regions at the miR-30a whole-genome (Figure 4A, Middle). In addition, MethPrimer software (http://www.urogene.org/methprimer/) analysis showed that the potential methylation sites in the miR-30a promoter (−2,000 to +200) contained several CpG sites (red vertical bars) (Figure 4A, Bottom). Based on these predictions, 4 synthesized fragments (−2,000/+71, −1,071/+71, −761/+71, −216/+71) were inserted into the firefly luciferase vector, pGL3. Among of them, the (−761/−216) fragment of miR-30a was the basic core regulation region, as it provided the structural basis for methylation (Figure 4B). In addition, bisulfite sequencing (BSP) indicated that 2 core regions of the promoter (−761 to −421, −143 to +80) contain 8 CpG sites relative to the miR-30a’s transcription start site (TSS). Compared with H/R, HPostC only decreased miR-30a DNA methylation level at the −761 to −421 region, but not at the −143 to +80 region. These data demonstrated that −261 to −761 was the regulating region of miR-30a transcription (Figure 4C
and
Supplementary Figure D). Next, in order to examine whether DNA methylation plays a direct role in regulating miR-30a promoter activity, we methylated the miR-30a promoter fragments using M.SssI, M.HhaI and M.HpaII. Among them, SssI methylated all 5′-CpG-3′ sites, HhaI only methylated the CpG site of 5′-GCGC-3′, and HpaII methylated the sequence of 5′-CCGG-3′. The successful methylation of these sites was confirmed by enzymatic digestion using enzymes of McrBC, HhaI and HpaII (Figure 4D). By using the luciferase activity assay, miR-30a promoter activity was found to be markedly blocked after with M.SssI treatment (methylates 15 CpG sites), which was found to reduce miR-30a promoter activity the most (Figure 4E). These results implied that DNA methylation contributes to the suppression of miR-30a promoter activity in H/R injury, which can be reversed by HPostC.
DNA methylation is a major epigenetic modification catalyzed by DNMTs.26
To determine which DNMTs contributed to the HPostC-induced hypomethylation of miR-30a, we treated aged cardiomyocytes with DNMTs inhibitors.
Figure 5A
indicated that Nanaomycin A (NA), a DNMT3b inhibitor, significantly enhanced the expression of miR-30a, while no significant effect was observed for DC-05 and Theaflavin-3 (DNMT1- and DNMT3a-specific inhibitors) treatment. These data demonstrated that DNMT3b is the key methyltransferase in the regulation of miR-30a expression under HPostC. Meanwhile, HPostC strongly decreased DNMT3b expression in the aged cardiomyocytes compared to H/R injury (Figure 5B,C). Accordingly, knockdown of DNMT3b inhibited the methylation of miR-30a, while enhanced miR-30a level in aged cardiomyocytes (Figure 5D,E). Additionally, DNMT3b knockdown significantly suppressed the LC3B-II and
BECN1
level, whereas it upregulated p62 expression under HPostC treatment (Figure 5F–H). And cell viability staining elucidated that NA treatment can increase aged cardiomyocytes viability under H/R or HPostC conditions, which implied that NA promotes protection afforded by IPostC against H/R injury (Figure 5I). Collectively, it concluded that reduction of DNMT3b expression induced by HPostC could result in the miR-30a DNA hypomethylation and lead to miR-30a activation in the aged cardiomyocytes.
Discussion
MI is the leading cause of human death and disability worldwide, and is caused by myocardial ischemia, or hypoxia due to vascular stenosis.27
Currently, the most effective treatment of MI patients is to restore myocardial perfusion and save the viable myocardium. However, the myocardium is susceptible to another form of injury stemming from reperfusion of the previously ischemic tissue once coronary flow is restored.28
Fortunately, IPostC performed at immediate reperfusion has been successfully applied to patients with acute MI.29
Although some clinical studies have indicated the significant benefit of IPostC for the prognosis of aged patients suffering from acute MI,30
the effect of IPostC on the aged myocardium is still paradoxical. In this study, using H/R and HPostC models to mimic the pathological environment of I/R and IPostC, we explored the protective effect of HPostC on H/R injury in aged cardiomyocytes and the shielding molecular mechanism. Based on the observation, we found that HPostC could effectively relieve H/R injury in aged cardiomyocytes, which is consist with a previous clinical report.30
We then elucidated the underlying molecular mechanisms of HPostC alleviating H/R injury, which may contribute to the discovery of therapeutic targets for aged people with MI.
