I. General Remarks on Amyloidosis
1. Concept and Classification of Amyloidosis
1.1 Disease Concept and Classification of Amyloidosis
Amyloidoses are diseases in which misfolding proteins form β-sheet structured amyloid fibrils, that are deposited in multiple organs throughout the body, leading to organ dysfunction. Amyloidosis is broadly divided into systemic amyloidosis, in which amyloid is deposited in multiple organs, and localized amyloidosis, in which amyloid is localized in specific organs. It is classified further by precursor proteins and their corresponding clinical disease types4
(Table 5).
Table 5.
Classification of Amyloidosis
| |
Fibril protein |
Precursor protein |
Clnical disease name |
| Systemic amyloidosis |
| Hereditary |
ATTRv |
Transthyretin, variants |
Hereditary transthyretin amyloidosis |
| Agel |
Gelsolin, variants |
Hereditary gelsolin amyloidosis |
| AApoAI |
Apolipoprotein A-I, variants |
Hereditary apolipoprotein A-I amyloidosis |
| AApoAII |
Apolipoprotein A-II, variants |
Hereditary apolipoprotein A-II amyloidosis |
| AApoCII |
Apolipoprotein C-II, variants |
Hereditary apolipoprotein C-II amyloidosis |
| AApoCIII |
Apolipoprotein C-III, variants |
Hereditary apolipoprotein C-III amyloidosis |
| ALys |
Lysozyme, variants |
Hereditary lysozyme amyloidosis |
| AFib |
Fibrinogen α, variants |
Hereditary fibrinogen amyloidosis |
| ACys |
Cystatin C, variants |
Hereditary cystatin C amyloidosis |
| Aβ2M |
β2-Microglobulin, variants |
Hereditary β2-microgloblin amyloidosis |
| APrP |
Prion, variants |
Prion protein systemic amyloidosis |
| Non-hereditary |
ATTRwt |
Transthyretin, wild type |
Wild-type transthyretin amyloidosis |
| AL |
Immunoglobulin light chain |
AL amyloidosis |
| AH |
Immunoglobulin heavy chain |
AH amyloidosis |
| AA |
Serum amyloid A |
AA amyloidosis |
| Aβ2M |
β2-Microglobulin, wild type |
Dialysis-related amyloidosis |
| Localized amyloidosis |
| Brain |
Aβ |
Amyloid-β precursor protein
(wild-type, variants) |
Alzheimer disease, Cerebral amyloid angiopathy
(CAA) |
| APrP |
Prion protein (wild-type, variants) |
Creutzfeldt-Jakob disease |
| ACys |
Cystatin C, variants |
Hereditary cerebral amyloid angiopathy |
| ABri |
ABri precursor protein, variants |
Familial British dementia |
| ADan |
ADan precursor protein, variants |
Familial Danish dementia |
| Endocrine |
ACal |
(Pro)calcitonin |
Associated with medullary thyroid cancer |
| AIAPP |
Amylin |
Associated with type II DM |
| AANP |
ANP |
Isolated atrial amyloidosis |
| APro |
Prolactin |
Prolactin-producing tumor |
| Cornea |
ALac |
Lactoferrin |
Corneal amyloidosis |
| AKer |
Kerato-epithelin |
Corneal amyloidosis |
| Others |
AMed |
Lactadherin |
Aortic medial amyloidosis |
| AIns |
Insulin |
Insulin-derived amyloidosis (iatrogenic) |
| AL |
Immunoglobulin light chain |
Localized nodular amyloidosis |
(Modified from Benson MD et al. 20184)
More than 30 amyloid precursor proteins have been identified. Of these, amyloid fibrils, which are formed by immunoglobulin light chains, TTR, or amyloid A protein (AA) accumulate in the heart and cause cardiac dysfunction (Table 6). ATTR can be either ATTRv amyloidosis (formerly called FAP) with a pathogenic mutation in the
TTR
gene, or systemic ATTRwt amyloidosis without a mutation (formerly called SSA).8
AA amyloidosis is associated with chronic inflammatory diseases such as rheumatoid arthritis, vasculitis syndrome, and autoimmune diseases, and mainly presents with renal disorders such as proteinuria and renal failure, but rarely with cardiac involvement.9
Table 6.
Classification of Cardiac Amyloidosis
| |
Precursor
protein |
Underlying
disorder |
Organ involvement |
Treatment |
| Heart |
Kidneys |
Liver |
PN (AN) |
Other |
| AL |
Monoclonal
immunoglobulin
light chain |
Plasma cell
dyscrasia |
+++ |
+++ |
++ |
+ (+) |
Soft tissue,
gastrointestinal |
Chemotherapy or
ASCT |
| ATTRwt |
Wild-type TTR |
Aging |
+++ |
+ |
− |
+ |
Carpal tunnel
syndrome |
TTR stabilizer |
| ATTRv |
Mutant TTR |
Mutations in TTR
gene |
++ |
+ |
− |
+++
(+++) |
Gastrointestinal,
retina |
Liver transplant
TTR stabilizer
Oligonucleotide
therapy |
| AA |
SAA |
Inflammatory
disorders (RA, JIA) |
−/+ |
+++ |
+ |
− |
Gastrointestinal |
Suppression of
inflammation |
(Modified from Wechalekar AD, et al. 20168)
1.2 Concept of Cardiac Amyloidosis (CA)
CA is a clinical disorder in which the interstitial deposition of amyloid fibrils causes morphological and functional abnormalities of the heart. As mentioned above, CA is divided into three main types, AL, ATTRv, and ATTRwt amyloidosis, which share common features in terms of clinical signs and laboratory findings. In general, CA presents with heart failure mainly due to diastolic dysfunction with cardiac hypertrophy, in addition to left ventricular (LV) systolic dysfunction, atrioventricular (AV) conduction disorder, atrial fibrillation (AF), and fatal arrhythmias. Prognosis depends on the extent of cardiac involvement and the subtype of amyloid precursor protein. In recent years, effective treatments and drugs have been developed for AL and ATTR. The clinical practical guideline for cardiomyopathy compiled by the JCS/Japan Heart Failure Society also classifies CA as one of the secondary cardiomyopathies clearly associated with a specific cause or systemic disease that needs to be differentiated, and emphasizes the importance of appropriate diagnosis.6
1.2.1 Systemic AL Amyloidosis
AL amyloidosis is a plasma cell dyscrasia characterized by the pathologic production of amyloid fibrils formed by misfolded monoclonal light chains that are deposited in tissues and cause organ dysfunction. Although the major organs involved are the kidney, heart, liver, gastrointestinal tract, and peripheral nerves, any organ throughout the body can be damaged; AL amyloidosis can damage multiple organs, or only a single organ (e.g., heart, kidney, liver). Monoclonal immunoglobulin light chains are mainly produced from plasma cells in the bone marrow. Amyloidosis caused by abnormal production of free light chains from bone marrow plasma cells that does not meet the conditions for multiple myeloma is called “primary AL amyloidosis”, and amyloidosis associated with B-cell malignancy such as multiple myeloma is called “secondary AL amyloidosis”. In each case, monoclonal immunoglobulin-producing cells are the treatment target.
1.2.2 Systemic ATTRwt Amyloidosis (Formerly Called Senile Systemic Amyloidosis [SSA])
ATTRwt amyloidosis is caused by wild-type TTR, which mainly damages heart, tendon, and ligament tissue (e.g., carpal tunnel, ligamentum flavum), the kidney, thyroid, peripheral nerves, and lungs. It is common in men over 60 years of age. Although aging is thought to be involved in the pathogenesis, the underlying mechanism remains unclear. In Japan, this disease is considered to be misdiagnosed frequently. TTR tetramer stabilizer has been reported to improve the prognosis of cardiac involvement in patients with ATTR.3
1.2.3 Hereditary ATTR (ATTRv Amyloidosis; Formerly Called Familial Amyloid Polyneuropathy [FAP])
ATTRv amyloidosis is a hereditary disease in which TTR forms amyloid that gets deposited in the interstitium of tissues due to a mutation in the
TTR
gene, causing damage to the nerves, heart, digestive tract, kidneys, and eyes. In Japan, there are large numbers of patients with ATTRv amyloidosis (V30M variant) in Kumamoto and Nagano Prefectures. Although ATTRv amyloidosis is an autosomal dominant disease, about half of the patients in areas other than Kumamoto and Nagano Prefectures show no clear family history. The age of onset (20–70 years or older) and symptoms vary. Early diagnosis is important because the effects of various treatments (e.g., liver transplantation, TTR stabilizers, oligonucleotide therapy) are expected to be seen in the early stage of the disease.
2. AL Amyloidosis
Amyloidoses are protein conformational diseases caused by the misfolding and aggregation of autologous proteins that are deposited in tissues in the form of amyloid fibrils. Amyloid light-chain (AL) amyloidosis is a multi-system disorder caused by a malignant plasma cell clone that results in insoluble fibrillary deposition. AL amyloidosis is seen in 10 to 15% of patients who are diagnosed with multiple myeloma (MM).10
MM is defined as plasma cells ≥10% with serum monoclonal (M-) protein ≥3 g/dL or 24-h urinary M-protein ≥500 mg. Patients are diagnosed as having symptomatic MM when they show any of the following myeloma-defining events (MDEs): hypercalcemia, renal dysfunction, anemia, or bone lesions. AL amyloidosis not associated with MM is called primary AL amyloidosis. The epidemiology and prognosis of primary AL amyloidosis are discussed.
AL amyloidosis is known to be associated with MM, but the characteristics of plasma cells were reported to be different between primary AL amyloidosis and MM.11,12
Translocation t(11;14), which is known to be found in about 15% of MM cases, is found more frequently (about 50%) in patients with AL amyloidosis.13
According to these findings, the treatment of AL amyloidosis should be considered differently from that of MM.
Epidemiologic data on primary AL amyloidosis are limited. An analysis in the United States of America between 2007 and 2015 showed that the incidence was 9.7 to 14.0 cases per million person-years, and the prevalence increased significantly from 15.5 in 2007 to 40.5 cases per million in 2015, an annual percentage change (APC) of 12%.14
It is suspected that more patients can survive with the advances in diagnosis and treatment. In this report, prevalent patients had a mean age of 63 years, and 55% were male. In Japan, the prevalence of patients with amyloidosis (not only AL amyloidosis, but also ATTR amyloidosis) was reported as 6.1 per million persons using the data on intractable disease of the Japanese Ministry of Health and Welfare. There is little information available concerning the status of AL amyloidosis, such as its incidence and the demographic features of AL amyloidosis patients in Japan.
The prognosis of patients with AL amyloidosis is highly dependent on the involved organs and the severity of organ damage. The revised Mayo Clinic staging system is frequently used for predicting prognosis.15
In this staging system, patients are assigned a score of 1 for each difference in free light chain (FLC; difference between involved and uninvolved FLC) ≥18 mg/dL (180 mg/L), cTnT ≥0.025 ng/mL, and NT-proBNP ≥1,800 pg/mL, creating stages I to IV with scores of 0 to 3 points. The median OS from diagnosis of patients with stages I, II, III, and IV was 94.1, 40.3, 14, and 5.8 months, respectively (Figure 1).15
3. ATTRwt Amyloidosis (Formerly Called SSA)
3.1 Concept of ATTRwt Amyloidosis
ATTRwt amyloidosis is caused by the deposition of wild-type TTR-derived amyloid fibrils. TTR is stable in the bloodstream if it forms tetramer, but upon aging, it becomes unstable, develops a misfolded monomer, and is ultimately deposited as amyloid fibrils. Because ATTRwt amyloidosis is often diagnosed in older patients, it has traditionally been known as SSA. However, the term ATTRwt amyloidosis is now recommended because amyloidosis was recently classified according to a precursor protein.16
3.2 Pathophysiology of ATTRwt Amyloidosis
In 1990, TTR was reported as a precursor protein of ATTR.17
TTR, which is mainly produced and secreted from the liver, binds and transfers thyroid hormone and retinol-binding protein with vitamin A. TTR is stable in circulation as long as it forms a tetramer, but in the case of aging, TTR becomes unstable, develops a misfolded monomer, and becomes a substrate of amyloid fibrils. Although the precise mechanism is unknown, posttranslational biochemical changes in hepatic TTR or chaperone protein are suspected as a cause of instability of the TTR tetramer.18,19
TTR-derived amyloid fibril is deposited in various tissues, such as the heart, lung, kidney, intestinal tract, adipose tissue, joints, and ligaments. However, the joints, ligaments, and heart are the predominant tissues in which symptoms become apparent (e.g., carpal tunnel syndrome, spinal canal stenosis, ventricular hypertrophy, arrhythmia, heart failure).20
Bilateral carpal tunnel syndrome is one of the earliest symptoms that appears in patients with ATTRwt amyloidosis, usually before cardiac symptoms when they are in their 50 s to 70 s. In patients undergoing carpal tunnel release surgery (median age, 68 years), 10.2% had a positive biopsy for amyloid and 2% had cardiac involvement.21
A study that compared 56,032 patients who underwent surgical treatment for carpal tunnel syndrome and a sex- and age-matched cohort from the general population demonstrated that carpal tunnel syndrome was associated with a higher incidence of heart failure.22
It was also reported that the carpal tunnel syndrome symptoms appeared about 7 years before cardiac symptoms.23
These results suggest that, in contrast to AL amyloidosis, ATTRwt amyloidosis develops over the long term.
3.3 Epidemiology of ATTRwt Amyloidosis
The deposition of TTR-derived amyloid fibrils progresses gradually with aging. By analyzing autopsy samples, amyloid deposition was detected in about 25% and 37% of those over 80 and 90 years of age, respectively.24,25
In Japan, wild-type TTR-derived amyloid fibrils were detected in 12% of autopsy samples.26
If the included patients were limited to those with heart failure with preserved ejection fraction (HFpEF), amyloid fibrils were documented in about 40% of those over 80 years of age.27
However, these data do not reflect the precise prevalence of ATTRwt amyloidosis because most of these patients had not been diagnosed before death because of the small amounts of deposition and lack of symptoms.
It has been shown that scintigraphy using a tracer with a high affinity for calcium has a high positive predictive value for ATTR based on a comparison of histology samples.1
Among patients aged over 60 years admitted due to HFpEF and LV hypertrophy (≥12 mm), 13.3% showed moderate-to-severe uptake on 99 mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99 mTc-DPD) scintigraphy.28
ATTRwt amyloidosis, diagnosed using 99 mTc-PYP scintigraphy, is prevalent in 16% of patients with severe calcific aortic stenosis undergoing transcatheter aortic valve implantation.29
It has been reported that the prevalence of ATTRwt amyloidosis does not differ by race.30
On the other hand, a remarkable sex difference has been reported. Significantly higher numbers of male compared with female patients are diagnosed with ATTRwt amyloidosis by histology or 99 mTcPYP scintigraphy.31–33
However, the mechanism underlying this sex difference remains unknown.
3.4 Diagnosis of ATTRwt Amyloidosis
Conduction system disturbances, such as complete left bundle branch block and AF, are more frequently observed in patients with ATTRwt amyloidosis compared with those with AL amyloidosis. Echocardiography and cardiac magnetic resonance imaging (MRI) have revealed that the extent of cardiac hypertrophy is more robust in patients with ATTRwt amyloidosis.31,34,35
The value of high-sensitive troponin T is usually higher in patients with ATTRwt amyloidosis than in those with hypertensive heart disease or hypertrophic cardiomyopathy, but lower than in those with AL amyloidosis.36
To confirm diagnosis, the detection of TTR-derived amyloid fibrils in biopsy specimens is necessary. In addition, genetic testing is needed to differentiate ATTRwt amyloidosis and ATTRv amyloidosis. Because cardiac amyloid fibril deposition can be detected in patients with ATTRwt cardiomyopathy with high probability, endomyocardial biopsy is regarded as the gold standard for confirming the diagnosis.37
However, due to the relatively invasive procedure, endomyocardial biopsy cannot be performed for all suspected patients. A diagnosis of ATTRwt amyloidosis is acceptable if the echocardiography and cardiac MRI (CMR) are highly suggestive of ATTRwt amyloidosis and amyloid deposition is detected in other tissues (see “
Chapter I, Section 5. Diagnostic Criteria
”).
3.5 Prognosis of ATTRwt Amyloidosis
The prognosis of ATTRwt amyloidosis is better than that of AL amyloidosis, but it is not so good compared with heart failure with other etiologies. The survival of patients with ATTRwt amyloidosis was thought to be over 5 years,38,39
but recent reports suggest that the median survival after diagnosis is 43–47 months.31,33
In 2018, tafamidis, a TTR stabilizer, was reported to be associated with reductions in all-cause mortality and cardiovascular-related hospitalizations, and to reduce the decline in functional capacity and quality of life (QOL) compared with placebo in patients with ATTR cardiomyopathy.3
Based on these results, tafamidis has been approved for patients with ATTR cardiomyopathy in Japan since 2019. Further evaluation is needed to evaluate the effect of tafamidis on the prognosis of ATTR cardiomyopathy in clinical practice.
4. ATTRv Amyloidosis (Formerly Called TTR-FAP)
4.1 Disease Concept and Pathology
ATTRv amyloidosis is the most frequent and representative form of autosomal dominant heritable systemic amyloidosis.4,40
As treatment for ATTRv amyloidosis is different from that for ATTRwt amyloidosis, it is very important to distinguish the two. Furthermore, when a single case of ATTRv amyloidosis is diagnosed, an active search should be considered because there can be multiple carriers of the mutant
TTR
gene in the patient’s family, and early diagnosis and treatment are possible. “
Chapter II, Section 10. Genetic Testing and Counseling
” details the genetic testing and counselling available for this condition.
TTR, the causative molecule of this disease, is a plasma protein primarily produced in the liver and secreted into the blood. Other known sites of TTR production include the choroid plexus, retinal pigment epithelium of the eyes, and α cells of the pancreatic islets of Langerhans. Although the
TTR
gene encodes 147 amino acids, the first 20 comprise a signaling peptide that is cut off prior to being expelled from the cell, and the remaining 127 comprise the TTR found in the blood. It is recommended that the mutation sites of the secretory TTR (127 amino acid residues) and the TTR coding sequence (147 amino acid residues) be written together (e.g., Val30Met [p.Val50Met] to describe the TTR mutation that causes this condition.4
TTR forms a tetramer in the blood and is responsible for the transport of thyroid hormone (thyroxine: T4) and vitamin A (retinol) through the binding of retinol-binding protein (RBP).
Over 150 types of mutants have been reported for the
TTR
gene, the majority of which are amino acid mutations by means of single residue replacement.41
The Val30Met (p.Val50Met) mutation, where the 30th amino acid (50th amino acid in the TTR coding sequence) residue, valine, is replaced with a methionine residue, was the first to be reported, and has been observed most frequently.40
With respect to ATTRv amyloidosis, which is caused by the same Val30Met (p.Val50Met) mutant, the endemic areas in Japan (Kumamoto and Nagano Prefectures) have reported numerous instances of early onset, and the non-endemic areas (other regions) numerous instances of late onset. In addition to the age at onset, differences have been reported in regard to sex, presence/absence of a family history the disease, and symptoms (Table 7).42–44
Many other TTR mutations (non-Val30Met [p.Val50Met]) have been reported, and cardiac symptoms are relatively frequent first symptoms (Table 7).44
Approximately 3.4% of African-Americans are known to have the Val122Ile (p.Val142Ile) TTR variant and be at increased risk of developing TTR amyloid cardiomyopathy (ATTR-CM).45
Table 7.
Clinical Features of ATTRv Amyloidosis in Japan
| |
Val30Met (p.Val50Met) type
(endemic areas) |
Val30Met (p.Val50Met) type
(non-endemic areas) |
Non-Val30Met (p.Val50Met) type
(non-endemic areas) |
| Onset age |
Generally early (20–60 years old) |
Generally late (35–90 years old) |
Varied (20–85 years old) |
| Family history of the disease |
High (80–100%) |
Low (about 30%) |
Low (about 30%) |
| Sex |
Equal frequency for both sexes |
Male-dominant (male: about 80%) |
Male-dominant (male: about 60%) |
| Survival after the onset |
About 10 years |
About 7 years |
About 10 years |
Types of ATTR amyloid fibrils
deposited in patients |
Type B |
Type A |
Type A |
| 99mTc PYP scintigraphy |
Negative |
Positive |
Positive |
| Initial symptoms |
|
|
|
| Polyneuropathy |
About 60% |
About 80% |
About 30% |
| Autonomic symptoms |
30–60% |
2–10% |
About 25% |
| Cardiac symptoms |
0% |
2–4% |
About 35% |
| Carpal tunnel syndrome |
0–10% |
About 5% |
0% |
| Renal symptoms |
0% |
0–2% |
0% |
| Ocular symptoms |
0% |
6–13% |
About 10% |
The fact that the TTR tetramer destabilizes and dissociates into monomers in the event of a
TTR
gene mutation is important for the process of amyloid formation.46
In a basic study involving various TTR mutants, it was reported that the greater the instability of TTR according to the type of mutation, the more common are symptoms such as oculo-leptomeningeal and peripheral nervous system symptoms, while the greater the stability of TTR, the greater is the tendency to experience cardiac symptoms.47
Biochemical analyses of amyloid fibrils that deposit in the tissues in this condition have shown that there are cases in which both full-length TTR (127 amino acid residues) and C-terminus TTR fragments are detected (Type A), as well as cases in which only full-length TTR is detected (Type B).48
The relationship between the pathogenesis of this condition and TTR and C-terminus fragmentation has also been discussed.
4.2 Epidemiology
Previously, large families of ATTRv amyloidosis (Val30Met [p.Val50Met] type) were thought to exist only in Portugal, Japan (Kumamoto and Nagano Prefectures), and Sweden, but in recent years, improved diagnostic techniques and widespread knowledge about the disease has led to more cases of ATTRv amyloidosis being reported around the world. Globally, it is estimated that approximately 10,000 people have this condition.49
Although many cases with the Val30Met (p.Val50Met) mutant have been reported in Japan, there have also been numerous reports of other family types of ATTRv amyloidosis caused by other TTR mutants (Figure 2).44
The results of an epidemiological investigation by the MHLW, titled “Research Team on Amyloidosis,” estimated that there were approximately 830 people in Japan who had the condition.50
As there may also be many undiagnosed cases, we believe that the actual number of such patients is even higher.
4.3 Symptoms
ATTRv amyloidosis exhibits various systemic organ symptoms, such as peripheral and autonomic nerve, cardiac, gastrointestinal, kidney, ocular, and central nervous system symptoms. During the advanced stage of the disease, patients tend to present with respiratory muscle paralysis, severe orthostatic hypotension, heart failure, fatal arrhythmia, nephrotic syndrome, kidney failure, protein-losing gastroenteropathy, and severe glaucoma, in addition to paralysis of the extremities. These details are described in “
Chapter II, Section 1.2
”. It is important to suspect this condition when observing peripheral neuropathy and heart failure, which are difficult to explain from other diseases and pathologies.
4.4 Prognosis
When the condition is advanced, it can often lead to death as a result of severe peripheral neuropathy and cardiac, renal, and respiratory disorders. When left untreated, the mean life expectancy from onset for ATTRv amyloidosis is approximately 10 years (early-onset Val30Met [p.Val50Met]) in endemic areas,51
and approximately 7 years for late-onset disease (late onset Val30Met (p.Val50Met) in non-endemic areas.43
Even for ATTRv amyloidosis due to non-Val30Met (p.Val50Met) mutants, the mean life expectancy after onset is approximately 10 years if left untreated.52
Although the mean life expectancy for ATTRv amyloidosis is almost the same in the US and Europe,53
it is even shorter, at 2–3 years, in cases for which the main symptoms are cardiac symptoms,54
which may take a long time to be diagnosed because they lack subjective symptoms at the early stages of the disease.
4.5 Diagnosis
This condition is diagnosed by identifying the clinical signs specific to the condition, detecting TTR-derived amyloid deposition, and identifying pathogenic mutations in the
TTR
gene. Many cases of late-onset disease with no family history have been reported in non-endemic areas (other than Kumamoto and Nagano Prefectures). In many such cases, it takes several years or more from the time of onset for the condition to be diagnosed.44
The items in
Chapter II
describe in detail the images and biomarkers thought to be useful for diagnosing and evaluating the pathology of amyloid cardiomyopathy. Although nerve conduction tests are often conducted to evaluate the extent of nerve damage, they are not suitable for evaluating small fiber neuropathy. The evaluation of nerve density in the epidermis using skin punch biopsy55
and of simple sweat function56
is actively used to evaluate small fiber neuropathy (see “
Chapter I, Section 5. Diagnostic Criteria
”).
5. Diagnostic Criteria
In the diagnosis of amyloidosis, it is important not only to detect amyloid deposition histologically, but also to confirm clinical findings caused by organ damage due to amyloid deposition. Therefore, to diagnose amyloidosis, it is necessary to first suspect this disease from the clinical symptoms caused by organ damage, distinguish it from other diseases based on supporting laboratory findings or imaging tests, and determine the final diagnosis. Especially for the diagnosis of CA, since many nonspecific clinical findings, such as AF, occur in older individuals, regardless of the background of amyloidosis, a comprehensive assessment of clinical and laboratory findings suggesting amyloid deposition in the heart is required. It is necessary to determine amyloid deposition in tissues histologically. Clinical signs or laboratory findings due to each type of amyloidosis are present. Next, using specific antibodies against individual precursor proteins (κ chain, λ chain, TTR, AA, etc.), immunohistochemical staining is performed to categorize the disease type. For diagnosing AL, it is important to test blood/urine M protein and the free light-chain ratio to confirm the clonal proliferation of plasma cells in bone marrow. In ATTRv amyloidosis, it is necessary to examine
TTR
gene mutations using genetic testing. In recent years, evidence that 99 mTc-PYP is useful for the diagnosis of ATTR has been accumulated,57,58
and diagnostic criteria using noninvasive imaging without biopsy have been proposed.1
Because it is sometimes difficult to make a definitive diagnosis and disease typing, each disease type has a diagnostic category of either “definite” or “probable”.
Few official guidelines in Japan or overseas provide clear diagnostic criteria for systemic amyloidosis. For AL amyloidosis, the diagnostic criteria published by the International Myeloma Working Group are widely used as international criteria.59
In Japan, the “Amyloidosis Clinical Practice Guidelines 2010”,60
which is published by a research group on amyloidosis that conducts a project for overcoming intractable diseases funded by the MHLW, details the diagnostic criteria for AL, ATTR, AA, Aβ2
M, and cerebral amyloidosis. However, given the recent advances in diagnostic technology and changes in the disease concepts, and the launch of new therapeutic agents, the revision of the diagnostic criteria for amyloidosis has become an unmet medical need. Therefore, the Japanese Research Group on Amyloidosis has revised the diagnostic criteria for AL, ATTRwt, and ATTRv amyloidosis based on a consensus of each society (http://amyloidosis-research-committee.jp). The following guideline describes the latest diagnostic criteria reflecting these contents.
5.1 Common Items of Diagnostic Criteria for Each Type of Amyloidosis
A, B, and C are common to each disease type.
A. Clinical signs and laboratory findings
Clinical signs or laboratory findings due to each type of amyloidosis are present.
B. Pathohistological findings
In biopsied tissue samples, there are amyloid deposits exhibiting positive staining with Congo-red under a light microscope and show apple-green birefringence under a polarizing microscope.
C. Amyloid typing
Amyloid deposits show positive staining for each type of precursor protein.
Although each item included in “A” is characteristic of each type of disease, it is not a finding specific to amyloidosis, and is considered to be an entry point for diagnosis. The findings of cardiac involvement are common to each disease type, as noted
Table 8.
Table 8.
Clinical and Laboratory Findings Suggesting Cardiac Amyloidosis (CA)
| Clinical signs |
Laboratory findings |
Symptoms of heart
failure
(e.g., shortness of
breath, edema),
dizziness, and
syncope |
Atrial fibrillation, conduction system disorder (e.g., atrioventricular block, bundle branch block, intraventricular conduction
disorder), ventricular arrhythmia, low voltage in limb leads, QS pattern in precordial leads (V1–3), ventricular wall
thickening (including right ventricle), atrial septal thickening, ventricular diastolic dysfunction (restrictive), granular
sparkling appearance, pericardial effusion, valve thickening, reduction in longitudinal strain at the base of left ventricle
(apical sparing), elevated B-type natriuretic peptides (BNP) and N-terminal pro-BNP, elevated cardiac troponin T/I,
global diffuse myocardial late gadolinium enhancement in the subendocardial layers on cardiac magnetic resonance
imaging, elevated native T1 and extracellular volume fraction in T1 mapping |
Since the diagnostic criteria are mutually exclusive for each type, differential diagnosis is essential. The diagnosis flowchart is shown in
Figure 32
(“
Chapter IV. CA Diagnostic Algorithm
”).