Wang et al31
proved that IPostC can reduce I/R injury and improve cell survival by inhibiting autophagy activation and maturation in vivo and in vitro. Although autophagy is related to the protective effect of IPostC,32
the exact role of this effect in the ischemic myocardium is still controversial. In this study, we found that inhibition of autophagy contributed to the HPostC-elicited cardioprotection against H/R injury in aged cardiomyocytes, which is in agreement with a previous study.31
However, several other studies have shown that autophagy activation, but not inhibition, is associated with protective effects of IPostC. For example, Wei et al33
reported that IPostC protects cardiomyocytes against I/R injury by increased autophagy via the upregulation of LC3B-II and
BECN1
expressions. These controversial results may be due to the differences between ischemic models, the duration of ischemic injury and animal age. In any case, it is necessary to clarify the molecular mechanisms of autophagy inhibition in the protection of aged cardiomyocytes from HPostC.
Post-transcriptional regulation of miRNAs has been extensively investigated in many diseases, including heart failure, cardiac hypertrophy, and MI.34
However, the mechanism responsible for miRNA regulation and the contributions of HPostC-regulated miRNAs in towards cardioprotection of aged cardiomyocytes is still limited. Tu et al35
demonstrated that miR-21 is involved in the IPostC-mediated cardiac protection against I/R injury through the PTEN/Akt-signaling pathway. Others have reported that miR-1 and miR-133a may attenuate cardiomyocyte apoptosis by targeting CASP9.36
In this study, we demonstrated that the elevated expression of miR-30a in HPostC-treated aged cardiomyocytes enhanced protective effects against H/R injury through inhibiting
BECN1
expression. Moreover, we found that miR-30a can directly promote degradation of
BECN1
by binding with its 3′-UTR region, which is consistent with findings from a previous report.37
As a mammalian autophagic gene,
BECN1
is essential for autophagy induction and regulation.38
Maejima et al39
has shown that
BECN1
inhibition can reduce cell death of cardiomyocytes induced by I/R injury through inhibiting autophagy. The present study also disclosed that HPostC could suppress
BECN1
expression, thereby inhibiting autophagy in the aged cardiomyocytes.
In addition, miRNAs harboring CpG islands can undergo DNA methylation-associated repression with a similar chromatin context to coding genes.40
Andolfo et al41
reported that methylation of the miR-199b-5p promoter can downregulate its expression in the medulloblastomas. Lopez-Bertoni et al42
reported that DNMT-dependent suppression of miRNAs regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. DNMT3b functions as a de novo methyltransferase that establishes new DNA methylation patterns,43
over-expression and hyperactivation of DNMT3b, which can result in hypermethylation of promoter CpG islands and gene silence.44
In the present study, we disclosed that HPostC can induce DNA hypomethylation of the miR-30a promoter and upregulate its expression through inhibiting DNMT3b expression in the aged cardiomyocytes. These data supported the point that DNA hypomethylation contributes to genome instability and transcriptional activation, while DNA hypermethylation leads to gene silence.45
This finding reasonably explained why the inhibition of DNMT3b activity can result in the loss of DNA methylation during miR-30a replication. Our observation provided an additional regulatory pathway of miR-30a expression in HPostC-treated aged cardiomyocytes.