5.2 Diagnostic Criteria for Systemic AL Amyloidosis
A. Clinical signs and laboratory findings
Clinical signs or laboratory findings due to each type of amyloidosis are present (Table 9).
Table 9.
Clinical Signs and Laboratory Findings Suspecting AL Amyloidosis
| |
Clinical symptoms |
Laboratory findings |
| Cardiac amyloidosis |
See Table 8 |
| Renal amyloidosis |
Edema, weight gain |
Proteinuria (>0.5 g/day, mainly albumin)
Increased serum creatinine, decreased eGFR, increased
urinary NAG, increased urinary β2-microglobulin |
| Hepatic amyloidosis |
Hepatomegaly |
Maximum liver longitudinal diameter >15 cm (excluding
heart failure), increased alkaline phosphatase (>1.5
times the upper limit of normal) |
Peripheral nerve
amyloidosis |
Small fiber predominant polyneuropathy (a decline mainly
in pain and temperature sensations in the extremities) |
Nerve conduction abnormalities due to axonal disorders |
Autonomic
amyloidosis |
Orthostatic hypotension, diarrhea, constipation, dysuria |
|
Gastrointestinal
amyloidosis |
Melena, nausea, loss of appetite, intestinal obstruction,
malabsorption syndrome |
|
Tendon/ligament
amyloidosis |
Carpal tunnel syndrome (pain and numbness of the
hand) |
A delay in conduction latencies at wrist in nerve
conduction tests |
| Joint amyloidosis |
Shoulder pad sign, joint swelling |
|
| Tongue amyloidosis |
Macroglossia |
|
Cutaneous
amyloidosis |
Scleroderma-like thickening, nodules, purpura |
|
Amyloidosis of other
organs |
Hard swelling of the thyroid gland, salivary glands, lymph
nodes, etc., claudication (due to vascular amyloid),
myopathy (pseudohypertrophy) |
Pattern of diffuse interstitial lung disease on computed
tomography |
• B-cell malignancies such as multiple myeloma should be excluded.
• The underlined text in the table shows the indicators of major organ damage described in the International Consensus Opinion.61 In this Consensus Opinion, the index of CA is “thickness of the ventricular septum and posterior wall (>12 mm)”.
• AL amyloidosis is often negative on 99 mTc-pyrophosphate scintigraphy.
• Monoclonal gammopathy of renal significance (MGRS) is due in part to renal amyloidosis.
B. Pathohistological findings
In biopsied tissue samples, there are amyloid deposits exhibiting positive staining with Congo-red under a light microscope and showing apple-green birefringence under a polarizing microscope are observed (Note 1).
C. Amyloid typing
Amyloid deposits show positive staining for immunoglobulin light chains (Note 2).
D. M protein
M proteins are detected in blood or urine by immunoelectrophoresis, immunofixation, or free light chain (Note 3).
E. Differential diagnosis
Other conditions that may cause the clinical signs and laboratory findings of “A” are excluded. Especially in the case of CA, since ATTRwt amyloidosis with monoclonal gammopathy of undetermined significance cannot be ruled out, ATTRwt should be ruled out using the ATTRwt amyloidosis diagnostic criteria/diagnosis flowchart.
<Diagnosis Category>
Definite:
At least one item of “A” and +B+C+D+E. Or, two or more items of “A” and +B+C+E. However, when the relevant item of “A” is in the column of heart, kidney, or liver, and B+C+E is satisfied, the case is diagnosed as systemic, even if only one organ is damaged.
Probable:
At least one item of “A” and +B+D+E.
*It is difficult to stain with immunohistochemistry using specific antibodies against κ- or λ-light chain. Therefore, even if no clear result is acquired in immunostaining for immunoglobulin light chain, a “Probable” diagnosis is established assuming a case in which treatment should be started immediately after ruling out ATTR.
(Note 1) Biopsy samples can be obtained from the abdominal wall fat, labial salivary glands, digestive tract, bone marrow, etc., and do not necessarily need to be collected from damaged organs. If there are no symptoms with only gastrointestinal lesions, localized gastrointestinal amyloidosis may be present. The occurrence/exacerbation of symptoms such as melena, diarrhea, and constipation needs to be monitored regularly. Localized AL amyloidosis is often asymptomatic or without M protein, so the diagnosis is confirmed by B+C.
(Note 2) For coexisting cases of monoclonal gammopathy of undetermined significance and ATTRwt amyloidosis, it is necessary to prove the type of immunoglobulin light chain that matches the type of M protein in the Congo red-positive site. ALκ or ALλ (+), ATTR (−), and AA (−) should be confirmed by immunostaining, or the amyloid precursor protein should be identified using laser microdissection (LMD) and liquid chromatography-tandem mass spectrometry (LC-MS/MS) in biopsy tissue samples. If it is difficult to perform the test at your own facility, an analysis request can be made to the Research Group on Amyloidosis (http://amyloidosis-research-committee.jp/).
(Note 3) For the detection of M protein, it is recommended to examine serum immunofixation, serum free light chain (FLC), and urine immunofixation. Immunofixation is more sensitive than immunoelectrophoresis. Serum FLC should be assessed based on the difference between elevated pathological light chain-type and non-elevated light chain-type (difference FLC: dFLC) rather than by the absolute values of k and l chains or their ratio. Especially when dFLC ≥50 mg/L, it is useful also as evaluation of treatment effects.
5.3 Diagnostic Criteria for Systemic ATTRwt Amyloidosis
A. Clinical signs and laboratory findings
Clinical signs or laboratory findings due to each type of amyloidosis are present (Table 10).
Table 10.
Clinical Symptoms and Laboratory Findings Suggesting ATTRwt Amyloidosis
| |
Clinical symptoms |
Laboratory findings |
| Cardiac amyloidosis |
See Table 8 |
Peripheral nerve
amyloidosis |
Small fiber predominant polyneuropathy (a decline mainly
in pain and temperature sensations in the extremities) |
Decreased intraepidermal nerve fiber density in skin
biopsy |
Tendon and ligament
amyloidosis |
Carpal tunnel syndrome (numbness and pain of the
hand), spinal canal stenosis (lumbago, gait disturbance),
tendon rupture |
A delay in conduction latencies at wrist in nerve
conduction tests, spinal MRI |
• If an atypical case is suspected, the above findings are not applicable.
B. Pathohistological findings
In biopsied myocardium or other tissues, there are amyloid deposits exhibiting positive staining with Congo-red under a light microscope and showing apple-green birefringence under a polarizing microscope are observed (Note 1).
C. Amyloid typing
Amyloid deposits show positive staining for TTR (Note 2).
D. Scintigraphy
Intensely diffuse myocardial uptake on 99 mTc-PYP scintigraphy is confirmed (Note 3).
E. Genetic testing
No pathogenic mutations involve amino acid alterations in the
TTR
gene.
F. M protein is not detected (Note 4).
G. Differential diagnosis
1. Localized ATTRwt amyloidosis, which is limited to tendon/ligament tissues, should be excluded.
2. Other diseases that cause clinical signs or laboratory findings of “A” should be excluded. However, it should be noted that ATTRwt amyloidosis can be complicated with other diseases causing cardiac hypertrophy.
<Diagnosis Category>
Definite:
A+B+C+E+G1.
Probable:
A+D+E+F+G2.
*“Probable” is set for the noninvasive diagnosis of systemic ATTRwt amyloidosis without biopsy using 99 mTc-PYP scintigraphy.
(Note 1) In the case of ATTRwt amyloidosis, the detection rate is low for amyloid in abdominal fat pad needle aspiration biopsy, skin biopsy, gastrointestinal biopsy, lip biopsy, etc. Therefore, if amyloid is not detected at these biopsy sites, a myocardial biopsy should be planned. If the disease is strongly suspected due to clinical signs or other laboratory findings, it needs to be detected by repeated biopsy from each tissue site. Amyloid deposition in ATTRwt amyloidosis shows weak staining with Congo-red and weak apple-green birefringence under polarized light. If it is difficult to judge at your own facility, it is recommended to consult with the research group or specialized facilities.
(Note 2) ATTR (+), ALκ (−), ALλ (−), and AA (−) should be confirmed by immunostaining, or the amyloid precursor protein should be identified using LMD and LC-MS/MS in biopsy tissue samples. If it is difficult to perform the test at your own facility, an analysis request can be made to the Research Group on Amyloidosis (http://amyloidosis-research-committee.jp/).
(Note 3) Visual grading method using frontal planar images taken 3 h later (Grade 0: No accumulation in the heart; Grade 1: Mild accumulation in the heart weaker than the ribs; Grade 2: Moderate degree in the heart equivalent to the ribs accumulation; Grade 3: Higher accumulation in the heart than in the ribs; Grade 2 or higher is positive), or a quantitative validation method using planar images taken 1 h later (a heart-to-contralateral [H/CL] ratio ≥1.5) should be used for evaluation. (For the details of myocardial scintigraphy, please refer to “
Chapter II, Section 6. Nuclear Medicine Examination
”.)
(Note 4) No abnormalities in the immunoglobulin free light-chain κ/λ ratio. In addition, no M protein in serum or urine immunofixation electrophoresis.
5.4 Diagnostic Criteria for Systemic ATTRv Amyloidosis
A. Clinical signs and laboratory findings
Clinical signs or laboratory findings due to each type of amyloidosis are present (Table 11).
Table 11.
Clinical Symptoms and Laboratory Findings Suggesting ATTRv Amyloidosis
| |
Clinical symptoms |
Laboratory findings |
Peripheral nerve
amyloidosis |
Sensory/motor polyneuropathy, small fiber
predominant polyneuropathy (a decline mainly in pain
and temperature sensations in the extremities, atrophy
and weakness of the extremities) |
Nerve conduction abnormalities due to axonal
disorders, decreased intraepidermal nerve fiber
density in skin biopsy, enlargement of dorsal root
ganglion and proximal sciatic nerve in MR neurography |
| Autonomic amyloidosis |
Orthostatic hypotension, vomiting, diarrhea,
constipation, dysuria, dysgenesis, dysphoria, etc. |
Decreased cardiac uptake in MIBG myocardial
scintigraphy, laser Doppler imaging of skin blood flow,
sweat function test, R–R interval fluctuation,
Schellong test, EGG, etc. |
Tendon/ligament
amyloidosis |
Carpal tunnel syndrome (numbness and pain of the
hand) |
A delay in conduction latencies at wrist in nerve
conduction tests |
| Cardiac amyloidosis |
See Table 8 |
| Renal amyloidosis |
Edema, weight gain |
Proteinuria, etc. |
| Ocular amyloidosis |
Dry eyes, vitreous opacity, glaucoma, irregular pupils,
etc. |
Increased intraocular pressure, etc. |
Central nervous system
amyloidosis |
Transient focal neurological episodes, disturbance of
consciousness, cerebral hemorrhage, etc. |
Imaging of meninges in contrast-enhanced head or
spine MRI, cerebral hemorrhage including
microhemorrhage, etc. |
| Other organ amyloidosis |
Hypoglycemia, hypothyroidism, etc. |
|
• Age of onset: Most cases for the Japanese focus area of FAP (Kumamoto and Nagano Prefectures) tend to occur in patients in their 20 s to 40 s. However, in other areas, onset is more likely after the age of 50 years. About half of patients do not have a clear family history.
• Inheritance pattern: Autosomal dominant inheritance, but family history may not be known by interviews alone.
• It should be noted that various symptoms (e.g., peripheral nerve type, cardiac type, cerebral meningeal vascular type, ocular type) are exhibited depending on the TTR gene variant. Even within the same family, the age of onset can vary greatly.
B. Pathological examination findings
In biopsied tissue, there are amyloid deposits exhibiting positive staining with Congo-red under a light microscope and showing apple-green birefringence under a polarizing microscope are observed (Note 1).
C. Amyloid typing
Amyloid deposits show positive staining for TTR (Note 2).
D. Scintigraphy
Intensely diffuse myocardial uptake on 99 mTc-PYP scintigraphy is confirmed (Note 3).
E. Genetic testing
A pathogenic mutation leading to a change of amino acids in the
TTR
gene.
<Diagnosis Category>
Definite:
A+B+C+E.
Probable:
A+B+E or A+D+E.
*Considering FAP patients with young onset in the focus area (Kumamoto and Nagano Prefectures) as a typical clinical picture of ATTRv amyloidosis in Japan, monoclonal gammopathy of undetermined significance (MGUS) is seldom concurrent, and there is little need to check M protein. By contrast, ATTRwt amyloidosis is reportedly frequently associated with MGUS (10–18%), and 39% of the patients with ATTRwt amyloidosis have abnormal free light-chain values. Therefore, the diagnosis of ATTRwt amyloidosis requires the absence of M protein, but the item related to M protein is excluded from the diagnostic criteria of ATTRv amyloidosis.
(Note 1) Amyloid can be detected in abdominal fat pad needle aspiration biopsy, skin biopsy, gastrointestinal biopsy, lip biopsy, peripheral nerve biopsy, myocardial biopsy, etc. Since amyloid deposits are often found on the vascular wall of the gastrointestinal submucosa, it is recommended to perform gastrointestinal biopsy down to the submucosal layer. If the disease is strongly suspected due to clinical signs or other laboratory findings, it needs to be detected by repeated biopsy from each tissue site.
(Note 2) ATTR (+), ALκ (−), ALλ (−), and AA (−) should be confirmed by immunostaining, or the amyloid precursor protein should be identified using LMD and LC-MS/MS in biopsy tissue samples. If it is difficult to perform the test at your own facility, an analysis request can be made to the Research Group on Amyloidosis (http://amyloidosis-research-committee.jp/).
(Note 3) Visual grading method using frontal planar images taken 3 h later (Grade 0: No accumulation in the heart; Grade 1: Mild accumulation in the heart weaker than the ribs; Grade 2: Moderate degree in the heart equivalent to the ribs accumulation; Grade 3: Higher accumulation in the heart than ribs; Grade 2 or higher is positive), or a quantitative validation method using planar images taken 1 h later (H/CL ratio: ≥1.5) should be used for evaluation. (For the details of myocardial scintigraphy, refer to “
Chapter II, Section 6. Nuclear Medicine Examination
”.)
II. Diagnosis of CA
1. Medical History, Symptoms, and Physical Findings Indicative of CA
The key to diagnosing CA is to suspect it based on the patient’s medical history, symptoms, and physical findings. Amyloidosis is a systemic disease; therefore, it presents with cardiac and various other findings. These findings must not be overlooked.
1.1 AL Amyloidosis
Amyloid deposits can be found in multiple organs, including the heart, kidneys, gastrointestinal system, and nerves. As a result, AL amyloidosis presents with diverse clinical symptoms and must be suspected based on a combination of medical history and physical findings.
Heart failure associated with AL amyloidosis is typically refractory. In many cases, pedal edema and pleural effusion cannot be controlled, even with large doses of diuretics. Nephrotic syndrome, a complication of AL amyloidosis, results in hypoalbuminemia, which reduces plasma osmolality, attenuates the effects of loop diuretics, and pleural amyloidosis causes pleural effusion in a pathology separate from that of heart failure.62
Amyloid deposition in the atrial muscle can cause left atrial (LA) thrombus regardless of sinus rhythm, as well as cardiogenic embolism.63,64
Amyloid deposition in arterioles is known to result in jaw claudication, intermittent claudication, and anginal pain.65
AL amyloidosis can also easily result in His bundle–ventricular block, leading to complete AV block before death in many patients with severe AL amyloidosis and a history of syncope.66,67
Thus, AL amyloidosis may be present in patients with syncope and complete AV block.
Regarding noncardiac physical findings, macroglossia is observed in 10–20% of cases (Figure 3). Due to amyloid deposition, the tongue presents as diffuse puffiness or nodular, and tooth pressure marks are seen on the lateral border. Amyloid deposition in vascular walls makes them fragile, heightening bleeding tendency and making patients prone to purpura. A characteristic physical finding is periorbital purpura, colloquially called “raccoon eyes” or “panda eyes.” Amyloid deposition in the gastrointestinal tract causes malabsorption syndrome and diarrhea, leading to weight loss and general debility. Amyloid deposition in the liver and spleen results in hepatosplenomegaly, which feels harder than typical hepatosplenomegaly associated with right-sided heart failure. Other noncardiac physical findings in AL amyloidosis include the shoulder pad sign, scleroderma-like skin thickening and nodules, and hard swelling in the thyroid gland, salivary glands, and lymph nodes. Peripheral neuropathy manifests as sensory abnormalities, numbness, and muscle weakness. Sensory disturbances are typically symmetrical and often present in the lower extremities. Autonomic neuropathy manifests as dysuria, impotence, and orthostatic hypotension.60,68

1.2 Amyloid Transthyretin Amyloidosis (ATTRwt and ATTRv)
ATTRwt amyloidosis occurs most commonly in older men. In many cases, the initial symptoms consist of conduction disturbance or HFpEF, primarily right-sided heart failure (pretibial edema, pleural effusion, etc.).32,69
In addition, a previous study reported that 35% of patients diagnosed with ATTRwt amyloidosis were previously misdiagnosed with another disease, such as hypertrophic cardiomyopathy or hypertensive heart disease.32
Another problem is that ATTRwt amyloidosis is not diagnosed until 1–2 years after symptoms manifest.31,70
Thus, many patients receive treatment for an incorrect diagnosis or remain undiagnosed for a long period of time, with ATTRwt amyloidosis not even being suspected.
Many patients with ATTRwt amyloidosis (40–50%) have a history of carpal tunnel syndrome;31,32,69,70
in fact, comorbid bilateral carpal tunnel syndrome is characteristic of ATTRwt amyloidosis. The mean length of time from the onset of carpal tunnel syndrome to a diagnosis of ATTRwt amyloidosis is reportedly 6.9 years.23
A history of numbness and pain in the median nerve region (radial side of the thumb, index, middle, and ring fingers) is often indicative of carpal tunnel syndrome. Pain in carpal tunnel syndrome is characteristically enhanced at night (particularly at dawn), and is often severe enough to awaken the patient during the night. Carpal tunnel syndrome is also characterized by the patient shaking their hands or changing the positions of their limbs to relieve the pain. A diagnosis of carpal tunnel syndrome is supported by a positive Phalen’s test (exacerbated dysesthesia due to increased carpal tunnel pressure after keeping the wrists flexed for 1 min) (Figure 4), a positive reverse Phalen’s test (the same sign but with the wrists extended), and a positive Tinel’s sign (tingling pain when the carpal tunnel is tapped with a hammer) (Figure 5).71
The progression of carpal tunnel syndrome can result in atrophy of the thenar eminence, difficulties in fine motor skills (buttoning buttons, etc.), the inability to create a neat circle with the thumb and index finger (perfect O sign) (Figure 6). Other comorbidities reported in ATTR amyloidosis include spinal canal stenosis (14–22%), cubital tunnel syndrome, rotator cuff tear, biceps tendon rupture, and quadriceps tendon rupture.69,72–74
Therefore, a history of these conditions should be confirmed for the diagnosis of ATTR amyloidosis.

ATTRv amyloidosis may lead to symptoms, such as peripheral neuropathy, dysautonomia, carpal tunnel syndrome, cardiac conduction defect, heart failure, digestive symptoms, nephropathy and eye symptoms, that affect various organs in the body (Figure 7).60,68,75
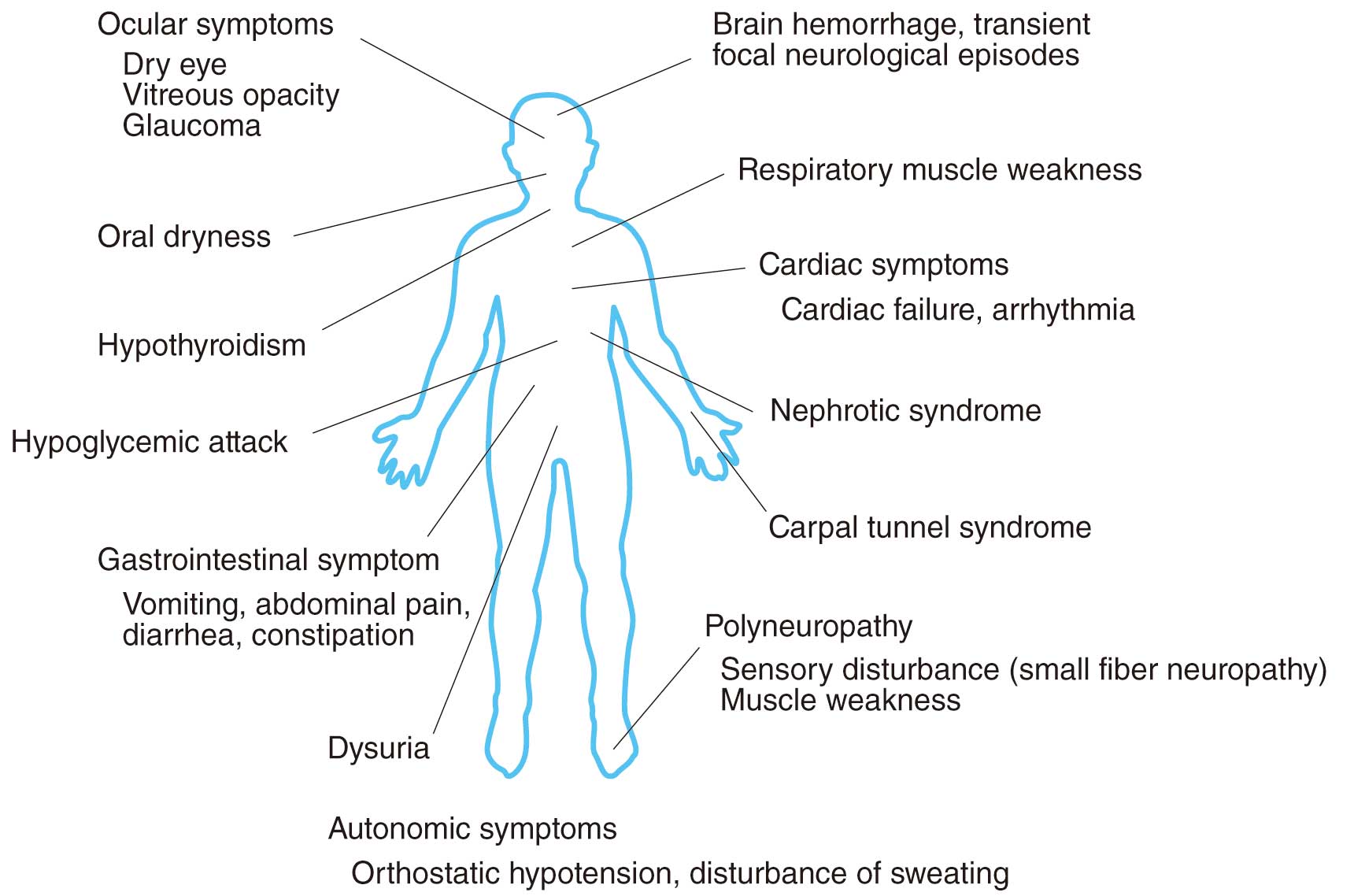
The initial symptoms of peripheral neuropathy begin in the distal part of both lower limbs where the axons are farthest away from the cell bodies and axonopathy is the underlying disease. Generally, the onset of neurological symptoms follows the order of dysautonomia, sensory disorders (thermal hypoalgesia), and movement disorders (bilateral peripheral superiority). This is because the disorder progresses from small-diameter unmyelinated fibers to large-diameter myelinated fibers following amyloid deposition. During the initial stage of the illness, while patients retain a normal sense of touch, a dissociated sensory loss is frequently observed, with thermal hypoalgesia starting at the feet or ankles. Thermal hypoalgesia makes it difficult for the patients to recognize burns and injuries to their feet.
Dysautonomia presents with symptoms such as orthostatic hypotension, erectile dysfunction, dysuria, nausea/vomiting, diarrhea, constipation, dyshidrosis, dry eye, and dry mouth. In cases of juvenile-onset ATTRv amyloidosis (Val30Met (p.Val50Met) mutation), features of dysautonomia are noticeable during the initial stages in areas with a high disease concentration but may be unrecognizable in areas with low disease concentration.
The onset of motor neuron symptoms such as muscular atrophy and muscle weakness is often delayed because of the sensory disorders, but there are also some cases in which there is mild sensory disorder during the initial illness stage with motor neuron disease as the underlying disease. In advanced stages, patients present with sensory disorders, muscle weakness, respiratory muscle paralysis, etc. in the limbs as a result of advanced peripheral neuropathy.
Amyloid deposition may sometimes lead to bilateral carpal tunnel syndrome. In a few cases, amyloid deposition in the cranial meninges and cerebral blood vessels may lead to central nervous system-related symptoms such as disturbance of consciousness and cerebral hemorrhages.
For cases where the family history is not particularly clear, caution is required because many patients are misdiagnosed in the initial stages as having chronic inflammatory demyelinating polyneuropathy (CIDP), diabetic neuropathy, lumbar spinal canal stenosis, etc.
Examples of cardiovascular symptoms include sick sinus syndrome, atrial fibrillation, atrial block due to amyloid deposition in the heart, and early orthostatic hypotension due to dysautonomia.76,77
The onset of ventricular diastolic dysfunction occurs prior to systolic dysfunction due to heart failure caused by amyloid deposition in the myocardium. There are some types of TTR mutations where a heart disorder will be the underlying cause of disease and peripheral neuropathy will not be noticeable.76,77
In particular, it is important to perform careful and frequent evaluations while adequately monitoring the risk of sudden death caused by conduction disorders. In many cases, orthostatic hypotension due to dysautonomia becomes severe as the illness progresses.78
Examples of digestive symptoms that may occur include gastrointestinal symptoms such as severe alternating diarrhea and constipation and nausea due to dysautonomia.79
Although severity differs from case to case, the patient’s daily lifestyle may be impaired drastically if symptoms are severe. In particular, persistent watery diarrhea might occur during the endstage of juvenile-onset ATTRv amyloidosis in areas of high disease concentration, leading to malabsorption and protein leakage.
Mutant TTR is produced not only by the liver but also by the retina and is responsible for eye symptoms. Vitreous opacities caused by amyloid deposition are also frequently observed in ATTRv patients and may present as an initial symptom in some cases. Glaucoma is also caused by amyloid deposition in the anterior part of the eye, which, on progression, can become a primary cause of blindness. Lacrimal hyposecretion might lead to dry eye.
Kidney symptoms such as nephrotic syndrome or kidney failure may also be caused by amyloid deposition with severity varying from case to case. Often, kidney symptoms are not noticeable during the initial stage of illness.
Patients may also experience other symptoms such as hypothyroidism, hypoglycemic attacks, and dry cough.
2. Blood Sampling Test
2.1 AL Amyloidosis
The most important point in diagnosing AL amyloidosis is to suspect it from clinical symptoms, physical findings, and electrocardiographic, chest X-ray, and echocardiographic findings. When we suspect AL amyloidosis, serum immunoglobulin electrophoresis, serum and urine immunofixation (or electrophoresis), and serum free light chain (FLC) measurements should be performed.
There has been a report on screening panels for detection of monoclonal gammopathies including 581 patients with AL amyloidosis.80
In the paper, the detection sensitivity for AL amyloidosis was 88.3% by serum FLC, 96.2% by serum FLC+serum immunoglobulin electrophoresis, 97.1% by serum FLC+serum immunoglobulin electrophoresis+serum immunofixation, and 98.1% by serum FLC+serum immunoglobulin electrophoresis+serum and urine immunofixation.80
Another paper showed that the combination of serum and urine electrophoresis immunofixation with serum FLC had sensitivity of 100% for amyloidogenic light chain detection.81
Thus, it is not enough to judge by serum FLC alone; at least one additional modality with serum/urine immunofixation is necessary.
To evaluate cardiac findings in AL amyloidosis, plasma natriuretic peptide (BNP or NT-proBNP) and high-sensitivity cardiac troponins are measured. If renal amyloidosis is suspected, urine protein, microalbuminuria, and urinary Bence-Jones protein are measured to evaluate the type and amount of proteinuria. Bone marrow aspiration and biopsy can confirm the presence of clonal plasma cells and evaluate the presence or absence of amyloid deposits in the bone marrow.
Serum FLC measurement is very useful for the diagnosis of AL amyloidosis.
Figure 8
shows serum FLC values in each disease.82
In AL amyloidosis, about 80% of cases are of type λ, and the measured values are often distributed in the range of 50–1,000 mg/L. Serum FLC is elevated due to decreased renal function. Therefore, not the FLC value itself or the k/λ ratio, but the FLC difference (difference FLC: dFLC) is often used. The dFLC is one of the factors associated with a poor prognosis in the Revised Mayo stage 2012, and it is an important index used to determine the efficacy of treatment.