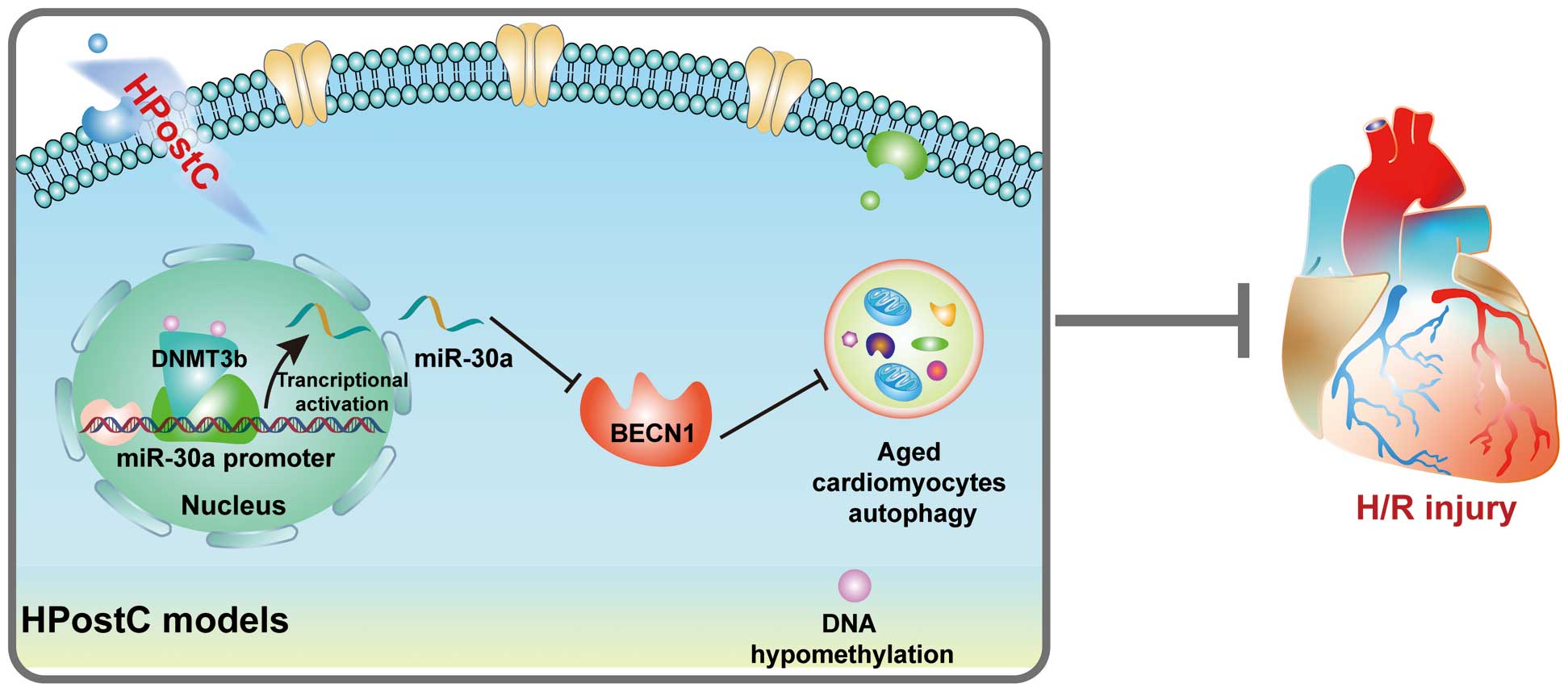
In conclusion, HPostC showed a protective effect on the aged cardiomyocytes from H/R injury via DNMT3b-mediated DNA hypomethylation at the miR-30a promoter and thereby upregulated miR-30a transcription, which accordingly suppressed
BECN1
expression and attenuated autophagy (Figure 6). As a result, the discovery of aberrant DNA hypomethylation of miR-30a during HPostC treatment not only showed a new protective mechanism of IPostC on the aged cardiomyocytes from I/R injury, but also provided a potential target for precise therapy to ameliorate aged myocardial injury from I/R as well.
Conflict of Interests
None.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (No. 81560242, 81560084, 81570452, 81860044, 81870225), Natural Science Foundation of Ningxia Province (No. 2018A0151), Ningxia Science and Technology Leading Talents Project (No. KJT2015019), the Key Research and Development Projects in Ningxia Province (No. 2018BEG02004) and Ningxia High School first-class Disciplines (West China first-class Disciplines Basic Medical Sciences at Ningxia Medical University No. NXYLXK2017B07).
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-19-0915
References
- 1.
Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res 2018; 123: 825–848.
- 2.
Christiansen CF, Thomsen RW, Schmidt M, Pedersen L, Sorensen HT. Influenza vaccination and 1-year risk of myocardial infarction, stroke, heart failure, pneumonia, and mortality among intensive care unit survivors aged 65 years or older: A nationwide population-based cohort study. Intensive Care Med 2019; 45: 957–967.
- 3.
Tian Y, Miao B, Charles EJ, Wu D, Kron IL, French BA, et al. Stimulation of the Beta2 adrenergic receptor at reperfusion limits myocardial reperfusion injury via an interleukin-10-dependent anti-inflammatory pathway in the spleen. Circ J 2018; 82: 2829–2836.
- 4.