2.2 ATTR Amyloidosis
Unlike serum FLC in AL amyloidosis, no biomarkers in ATTR amyloidosis are directly linked to its diagnosis or the assessment of its therapeutic effects. However, the following biomarkers are reportedly useful aids in the diagnosis of ATTR amyloidosis: cardiac troponin, a marker of myocardial damage; B-type natriuretic peptides (BNP) and N-terminal pro-BNP (NT-proBNP), biomarkers of heart failure; and RBP4, a plasma transport protein for vitamin A (retinol).36,83,84
2.2.1 Cardiac Troponin
Elevated cardiac troponin is used to diagnose acute myocardial infarction. However, a persistently elevated cardiac troponin level is a red flag for CA. One study reported that, compared with a group of patients with LV hypertrophy for whom endomyocardial biopsy confirmed the absence of CA (control group), the CA group (including AL and ATTR) demonstrated significantly elevated high-sensitivity cardiac troponin T levels (control group [median]: 0.018 ng/mL; CA group [median]: 0.048 ng/mL).36
When the cutoff for high-sensitivity cardiac troponin T levels was set at 0.031 ng/mL, the sensitivity and specificity for diagnosing CA based on LV hypertrophy were 74% and 79%, respectively (area under the curve: 0.787).
Another study reported that an elevated high-sensitivity cardiac troponin T level (>0.05 ng/mL) combined with an elevated NT-proBNP level (>3,000 pg/mL) is useful for assessing prognosis in ATTRwt CA. In that study, the median survival times for Stage 1 (no elevation in either biomarker), Stage 2 (elevation in one of the two biomarkers), and Stage 3 (elevation in both biomarkers) were 66, 42, and 20 months, respectively.33
Assessing cardiac troponin requires the consideration of factors that affect its elevation, such as myocardial ischemia, renal dysfunction, pulmonary embolism, and sepsis. Multiple assessments are recommended to confirm persistent elevation. Since cardiac troponin levels are also elevated in acute heart failure, they should be measured in a compensated state.85,86
2.2.2 Natriuretic Peptides
BNP and NT-proBNP are widely used to screen for, diagnose, and predict prognosis in heart failure. In CA (particularly AL amyloidosis), the elevation in NT-proBNP is pronounced compared with heart failure severity and hemodynamics.87
In ATTRv amyloidosis, since NT-proBNP levels increase before cardiac symptoms manifest, elevated NT-proBNP levels are reportedly useful for detecting cardiac involvement.83
2.2.3 Retinol-Binding Protein 4 (RBP4)
RBP4, a plasma transport protein for vitamin A (retinol), is thought to help stabilize TTR, inhibit misfolding and aggregation, and suppress amyloid fibril formation. A group of patients with ATTRv amyloidosis involving a mutation in the
V122I
gene reportedly demonstrated significantly lower RBP4 levels than did a group of patients with non-amyloid heart failure, suggesting that RBP4 could be useful in screening for CA.84,88
2.2.4 Important Points
In the diagnosis of ATTR, serum FLCs confirm the absence of immunoglobulin abnormalities. Monoclonal gammopathy of undetermined significance, which is assessed to rule out AL amyloidosis, is observed in 5.3% of patients aged ≥70 years89
and in 40–50% of those with ATTR.90
In patients with suspected CA who demonstrate immunoglobulin abnormalities, amyloid precursor protein must be identified in a biopsy specimen to differentiate ATTR from AL amyloidosis.
Recommendations and levels of evidence for blood testing for suspected cardiac amyloidosis are summarized in
Table 12.
Table 12.
Recommendations and Levels of Evidence for Blood Testing for Suspected Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Measurement of high-sensitivity cardiac troponin to aid the
diagnosis of amyloidosis* |
IIa |
C |
C1 |
IVb |
Measurement of BNP/NT-proBNP to aid the diagnosis of
amyloidosis |
IIa |
C |
C1 |
IVb |
Measurement of high-sensitivity cardiac troponin to assess
prognosis in ATTRwt amyloidosis* |
IIa |
C |
C1 |
IVb |
Measurement of BNP/NT-proBNP to assess prognosis in ATTRwt
amyloidosis |
IIa |
C |
C1 |
IVb |
Measurement of serum immunoglobulin to diagnose AL
amyloidosis |
I |
A |
A |
IVb |
Serum and urine immunofixation (or electrophoresis) to diagnose
AL amyloidosis |
I |
A |
A |
IVb |
Measurement of serum free-light chains to diagnose AL
amyloidosis |
I |
A |
A |
IVb |
*Not covered by insurance in Japan for diagnosing cardiac amyloidosis. COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS); AL, amyloid light-chain; ATTRwt, wild-type amyloid transthyretin; BNP, b-type natriuretic protein; NT-proBNP, N-terminal pro-BNP.
Electrocardiogram (ECG) is a noninvasive test often used to screen cardiac diseases. Abnormal ECG findings often lead to a diagnosis of CA. Amyloid deposits in the myocardial interstitium or conduction system may cause abnormal ECG findings such as low voltage, pseudoinfarct pattern, conduction disorders, and AF (Table 13). However, most of these ECG findings are not specific to CA, and the diagnostic values of these findings may differ among the types of amyloidosis or by the degree of the disease. Thus, ECG may be normal at the early stage of the disease. As CA is generally a progressive disease, it is important to evaluate chronological changes, even in patients without ECG abnormalities.
Table 13.
Rate of Electrocardiogram Findings Among Different Types of Cardiac Amyloidosis
| Types |
Low voltage |
Pseudoinfarct pattern |
Atrioventricular block |
Atrial fibrillation |
| AL |
23–64% |
15–69% |
15–26% |
6–32% |
| ATTRwt |
13–40% |
18–71% |
11–33% |
27–67% |
| ATTRv |
23–38% |
18–69% |
25–45% |
5–17% |
3.1 Low Voltage and Decreased Voltage/Mass Ratio
Low voltage on ECG despite LV hypertrophy is a classical finding in CA, and relatively common in AL amyloidosis (23–64%); however, it is only observed in 13–40% of patients with ATTRwt CA and 23–38% of those with ATTRv amyloidosis.31,32,34,54,69,70,91–93
Thus, CA cannot be ruled out by the absence of low voltage. Low voltage is often defined as “limb leads ≤0.5 mV” or “precordial leads ≤1.0 mV”. The Sokolow index (SV1+RV5 or SV1+RV6 <1.5 mV) may also be used. Low voltage has been reported as a worse prognostic predictor in studies including both AL and ATTR amyloidosis.91,94,95
Furthermore, a decreased voltage/mass ratio (Sokolow index divided by wall thickness) has also been reported as a useful finding when diagnosing CA.54,91,96
3.2 Pseudoinfarct Pattern (Abnormal Q Waves, Poor R Progression)
Abnormal Q waves and poor R progression in a patient without coronary artery disease is called a “pseudoinfarct” or “pseudonecrosis” pattern. The pseudoinfarct pattern is relatively common in all types of CA (15–69% of AL, 18–71% of ATTRwt, and 18–69% of ATTRv CA cases).32,34,91,92,94
Rates of the pseudoinfarct pattern are associated with the extent of late gadolinium enhancement (LGE) on CMR, suggesting that an amyloid burden is associated with the occurrence of the pseudoinfarct pattern.97
3.3 Conduction Disturbances (AV Block, Bundle Branch Block, Intraventricular Conduction Disturbance)
Amyloid deposits involving the conduction system may result in AV block, bundle branch block, or intraventricular conduction disturbances. AV block is observed in 15–26% of AL, 11–33% of ATTRwt CA, and 25–45% of ATTRv amyloidosis cases.32,34,70,91,98
Right bundle branch block is observed in 3–19% of AL and 12–16% of ATTR amyloidosis cases, and left bundle branch block is observed in 4–6% of AL and 7–40% of ATTR amyloidosis cases.32,34,70,93
Marume et al. reported that a QRS width ≥120 ms is significantly associated with positive findings on 99 mTc-PYP scintigraphy, suggesting that the presence of conduction disturbances may be useful in diagnosing CA.99
Conduction disturbances may also reflect disease progression, and intraventricular conduction disturbance in AL or prolonged PQ interval in ATTRv amyloidosis is associated with poor prognosis.100,101
3.4 Atrial Fibrillation (AF)
Although AF is not a specific finding for CA, it is frequently observed in ATTRwt amyloidosis (27–67%), whereas it is not as frequent in AL (6–32%) and ATTRv CA (5–17%).31–34,54,69,70,91–94,102–104
Thus, patients who have AF and LV hypertrophy should be carefully examined for other findings suggesting CA (especially ATTRwt CA). Previous studies have reported that AF is not associated with survival in CA,102,103,105
but is associated with heart failure events.103
The causes of AF in CA include amyloid deposits in the atrium or increased LA pressure secondary to LV filling pressure and/or LV diastolic dysfunction. Recent studies have also reported that amyloid deposits limited to the atrium, called “isolated atrial amyloidosis”, may be one of the causes of AF.106–109
Rocken et al. reported that amyloid deposits were found in 16% of the right atrial appendages of patients who had undergone cardiac surgery, and that these amyloids were derived from atrial natriuretic peptides (ANPs).109
It is believed that the excretion of ANPs due to the extension of the atrium or AF itself is associated with this phenomenon; however, the mechanism of ANPs forming amyloid has not been elucidated.106
3.5 Others (Ventricular Arrhythmias, Prolonged QT Interval)
Data regarding the incidence of ventricular arrhythmias in CA are limited; however, ventricular tachycardia or fibrillation may be the first presentation of CA. While non-sustained ventricular tachycardias and sudden deaths are observed in advanced CA, their clinical significance, as well as the usefulness of implantable cardioverter defibrillators in CA, has not been established (please also refer to
CQ3-3). Prolonged QT is also a common finding in CA.32,34,91,93
4. Echocardiography
Echocardiography is a noninvasive, reproducible method for assessing cardiac morphology and function in CA, and some echocardiographic indices are prognostic for amyloidosis.
4.1 M-Mode Echocardiography
Normal LV diastolic dimensions, increased systolic dimensions, diminished amplitude of excursion, and pericardial effusion are features of CA.110
The following findings are basic: 1) symmetric LV hypertrophy in the absence of hypertension or aortic valvular disease; 2) hypokinesia and decreased systolic thickening in the interventricular septum and LV posterior wall; and 3) small-to-normal LV cavity size. In addition to the finding of LV hypertrophy, the characteristics of infiltrate cardiomyopathy include a septal/posterior free wall thickness ratio <1.3, increased maximal LA transverse dimension, reduced mitral valve closure (E-F slope), and preserved EF >60%.111
The right ventricular (RV) anterior wall thickness is reportedly significantly increased in patients with clinically significant amyloid infiltrate cardiomyopathy, and this is consistent with pathologic findings.112,113
4.2 Two-Dimensional (2D) Echocardiography (Figures 9 and 10)

Two-dimensional (2D) echocardiography can reveal additional features such as thickened papillary muscles, thickened valves, better appreciation of the thickened RV wall, and a characteristic “granular sparkling” appearance of the thickened cardiac walls.114
Although increased echogenicity of the myocardium, particularly with a “granular sparkling” appearance, has been reported in several studies, sensitivity is not high with this pattern, which is seen in about 30% of CA cases. In addition, it should be noted that this granular pattern applies only to standard echocardiographic imaging, without tissue harmonics being applied, as this increases myocardial echogenicity in general. Newer echocardiographic image processing techniques may also reduce the granular appearance. Thus, although increased echogenicity is common in amyloid cardiomyopathies, its usefulness as a discriminating factor remains limited.115
Mohty et al. examined the prevalence and prognostic impact of left-sided valve thickening in systemic AL amyloidosis.116
In patients with AL amyloidosis, mitral and/or aortic valve thickening (>3 mm) was significantly associated with decreased 5-year survival. The electrocardiographic voltage tended to be low and the echocardiographic muscle cross-sectional area tended to be increased.96
When these two techniques were combined, an inverse correlation was observed between voltage and the muscle cross-sectional area (r=−0.79); moreover, a marked derangement of the voltage/cross-sectional area relation was associated with clinical symptoms and mortality. The combination of increased LV thickness and a low-voltage electrocardiographic pattern is highly specific for CA, and was found in 3/30 (10%) and 13/24 (54%) of secondary and amyloid-light chain (AL) amyloidosis patients, respectively.117
One model showed that low voltage was present and the interventricular septal thickness was >1.98 cm, and a diagnosis of CA could be made with a sensitivity of 72% and a specificity of 91%, with positive and negative predictive values of 79% and 88%, respectively.118
While all patients with amyloidosis show the presence of very bright or highly refractile echoes in the myocardium, this is also observed in patients with uncomplicated ventricular hypertrophy, chronic renal failure, hypertrophic cardiomyopathy, Pompe disease, hemochromatosis, and left heart hypoplastic syndrome, indicating that this finding is nonspecific for CA.119
The occurrence of clinical congestive heart failure correlated strongly with greater wall thickness and multiple other echocardiographic abnormalities, and survival was negatively influenced by greater wall thickness and reduced systolic function (fractional shortening), indicating that echocardiographic examinations are an important tool for identifying cardiac amyloid involvement and may be useful in estimating prognosis in systemic amyloidosis.120
A low voltage pattern on electrocardiography and a reduced LV ejection fraction (LVEF) help identify patients at high risk of death.94
Extensive CA results in atrial thrombi, even in patients with sinus rhythm.121
Severe atrial and ventricular infiltration by amyloids may lead to mechanical atrial standstill with resultant thrombus formation.122
AF, poor LV diastolic function, and lower LA appendage emptying velocity have been shown to be independent risk factors for intracardiac thrombosis, whereas anticoagulation has been found to be associated with a significantly decreased risk.
4.3 Doppler Echocardiography (Figure 11A,B)
Serial pulsed wave Doppler studies of LV inflow have identified changes from abnormal relaxation or “normal” patterns to restrictive patterns.123
Doppler-derived LV diastolic filling variables are also important predictors of survival in CA,124
with patients with a transmitral early filling wave deceleration time of ≤150 ms (indicating restrictive pattern) showing poor cardiac outcomes.125
4.4 Pulsed Tissue Doppler Imaging (Figure 11C,D)
CA is characterized by an initial impairment in early cardiac relaxation, whereas congestive heart failure is associated with an impairment of peak systolic wall motion velocities, most prominently seen in the longitudinal axis.126
Moreover, peak lateral and medial mitral annulus velocities and color M-mode tissue Doppler of the LV posterior wall (for measurements of mean myocardial velocities and myocardial velocity gradient) can differentiate patients with CA and controls with good overall accuracy.127
The longitudinal myocardial velocity gradients that indicate differences between the basal and mid-myocardial velocities using pulsed tissue Doppler imaging are reportedly significantly impaired in patients with compared with those without congestive heart failure.128
Furthermore, the LV long-axis function was depressed in all (100%) patients with CA compared with only 36% of those with idiopathic restrictive cardiomyopathy.129
4.5 Strain Doppler imaging
Longitudinal LV myocardial function as assessed by strain and strain rate tissue Doppler echocardiography can detect early impairments in systolic longitudinal LV function in patients with AL amyloidosis upon fractional shortening, but prior to this, it is not detectable on tissue velocity imaging.130,131
Strain Doppler imaging is a sensitive method that can detect differences in LV function between ATTRv and AL amyloidosis that cannot be distinguished by standard echocardiographic parameters, despite the severity of congestive heart failure and cardiac mortality being much lower in ATTRv amyloidosis.132
This method can also detect impaired LV systolic function in patients with AL amyloidosis who have no evidence of cardiac involvement upon standard 2D or Doppler echocardiography.133
Mean LV basal longitudinal strain (LS) was reported to be a powerful predictor of clinical outcomes, and found to be superior to standard 2D echocardiographic Doppler flow measurements and simple tissue velocity indices.134
Additionally, RV function as assessed by Doppler myocardial imaging can also identify early impairment of cardiac function and stratify mortality risk in patients with AL amyloidosis.135
4.6 Speckle Tracking Echocardiography (Figure 12)
Speckle tracking echocardiography (STE) of myocardial strain and strain rates can discriminate CA from other causes of cardiac hypertrophy. Compared with other causes of LV hypertrophy, cardiac amyloid profoundly alters all strain parameters (longitudinal, circumferential, and radial strain).136
Endocardial and epicardial longitudinal and circumferential strain and radial strain were found to be significantly lower in patients with hypertrophic cardiomyopathy and ATTR compared with controls.137
Further, epicardial circumferential strain was significantly lower in patients with ATTR than in those with hypertrophic cardiomyopathy. A systolic septal longitudinal base-to-apex strain gradient (septal apical/basal longitudinal systolic ratio >2.1), combined with a shortened diastolic deceleration time of early filling (deceleration time of early filling <200 ms) aids in differentiating CA from other causes of concentric LV hypertrophy.138
A higher ratio of septal apical to basal segmental longitudinal peak systolic strain ratio >2.1 is suggestive of CA (sensitivity: 88%; specificity: 85%; positive predictive value: 67%; negative predictive value: 96%).138
When using the strain polar map, the apical sparing of LS visual pattern (a pattern of regional differences in deformation in which LS in the basal and middle segments of the left ventricle is more severely impaired than the apical segments) is an easily recognizable and accurate means of differentiating CA from alternative causes of LV hypertrophy.139
The pathological mechanisms of apical sparing in patients with CA have not been clarified. The echocardiographic imaging parameters, mean tissue Doppler-derived and 2D global LS of the LV cardiac serological biomarkers, and comprehensive clinical characteristics were prospectively assessed in 206 consecutive patients with biopsy-proven systemic AL amyloidosis.140
In a multivariate analysis, only diastolic dysfunction and 2D global LS remained independent predictors of survival. In addition, 2D-STE predicted outcomes and provided incrementally better prognostic information compared with existing prognostic staging systems, especially in the group without CA.141
The EF was preserved while the longitudinal systolic strain was significantly reduced in both compensated and decompensated AL amyloidosis patients. The longitudinal systolic strains were similar in the apical segments and significantly reduced in the basal segments in both groups, and multivariate analysis showed that New York Heart Association (NYHA) functional class and mid-septum systolic LS were independent predictors for survival.142
Compared with ATTRv and AL amyloidosis, ATTRwt amyloidosis was characterized by greater LV wall thickness and lower EF, and the LV basal LS was more depressed in both ATTRwt and AL amyloidosis. TTR-related causes were favorable predictors of survival, whereas LS and advanced NYHA class were negative predictors.54
Twenty-one consecutive patients with biopsy-proven ATTRwt amyloidosis were compared with 21 patients with ATTRv amyloidosis from the database, matched by age and LV wall thickness.143
LVEF, LV basal circumferential/radial strain, and mid-radial strain were significantly lower in patients with ATTRwt than in those with ATTRv amyloidosis. No significant differences in the other parameters were observed between the two groups. In the receiver-operating characteristics curve analysis, LVEF and LV basal mean radial strain were the best parameters for distinguishing between the two groups.
LA function used to be a focus in CA. In ATTR cardiomyopathy with increased LV wall thickness, LA myocardial function is abnormal, irrespective of atrial cavity size. Reduced LA myocardial strain rate during atrial systole, irrespective of cavity volume, E/e’, and LV deformation, is also a strong predictor of atrial arrhythmic events.144
LA function was severely impaired and highly correlated with LV deformation in CA. Differences in LA function between amyloid subtypes suggest that amyloid etiology plays a role in the pathophysiology of cardiac dysfunction in CA.145
Three-dimensional STE (3D-STE) provides new insights into the mechanism of cardiac dysfunction in patients with CA. 3D-STE reveals regional and global biventricular dysfunction (LV basal LS, LV peak basal rotation, RV basal free-wall LS) in CA. The assessment of RV dysfunction has an additive value in differentiating CA from other causes of myocardial wall thickening.146
Functional LA parameters are progressively altered in patients with AL amyloidosis according to the Mayo Clinic staging system. A decrease in 3D-peak atrial LS is associated with worse outcomes, independent of LA volume (Table 14).147
Table 14.
Recommendations for Echocardiography in Patients With Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
M-mode echocardioraphy is recommended for the assessment of LVH, LA enlargement,
pericaridal effusion, LVEF, and RV anterior wall thickness |
I |
B |
B |
IVb |
B-mode echocardioraphy is recommended for the assessment of LVH, LA enlargement,
pericaridal effusion, LVEF, thickened pallilary muscle, thickeed RV wall, increased
myocardial echogenicity, and voltage/mass ratio |
I |
B |
B |
IVb |
Transesophageal echocardiography is recommended for the detection of intracardiac
thrombus |
I |
B |
B |
IVa |
Doppler echocardiography is recommended for the assessment of LV inflow, PV flow, LVOT
flow, and/or the prediction of the prognosis |
I |
B |
B |
IVa |
Tissue Doppler echocardiography is recommended for the assessment of LV systolic and
diastolic function |
I |
B |
B |
IVa |
Strain Doppler echocardiography is recommended for early detection of systolic dysfunction,
and/or the prediction of the prognosis |
I |
B |
B |
IVa |
Speckle tracking enchocardiography is recommended for the differentiation of cardiac
amyloidosis from other causes of cardiac hyperthrophy, and/or the prediction of the
prognosis |
I |
B |
B |
IVa |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS); LV (H), left ventricular (hypertrophy); LA, left atrium; EF, ejection fraction; RV, right ventricular; PV, pulmonary vein; LVOT, LV outflow tract.
4.7 ATTR in Patients With Aortic Stenosis
See other section.
5. Cardiac Magnetic Resonance Imaging (MRI)/Computed Tomography (CT)
5.1 Cardiac MRI (CMR)
CMR is excellent for diagnosing CA. Although CMR can be used to diagnose advanced CA easily, its ability to diagnose other early diseases and amyloid types is limited. In addition, CA often presents with atypical findings on CMR. Therefore, CMR findings should be assessed in conjunction with the results of other tests and clinical findings. CMR is also useful for differentiating CA from other diseases with similar clinical presentations, such as hypertrophic cardiomyopathy and Anderson-Fabry disease. CA is typically diagnosed with cine CMR and LGE on CMR (LGE imaging). However, myocardial T1 mapping is reportedly useful for the quantitative assessment of myocardial tissue characterization, and is currently recommended for diagnosing CA. Myocardial T2 mapping and myocardial strain assessment using CMR are also suggested to be effective. CMR assessment that integrates these imaging methods would be useful for diagnosing CA, differentiating it from other diseases, assessing its severity, predicting patient prognosis, and monitoring disease states.
Tables 15
and
16
show CMR findings in CA and recommendation levels, respectively.
Table 15.
CMR Findings in Cardiac Amyloidosis
| Imaging |
Significance and findings |
Notes |
| Cine CMR |
Significance: Assessment of cardiac morphology and function
· Left ventricular hypertrophy predominantly at the basal
segments
· Symmetrical or asymmetrical left ventricular hypertrophy
· Thickening of the right ventricular wall and/or atrial septum
(≥6 mm)
· Left ventricular ejection fraction is often preserved |
· Caution required in differentiation from
hypertrophic cardiomyopathy
· Diagnosis is difficult using cine CMR alone |
| LGE imaging |
Significance: Assessment of amyloid deposition and myocardial
fibrosis
· Diffuse left ventricular subendocardial LGE
· Transmural LGE in advanced stage
· LGE also present in the right ventricular wall, left atrial wall, or
atrial septum
· Dark blood pools |
· Atypical LGE patterns are often seen
· LGE findings change with disease stage
· Prone to errors in inversion time setting
· Phase-sensitive inversion recovery sequence is
recommended
· Correlated with prognosis |
| T1 mapping |
Significance: Assessment of amyloid deposition and myocardial
damage
· Significantly elevated native T1 and ECV values
· ECV ≥40% |
· Enables quantitative assessment
· Reference value for native T1 must be set
· Measurement should be performed in the
interventricular septum on the left ventricular
short-axis images at the basal or middle level
· Amyloid type is difficult to diagnose
· Correlated with severity and outcomes
· Applied to monitor progress and assessment of
therapeutic effects |
| T2 mapping |
Significance: Assessment of myocardial edema and inflammation
· Higher values than healthy controls
· Demonstrates especially high values in AL |
· May assist in diagnosis of amyloid type
· Insufficient evidence |
| Myocardial strain |
Significance: Assessment of myocardial strain
· Abnormalities in longitudinal and circumferential strains
· Reduced peak strain value (absolute value)
· Peak strain time variations |
· Low disease specificity
· Applied in disease state monitoring
· Correlated with severity and outcomes |
AL, amyloid light-chain; ECV, extracellular volume fraction; LGE, late gadolinium enhancement; CMR, cardiac magnetic resonance imaging.
Table 16.
Recommendations and Evidence for CMR in Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| Assessment of cardiac morphology and function with cine CMR |
I |
C |
A |
IVb |
| Differentiation from other cardiomyopathies with LGE imaging |
I |
C |
A |
IVb |
| Differentiation from other cardiomyopathies with T1 mapping |
I |
C |
A |
IVb |
| Differentiation from other cardiomyopathies with T2 mapping |
IIa |
C |
B |
IVb |
Differentiation from other cardiomyopathies with myocardial strain
assessment using CMR |
IIa |
C |
B |
IVb |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS); CMR, cardiac magnetic resonance imaging; LGE, late gadolinium enhancement.
5.1.1 Cine CMR
Cine CMR is a basic sequence for assessing cardiac morphology and function. CA often causes LV hypertrophy, predominantly at the basal segments. Although symmetrical and circumferential hypertrophy has been considered to be typical, a recent study reported that asymmetrical hypertrophy, particularly asymmetrical septal hypertrophy, is often observed148
(Figure 13). The degree of LV hypertrophy tends to be greater in ATTR than in AL amyloidosis.35
However, it should be noted that 3–8% of CA cases present with a normal geometry. Thickening of the RV wall and atrial septum is frequently observed in CA, with atrial septum thickening ≥6 mm considered to be relatively specific to CA.149
Based on cine CMR functional analysis, LVEF is often preserved, but may decline as the disease progresses. Advanced CA often involves pericardial and pleural effusions.35
Morphological and functional findings in cine CMR are nonspecific, making CA difficult to diagnose using this modality alone.

5.1.2 LGE Imaging
LGE imaging, the preferred method for diagnosing CA, involves taking images 10 min after the injection of a gadolinium contrast agent to visualize myocardial damage (primarily fibrosis). Imaging is generally conducted using inversion recovery sequences. The diagnostic performance of CA with LGE imaging has been reported to have a sensitivity and specificity of 85–90%.148,150
Typical findings of CA in LGE imaging include the following: 1) diffuse LV subendocardial LGE; 2) LGE in the RV wall, LA wall, and atrial septum; and 3) dark blood pools97
(Figure 14). LGE reflects both amyloid deposition in the myocardial interstitium and subendocardial ischemic changes (fibrosis) associated with microangiopathy.97
Dark blood pools, which originate from the contrast agent kinetics unique to amyloidosis, in which gadolinium is quickly washed away from blood pools, form as a result of the distribution of gadolinium contrast agent in interstitial amyloid depositions throughout the body. Patterns of LGE tend to be more prominent in ATTR than in AL amyloidosis, and frequently develop into transmural LGE.35
LGE mostly occurs in the basal segments.151
The presence of LGE is considered a powerful prognostic predictor of CA.152
Recent findings indicate that LGE transitions from subendocardial to transmural as CA progresses, and that CA presents with various atypical LGE patterns (patchy, subepicardial, and diffuse transmural LGE) according to disease stage.153
No more than 50% of all patients with CA present with typical LGE (subendocardial LGE).153

LGE imaging based on inversion recovery was developed to detect local myocardial damage such as myocardial infarction. Diffuse myocardial diseases, especially CA, are prone to inadequate contrast enhancement due to technical imaging errors, which can cause LGE underestimations. The use of phase-sensitive inversion recovery sequences has recently been recommended as a way to avoid such imaging errors.153
Although LGE imaging is effective for diagnosing CA, the following limitations and disadvantages must be recognized: 1) LGE imaging cannot detect early-stage lesions; 2) CA often presents with atypical LGE; 3) LGE patterns are underestimated in diffuse myocardial damage; 4) imaging errors are likely to occur; 5) quantitative assessment is difficult; and 6) gadolinium contrast agent cannot be used in patients with advanced renal dysfunction or those on dialysis. T1 mapping, discussed below, is an important method that complements the disadvantages of LGE imaging.