Petz A, Grandoch M, Gorski DJ, Abrams M, Piroth M, Schneckmann R, et al. Cardiac hyaluronan synthesis is critically involved in the cardiac macrophage response and promotes healing after ischemia reperfusion injury. Circ Res 2019; 124: 1433–1447.
- 5.
Heusch G, Musiolik J, Gedik N, Skyschally A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 2011; 109: 1302–1308.
- 6.
Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: Role of autophagy paradox and toxic aldehyde. Eur Heart J 2011; 32: 1025–1038.
- 7.
Derghal A, Djelloul M, Azzarelli M, Degonon S, Tourniaire F, Landrier JF, et al. MicroRNAs are involved in the hypothalamic leptin sensitivity. Epigenetics 2018; 13: 1127–1140.
- 8.
Wu H, Liu C, Yang Q, Xin C, Du J, Sun F, et al. MIR145-3p promotes autophagy and enhances bortezomib sensitivity in multiple myeloma by targeting HDAC4. Autophagy, doi:10.1080/15548627.2019.1635380.
- 9.
Huang J, Zhao L, Fan Y, Liao L, Ma PX, Xiao G, et al. The microRNAs miR-204 and miR-211 maintain joint homeostasis and protect against osteoarthritis progression. Nat Commun 2019; 10: 2876.
- 10.
Palma CA, Al Sheikha D, Lim TK, Bryant A, Vu TT, Jayaswal V, et al. MicroRNA-155 as an inducer of apoptosis and cell differentiation in Acute Myeloid Leukaemia. Mol Cancer 2014; 13: 79.
- 11.
Ju S, Liang Z, Li C, Ding C, Xu C, Song X, et al. The effect and mechanism of miR-210 in down-regulating the autophagy of lung cancer cells. Pathol Res Pract 2019; 215: 453–458.
- 12.
Zhang X, Fernandez-Hernando C. miR-33 regulation of adaptive fibrotic response in cardiac remodeling. Circ Res 2017; 120: 753–755.
- 13.
Panico C, Condorelli G. microRNA-132: A new biomarker of heart failure at last? Eur J Heart Fail 2018; 20: 86–88.
- 14.
Zhang C, Liao P, Liang R, Zheng X, Jian J. Epigallocatechin gallate prevents mitochondrial impairment and cell apoptosis by regulating miR-30a/p53 axis. Phytomedicine 2019; 61: 152845.
- 15.
Roman-Gomez J, Agirre X, Jimenez-Velasco A, Arqueros V, Vilas-Zornoza A, Rodriguez-Otero P, et al. Epigenetic regulation of microRNAs in acute lymphoblastic leukemia. J Clin Oncol 2009; 27: 1316–1322.
- 16.
Yang A, Jiao Y, Yang S, Deng M, Yang X, Mao C, et al. Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis 2018; 9: 169.
- 17.
Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, et al. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat Genet 2003; 33: 61–65.
- 18.
Ortiz I, Barros-Filho MC, Dos Reis MB, Beltrami CM, Marchi FA, Kuasne H, et al. Loss of DNA methylation is related to increased expression of miR-21 and miR-146b in papillary thyroid carcinoma. Clin Epigenetics 2018; 10: 144.
- 19.
Vera O, Jimenez J, Pernia O, Rodriguez-Antolin C, Rodriguez C, Sanchez Cabo F, et al. DNA methylation of miR-7 is a mechanism involved in platinum response through MAFG overexpression in cancer cells. Theranostics 2017; 7: 4118–4134.
- 20.
McLendon PM, Davis G, Gulick J, Singh SR, Xu N, Salomonis N, et al. An unbiased high-throughput screen to identify novel effectors that impact on cardiomyocyte aggregate levels. Circ Res 2017; 121: 604–616.
- 21.
Sun X, Ju T, Cummings RD. Differential expression of Cosmc, T-synthase and mucins in Tn-positive colorectal cancers. BMC Cancer 2018; 18: 827.
- 22.
Hu L, Wang J, Zhu H, Wu X, Zhou L, Song Y, et al. Ischemic postconditioning protects the heart against ischemia-reperfusion injury via neuronal nitric oxide synthase in the sarcoplasmic reticulum and mitochondria. Cell Death Dis 2016; 7: e2222.
- 23.
Ni HM, Bockus A, Wozniak AL, Jones K, Weinman S, Yin XM, et al. Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 2011; 7: 188–204.
- 24.
Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal 2011; 14: 2179–2190.
- 25.