5.1.3 T1 Mapping
T1 mapping is an imaging method that quantitatively measures myocardial T1 values (T1 relaxation times [ms]). Due to the ability of T1 mapping to assess myocardial damage quantitatively, it is expected to be applied for the detection of early-stage lesions, the assessment of severity and outcomes, and the evaluation of therapeutic effects. There are two parameters in T1 mapping: native T1, which does not use a contrast agent, and extracellular volume fraction (ECV), which involves the use of a contrast agent. ECV is calculated based on the myocardial and blood T1 values of pre- and post-contrast agent injection using the following formula and the most recent hematocrit (Hct) result within the last 24 h:
ECV (%) = (1 − Hct) × (1 / T1myo post − 1 / T1myo pre) / (1 / T1blood post − 1 / T1blood pre) × 100
where native T1 includes both intracellular and extracellular information, and ECV reflects changes in the extracellular space. Native T1 is easily affected by the magnetic field strength of the systems (1.5 T or 3.0 T scanners) and imaging sequences, thereby requiring the establishment of reference values for each system and imaging sequence.154
By contrast, ECV, which shows the percentage of extracellular space in which the contrast agent is distributed, is mostly unaffected by magnetic field strength or imaging sequences. Therefore, ECV enables the stable assessment of myocardial properties. The reference value for ECV is 23–28%. Imaging and measurements should be performed in the interventricular septum on the LV short-axis images at the basal or middle level.154
In CA, native T1 and ECV frequently yield extremely abnormally high values155
(Figure 15). Native T1 and ECV values in CA are reported to be significantly higher than those in morphologically similar hypertrophic cardiomyopathy and aortic stenosis. For native T1 alone, the diagnostic sensitivity and specificity of CA are 80–92% and 56–91%, respectively. For ECV alone, the diagnostic sensitivity and specificity are 93% and 82%, respectively.155–157
While native T1 and ECV demonstrate excellent diagnostic capacities, combining them with other parameters in a comprehensive assessment may further improve their diagnostic capacity. In CA, ECV is reportedly 44–61%.153,157–159
Thus, CA must be strongly suspected when ECV is ≥40%.160
Although T1 mapping findings differ by the type of CA (AL, ATTRwt, or ATTRv amyloidosis), there is a great deal of overlap, which hinders the diagnosis of CA type.159
T1 mapping detects lesions with greater sensitivity than does LGE imaging; therefore, its use may aid the early diagnosis of CA.161,162
In addition, native T1 and ECV demonstrate correlations with CA severity and outcomes, creating anticipation for their application in the assessment of risk and therapeutic effects, progress monitoring, and other aspects of clinical management.153,158,159,162
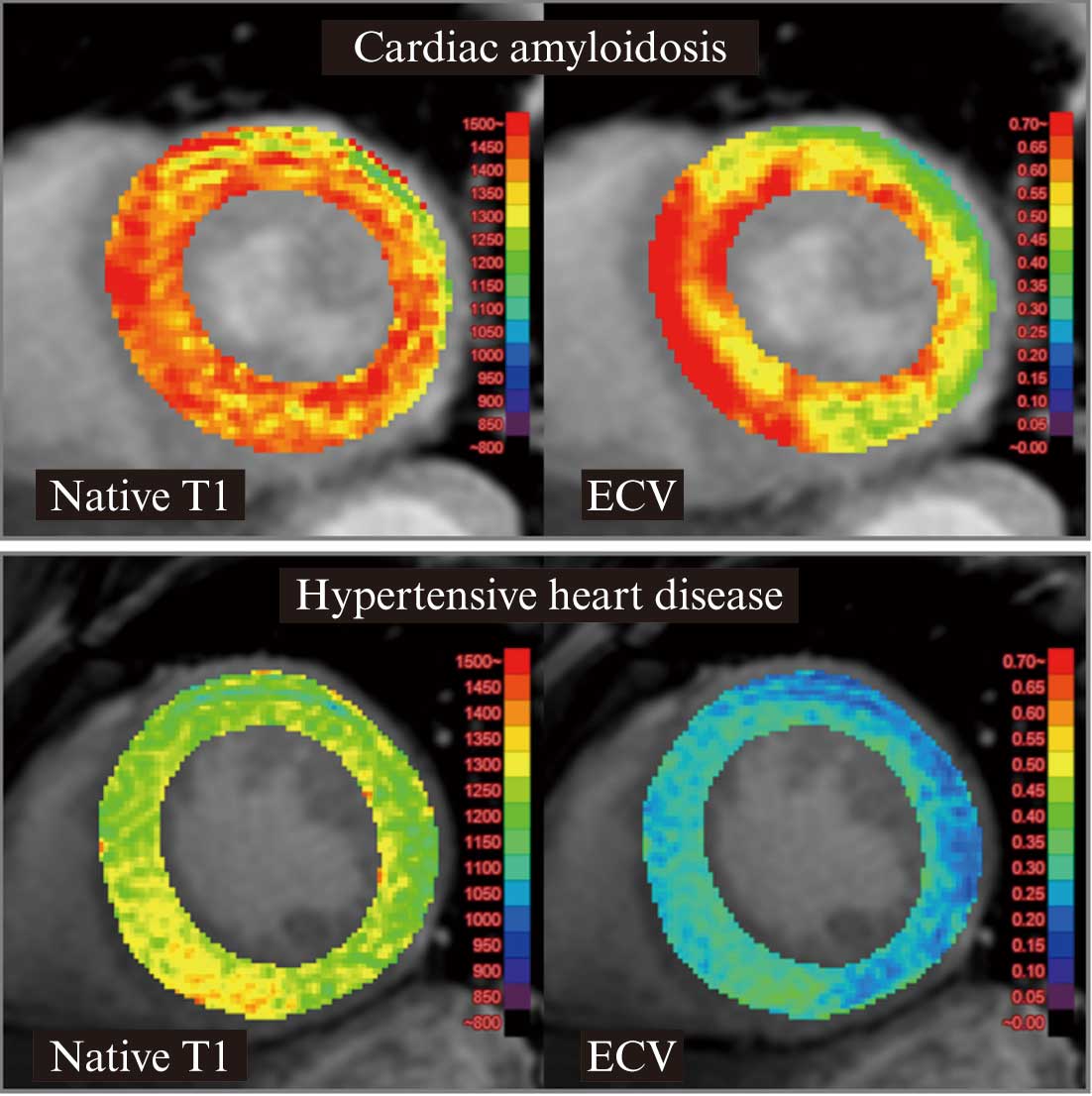
5.1.4 T2 Mapping
T2 mapping is an imaging method that measures myocardial T2 values (T2 relaxation times [ms]). T2 values reflect water in the tissue; thus, they are prolonged with the occurrence of myocardial edema or inflammation. Normal myocardial T2 values are reportedly 40–50 ms. However, as with T1 mapping, reference values must be established for each system and imaging sequence.154
According to recent studies, myocardial T2 values are significantly higher in patients with CA than in healthy controls, and values are particularly high in AL amyloidosis. Reported T2 values (with a 1.5 T MRI system) for AL amyloidosis, ATTR amyloidosis, and healthy controls were 56.6–63.2 ms, 54.2–56.2 ms, and 48.9–51.1 ms, respectively.163,164
These findings suggest that a degree of myocardial edema occurs in CA, particularly in AL amyloidosis (Figure 16). In addition, myocardial T2 is a significant predictor of prognosis in AL amyloidosis.163
5.1.5 Myocardial Strain Using CMR
Analysis of myocardial strain with 2D-STE has been established as an effective method for diagnosing CA. Myocardial strain can also be analyzed in a similar fashion in CMR. Imaging methods include the tagging and feature tracking techniques. As in echocardiography, LS has also been shown to be effective in CMR. Circumferential strain is also considered highly useful in CMR. In CA, peak circumferential strain is significantly reduced, and peak times in the myocardial segment vary (Figure 17). These findings are reportedly correlated with severity and associated with prognosis.151,165,166
Myocardial strain assessment using CMR may also be effective for detecting early-stage lesions and disease state monitoring.165
5.2 Cardiac CT
Cardiac CT can be used as an alternative to CMR for assessing myocardial late enhancement and ECV. The use of low tube voltage techniques and dual-energy CT-based virtual monochromatic imaging enables the assessment of myocardial late enhancement equivalent to that of CMR.167
In addition, ECV can be calculated based on myocardial contrast enhancement or iodine density.167,168
CT can be performed safely in patients with heart failure or in whom implantable devices have been inserted, i.e., cases in which CMR would be difficult to perform, as well as in patients undergoing hemodialysis, in whom gadolinium contrast agents are contraindicated. Thus, CT is highly practical in real-world clinical settings.
Tables 17
and
18
show cardiac CT findings in CA and recommendation levels, respectively.
Table 17.
Cardiac CT Findings in Cardiac Amyloidosis
| Imaging |
Significance and findings |
Notes |
Late enhancement
CT |
Significance: Assessment of myocardial fibrosis
· Diffuse left ventricular subendocardial late enhancement
· Transmural late enhancement in advanced stage |
· Enables three-dimensional assessment
· Iodinated contrast agents can be used in patients
undergoing hemodialysis
· Contrast resolution is inferior to that of CMR |
| CT-ECV |
Significance: Assessment of amyloid deposits and
myocardial damage
· Significantly elevated ECV value |
· Calculated with a subtraction method in conventional
single-energy CT systems
· Can be calculated with an iodine method in dual-energy
CT systems |
CT, computed tomography; ECV, extracellular volume fraction; CMR, cardiac magnetic resonance imaging.
Table 18.
Recommendations and Evidence for Cardiac CT in Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Myocardial late enhancement/ECV assessment with cardiac CT as
an alternative to CMR |
IIa |
C |
B |
IVb |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS); CT, computed tomography; ECV, extracellular volume fraction; CMR, cardiac magnetic resonance imaging.
5.2.1 Late Enhancement CT
The iodinated contrast agents used in contrast-enhanced CT demonstrate pharmacokinetics similar to those of gadolinium contrast agents in MRI. Therefore, myocardial late enhancement assessment is also theoretically possible in contrast-enhanced CT. However, the contrast resolution of CT is inferior to that of CMR. As a result, the use of late enhancement CT has been limited in clinical settings. Recently, low tube voltage techniques and dual-energy CT-based virtual monochromatic imaging have greatly improved the image quality of late enhancement CT. Although late enhancement CT may be capable of yielding the same findings as CMR for the diagnosis of CA,169
relevant evidence is currently insufficient (Figure 18).
5.2.2 CT-ECV
In late enhancement CT, myocardial contrast enhancement or iodine density values can be used to calculate ECV and assess it in a manner comparable to CMR T1 mapping.167
There are two methods for calculating ECV in CT, both of which are considered to yield comparable results: 1) a subtraction method based on myocardial contrast enhancement that can be obtained using a conventional CT system (single-energy CT system); and 2) an iodine method based on myocardial iodine density values using dual-energy CT systems.170
Similar to CMR, CT-ECV demonstrated significantly higher values in CA; thus, it appears useful for diagnosis169,171,172
(Figure 18). The typical CT imaging protocol is as follows: prospective ECG gating mode, CTDIvol, 11–25 mGy, 500–600 mgI/kg iodine contrast agent, iterative image reconstruction, and 7–10 min of scan delay time.
6. Nuclear Imaging
Nuclear imaging plays an important role in diagnosing CA. 99 mTc-PYP scintigraphy, a type of bone scintigraphy, is highly capable of diagnosing ATTR amyloidosis. 123I-metaiodobenzylguanidine (123I-MIBG) scintigraphy, a type of myocardial sympathetic innervation imaging used in the noninvasive diagnosis of disease stage, is able to detect denervation in CA and assess the state of heart failure. In addition, preclinical research has recently begun on amyloid positron emission tomography (PET), which uses amyloid-specific tracers.
Tables 19
and
20
show the nuclear imaging findings of CA and recommendation levels, respectively.
Table 19.
Nuclear Imaging Findings of Cardiac Amyloidosis
| Imaging |
Significance and findings |
Notes |
99mTc-PYP
scintigraphy |
Significance: Diagnosis of ATTR cardiac amyloidosis
· Grade 2 or 3 in visual assessment (3-h imaging)
· H/CL ratio > 1.5 (1-h imaging)
· H/CL ratio > 1.3 (3-h imaging) |
· Grade 2 may also be observed in AL cardiac
amyloidosis
· False positives may occur due to physiological
accumulation in blood pools
· Combination with SPECT is useful
· Useful for assessing prognosis |
123I-MIBG
scintigraphy |
Significance: Assessment of myocardial sympathetic
nerve function
· H/M ratio < 1.6 (delayed phase image) is a factor in
poor prognosis |
· Useful for assessing prognosis and risks
· Low disease specificity |
| Amyloid PET |
Significance: Assessment of myocardial amyloid deposits
· Significant myocardial accumulation |
· Preclinical stage
· Uses amyloid-specific tracers
· Can also be used to assess extracardiac amyloid loads |
AL, amyloid light-chain; ATTR, amyloid transthyretin; H/CL, heart/contralateral; H/M, heart/mediastinum; MIBG, metaiodobenzylguanidine; PET, positron emission tomography; PYP, pyrophosphate; SPECT, single-photon emission computed tomography.
Table 20.
Recommendations and Evidence for Nuclear Imaging for Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Diagnosis of ATTR cardiac amyloidosis with 99mTc-PYP
scintigraphy |
I |
C |
A |
IVb |
Assessment of myocardial sympathetic nerve function with
123I-MIBG scintigraphy |
IIa |
C |
B |
IVb |
| Diagnosis of cardiac amyloidosis using amyloid PET |
IIb |
C |
C1 |
V |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS); ATTR, amyloid transthyretin; MIBG, metaiodobenzylguanidine; PET, positron emission tomography; PYP, pyrophosphate.