Guo R, Hu N, Kandadi MR, Ren J. Facilitated ethanol metabolism promotes cardiomyocyte contractile dysfunction through autophagy in murine hearts. Autophagy 2012; 8: 593–608.
- 26.
Lyko F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat Rev Genet 2018; 19: 81–92.
- 27.
Ong SB, Samangouei P, Kalkhoran SB, Hausenloy DJ. The mitochondrial permeability transition pore and its role in myocardial ischemia reperfusion injury. J Mol Cell Cardiol 2015; 78: 23–34.
- 28.
Cooper HA, Eguchi S. Inhibition of mitochondrial fission as a novel therapeutic strategy to reduce mortality upon myocardial infarction. Clin Sci (Lond) 2018; 132: 2163–2167.
- 29.
Thuny F, Lairez O, Roubille F, Mewton N, Rioufol G, Sportouch C, et al. Post-conditioning reduces infarct size and edema in patients with ST-segment elevation myocardial infarction. J Am Coll Cardiol 2012; 59: 2175–2181.
- 30.
Zhang J, Zhang X, Cui Y, Ferdous M, Cui L, Zhao P. Different postconditioning cycles affect prognosis of aged patients undergoing primary percutaneous coronary intervention. Cardiol J 2018; 25: 666–673.
- 31.
Wang J, Han D, Sun M, Feng J. A combination of remote ischemic perconditioning and cerebral ischemic postconditioning inhibits autophagy to attenuate plasma HMGB1 and induce neuroprotection against stroke in rat. J Mol Neurosci 2016; 58: 424–431.
- 32.
Zhou B, Lei S, Xue R, Leng Y, Xia Z, Xia ZY. DJ-1 overexpression restores ischaemic post-conditioning-mediated cardioprotection in diabetic rats: Role of autophagy. Clin Sci (Lond) 2017; 131: 1161–1178.
- 33.
Wei C, Li H, Han L, Zhang L, Yang X. Activation of autophagy in ischemic postconditioning contributes to cardioprotective effects against ischemia/reperfusion injury in rat hearts. J Cardiovasc Pharmacol 2013; 61: 416–422.
- 34.
Wang L, Lv Y, Li G, Xiao J. MicroRNAs in heart and circulation during physical exercise. J Sport Health Sci 2018; 7: 433–441.
- 35.
Tu Y, Wan L, Fan Y, Wang K, Bu L, Huang T, et al. Ischemic postconditioning-mediated miRNA-21 protects against cardiac ischemia/reperfusion injury via PTEN/Akt pathway. PLoS One 2013; 8: e75872.
- 36.
He B, Xiao J, Ren AJ, Zhang YF, Zhang H, Chen M, et al. Role of miR-1 and miR-133a in myocardial ischemic postconditioning. J Biomed Sci 2011; 18: 22.
- 37.
Fu XT, Shi YH, Zhou J, Peng YF, Liu WR, Shi GM, et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett 2018; 412: 108–117.
- 38.
Peng Y, Miao H, Wu S, Yang W, Zhang Y, Xie G, et al. ABHD5 interacts with BECN1 to regulate autophagy and tumorigenesis of colon cancer independent of PNPLA2. Autophagy 2016; 12: 2167–2182.
- 39.
Maejima Y, Kyoi S, Zhai P, Liu T, Li H, Ivessa A, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med 2013; 19: 1478–1488.
- 40.
Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, et al. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat Commun 2014; 5: 4497.
- 41.
Andolfo I, Liguori L, De Antonellis P, Cusanelli E, Marinaro F, Pistollato F, et al. The micro-RNA 199b-5p regulatory circuit involves Hes1, CD15, and epigenetic modifications in medulloblastoma. Neuro Oncol 2012; 14: 596–612.
- 42.
Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero-Cazares H, Eberhart CG, et al. DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene 2015; 34: 3994–4004.
- 43.
Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, et al. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015; 520: 243–247.
- 44.
Joensuu EI, Nieminen TT, Lotsari JE, Pavicic W, Abdel-Rahman WM, Peltomaki P. Methyltransferase expression and tumor suppressor gene methylation in sporadic and familial colorectal cancer. Genes Chromosomes Cancer 2015; 54: 776–787.
- 45.
Unnikrishnan A, Freeman WM, Jackson J, Wren JD, Porter H, Richardson A. The role of DNA methylation in epigenetics of aging. Pharmacol Ther 2019; 195: 172–185.