6.1 99 mTc-PYP Scintigraphy (Bone Scintigraphy) (see CQ1)
Pyrophosphate, which possesses an affinity for calcium, has long been used to assess bone disease and acute myocardial infarction, and is generally known as a tracer in bone scintigraphy. 99 mTc-PYP scintigraphy has recently attracted attention due to the discovery that it is extremely effective for detecting ATTR (ATTRv and ATTRwt amyloidosis)1,2,173
(Figure 19). Planar imaging is acquired 1 or 3 h after the injection of 99 mTc-PYP. The 1-h imaging results in improved detection sensitivity, while the 3-h imaging results in improved detection specificity.2
The diagnosis of ATTR amyloidosis using 99 mTc-PYP scintigraphy has a sensitivity of 58–99% and a specificity of 79–100%.1,2
Most false positives are AL CA; however, if these cases are excluded, 99 mTc-PYP scintigraphy is considered to have a diagnostic specificity and positive predictive value of 100%.1
There are two methods of assessing 99 mTc-PYP scintigraphy: visual and quantitative assessment. Both of these methods are based on anterior planar images (Table 21, Figure 20).160
As the visual assessment has been established in the 3-h imaging, the same standards should not be applied in the 1-h imaging, which easily occurs high uptake in blood pools. A visual assessment of Grades 0, 1, and 2 requires differentiation of AL and/or physiological uptake in blood pools. Adding single-photon emission CT (SPECT) along with planar imaging is recommended at facilities when possible because it enables a more accurate assessment of uptake in the myocardium and blood pools.160
SPECT/CT fusion imaging is also effective as an aid in visual diagnosis. The cardiac uptake of 99 mTc-PYP has been demonstrated to be associated with prognosis. An H/CL ratio >1.6 is considered to be an independent predictor of poor prognosis.2
Although much remains unknown regarding the mechanism of uptake of pyrophosphate and other bone tracers in ATTR amyloidosis, it is suspected to be calcium-mediated. 99 mTc-hydroxymethylene diphosphonate (99 mTc-HMDP) and 99 mTc-DPD are other bone scintigraphy tracers considered to have diagnostic accuracy comparable to that of 99 mTc-PYP.1,174,175
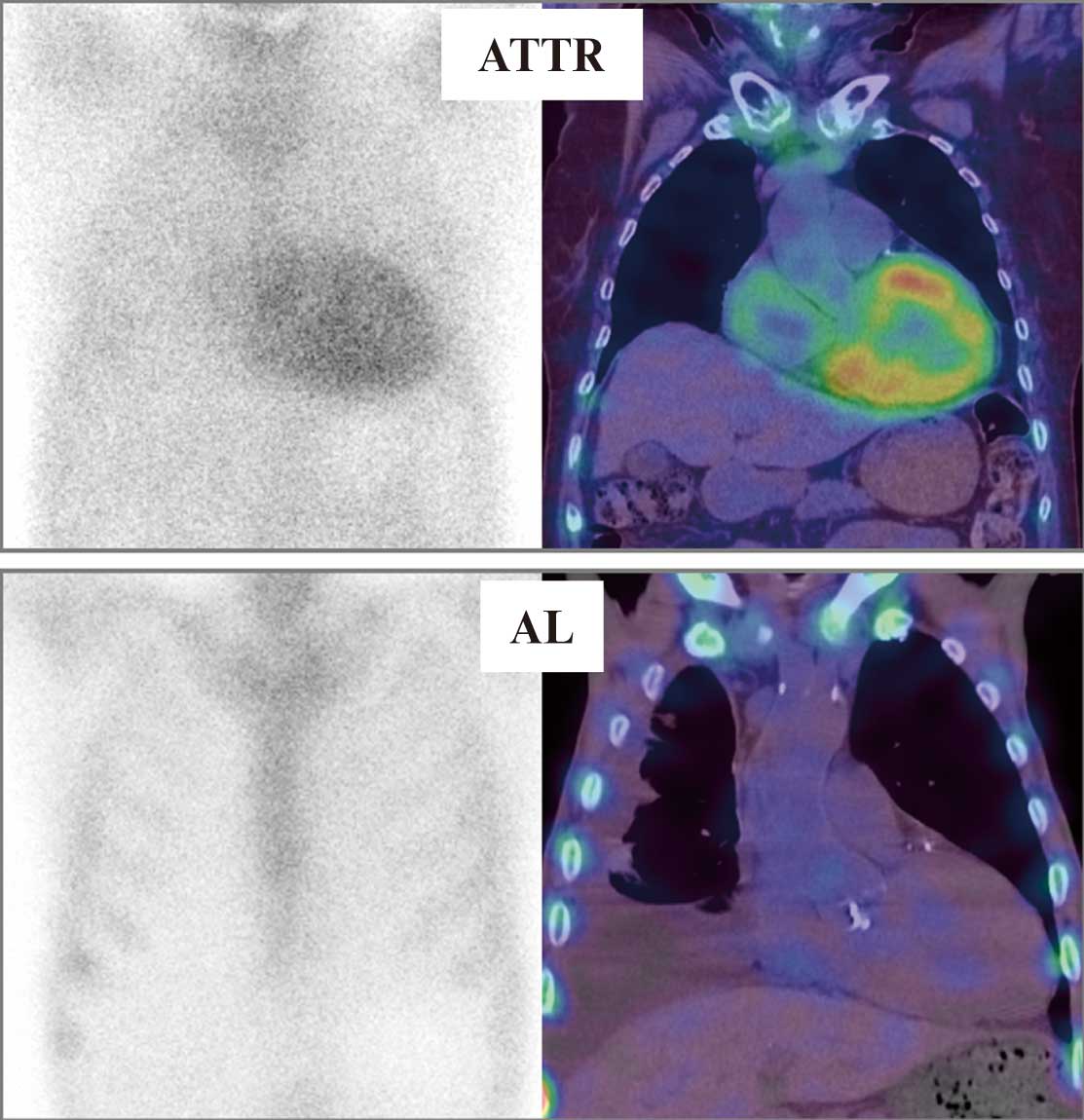
Table 21.
99mTc-PYP Scintigraphy Assessment Methods
| Visual assessment |
| Grade 0 |
No cardiac uptake |
| Grade 1 |
Mild cardiac uptake, less than rib uptake |
| Grade 2 |
Moderate cardiac uptake, equal to rib uptake |
| Grade 3 |
High cardiac uptake, greater than rib uptake |
| |
| Grades 2 and 3 are classified as positive. |
| (Note) Visual assessment should be performed using 3-h image. |
| Quantitative assessment |
| H/CL ratio > 1.5 (1-h imaging) |
| H/CL ratio > 1.3 (3-h imaging) |
| |
| H/CL: heart-to-contralateral |
The H/CL ratio is calculated by marking a region of interest in the site corresponding to the heart (H) on an anterior
planar image and another region of interest of the same size in the contralateral chest, lower lung field (CL). |
(Source: Prepared based on Dorbala S, et al. 2019160)
6.2 123I-MIBG Scintigraphy
123I-MIBG is a myocardial sympathetic nerve function imaging agent with a structure similar to that of the neurotransmitter norepinephrine. Myocardial uptake of 123I-MIBG reflects cardiac sympathetic innervation and can be used to assess myocardial denervation and other forms of sympathetic dysfunction. In addition, clearance of 123I-MIBG from the myocardium is thought to reflect sympathetic nerve activity. Imaging typically consists of anterior planar and SPECT imaging, which include early- and late-phase images taken 15 min and 3–4 h, respectively, after the injection of 123I-MIBG. These anterior planar images are used to calculate the heart/mediastinum (H/M) and washout ratios (Figure 21). 123I-MIBG can be used to assess myocardial denervation in CA. In ATTRv amyloidosis, a reduced late H/M ratio has been demonstrated to be associated with prognosis. A late H/M ratio <1.6 is considered an indicator of poor prognosis, with reported 5-year mortality rates for an H/M ratio <1.6 and ≥1.6 of 42% and 7%, respectively.176
In another study, an H/M ratio <1.6 in ATTRv amyloidosis was significantly associated with all-cause mortality, hospitalization for heart failure, pacemaker implantation, and cardiac resynchronization therapy.101

6.3 Amyloid PET
The application of amyloid PET, which uses amyloid-specific tracers, in CA has been the subject of preclinical research reports. Myocardial uptake of11
C-Pittsburgh compound B,177 18F-florbetapir,178
and 18F-florbetaben179
has been confirmed in AL and ATTR amyloidosis, and has been shown to be significantly higher in such patients compared with controls. The cardiac uptake of 18F-florbetapir and 18F-florbetaben tends to be higher in AL than in ATTR amyloidosis. Unlike the other modalities discussed here, amyloid PET can be used to assess amyloid deposition not only in the heart, but also throughout the body. Thus, amyloid PET may be capable of quantifying amyloid deposition throughout the body.
7. Cardiac Catheterization
Cardiac amyloidosis refers to a secondary form of cardiomyopathy caused by the extracellular and vascular deposition of amyloid fibrils presenting with cardiac dysfunction.27
It is characterized by left ventricular (LV) diastolic dysfunction. LV systolic dysfunction also appears at an advanced stage leading to arrhythmias, conduction disturbance, and heart failure. To make a definitive diagnosis, pathological evidence is required. On cardiac catheterization, endomyocardial biopsy (EMB) is primarily performed in patients with suspected cardiac amyloidosis. In addition, coronary angiography is performed to assess the presence of coronary stenosis if coronary artery disease is suspected. Left ventriculography can provide dynamic, volumetric, and morphologic assessments, and assess the severity of mitral valve regurgitation. In addition, LV pressure and the intraventricular pressure gradient can be measured. In patients with clinically suggested aortic valve stenosis, aortic valve area and the LV-aortic pressure gradient are measured. Pulmonary hemodynamics are assessed by right heart catheterization if needed. Electrophysiological examination is performed in patients with symptomatic bradycardia or tachyarrhythmia when considering a pacemaker or catheter ablation treatment.
7.1 Right Heart Catheterization
The importance of right heart catheterization has not been confirmed in patients with cardiac amyloidosis. The procedure is performed when it is difficult to evaluate hemodynamics on echocardiography in patients with pulmonary hypertension (PH) or refractory heart failure. PH is frequently observed in left heart disease (LHD). In fact, the number of patients with PH due to LHD is the largest within the clinical classification of PH. The pathogenesis of PH-LHD involves an increase in the pulmonary venous pressure related to increases in end-diastolic LV and left atrial pressures. In the guidelines for the classification of PH, PH-LHD is defined as group 2 PH. It is defined as post-capillary PH by an increase in mean pulmonary arterial pressure (mPAP) ≥25 mmHg and a pulmonary arterial wedge pressure (PAWP) >15 mmHg.180
To obtain accurate resting PAWP measurement, the 6th World Symposium on Pulmonary Hypertension in 2018 recommended that it could be measured at end-expiration and end-diastole181
(Figure 22). In sinus rhythm, this corresponds to the mean of the a-wave (B). In atrial fibrillation, it is appropriate to measure PAWP 130–160 ms after the onset of the QRS and before the v-wave (C) (Figure 22). Caution is needed in mitral valve regurgitation or stiff left atrial syndrome with a large v-wave, because PAWP during a whole cardiac cycle (A) can be overestimated.
7.2 Left Heart Catheterization
LV pressure can be measured directly by inserting a catheter into the LV, and LV compliance can be estimated from the LV end-diastolic pressure. Furthermore, if echocardiography suggests the presence of ventricular obstruction, the intraventricular pressure gradient can be accurately measured. In patients with clinically suggested aortic valve stenosis, the indication for surgical valve replacement or transcatheter aortic valve implantation can be assessed by directly measuring the intracardiac pressure.
7.3 Coronary Angiography
In cardiac amyloidosis, coronary angiography is indicated for patients with angina symptoms in whom revascularization may reduce myocardial ischemia and for those with suspected silent myocardial ischemia. Exercise testing, coronary computed tomography (CT), and/or myocardial perfusion scintigraphy may be considered in patients without symptoms and electrocardiographic or echocardiographic evidence of myocardial ischemia. If there is a low pre-test probability of coronary artery disease, coronary CT can evaluate coronary artery stenosis.182
Coronary angiography is recommended for symptomatic patients with a history of percutaneous coronary intervention or coronary artery bypass graft, a high pre-test probability, cardiopulmonary resuscitation for cardiac arrest, and fatal ventricular tachycardias. In patients with amyloidosis, myocardial ischemia might be present because of vascular compression by hypertrophic myocardium, and amyloid infiltration of the small intramyocardial vessels can induce angina. In such patients, the assessment of coronary lesions is limited using coronary angiography.183–185
7.4 Left Ventriculography
In patients with cardiac amyloidosis, the morphological findings are shared with many other cardiomyopathies. Misdiagnosis can occur frequently in cases with asymmetric LV hypertrophy.148
In patients with hypertrophic myocardium, dynamic, volumetric, and morphologic assessments are commonly evaluated by echocardiography or cardiac magnetic resonance imaging (MRI).186
Left ventriculography may be performed for assessment of LV characteristics in unevaluable patients using such modalities and in patients with a non-MRI-conditional pacemaker, implantable cardioverter-defibrillator, or cardiac resynchronization device. Some patients with cardiac amyloidosis present with normal cardiac contraction, and others have impaired contraction. In patients with cardiac amyloidosis, left ventriculography frequently indicates wall motion abnormalities that are mismatched to the coronary artery dominant areas and longitudinal dysfunction in mid and basal LV segments. In addition, left ventriculography can demonstrate the severity of mitral valve regurgitation.
7.5 Endomyocardial Biopsy (see also CQ2)
Despite imaging advances, EMB remains the gold standard technique to obtain pathological information on the myocardium, excluding surgical biopsy. To establish the diagnosis of cardiac amyloidosis, EMB is appropriate to differentiate from other cardiomyopathies, including hypertrophic cardiomyopathy (HCM).6,187
The incidence of serious complications related to EMB is reportedly less than 1%.188,189
However, the procedure is difficult in patients with s conduction disturbance and advanced age. For EMB, specimens are commonly collected from the right ventricular (RV) septum, although they are collected from the LV in some cases. It remains to be clarified which site of collection is better with respect to obtaining useful histological information or the incidence of complications.189
Because cardiac amyloid lesions are diffusely present through the LV and RV, there are few sampling errors in EMB.190
According to international guidelines, the indication for EMB is classified as class IIa in patients with heart failure associated with unknown restrictive cardiomyopathy such as cardiac amyloidosis and Fabry’s disease.191,192
As a common disease in cardiology, HCM is often diagnosed by familial inheritance, electrocardiography, echocardiography, and MRI without pathological evaluation by EMB. One to nine percent of suspected HCM patients were reported to have been diagnosed with cardiac amyloidosis based on the results of genetic testing, serum or urine protein electrophoresis, FLC testing, and bone scintigraphy.193–195
There is a report of the incidental diagnosis of amyloidosis in patients with HCM undergoing septal myectomy for LV outflow tract obstruction.196
Therefore, the diagnosis of patients with hypertrophic myocardium must be carefully performed, including information from EMB. The prognosis of patients with cardiac amyloidosis is markedly inferior to that of patients with HCM. EMB plays a crucial role in determining treatment strategies. The recommendations for EMB in cardiac amyloidosis and evidence levels are presented in
Table 22.
Table 22.
Recommendation of Endomyocardial Biopsy for Cardiac Amyloidosis and Evidence Levels
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Suspected cardiomyopathy with hypertrophic myocardium
undiagnosed by other modalities |
IIa |
C |
B |
II |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
8.1 Biopsy From Organs Other Than the Heart
In the pathological diagnosis of systemic amyloidosis, a high detection rate of amyloid in the organs or tissues exhibiting symptoms can be achieved. As an alternative to organs highly invasive for biopsy, such as the heart, nerves, and kidney, relatively less invasive biopsy sites, such as the abdominal wall fat, skin, digestive tract, and lips, can be used to detect amyloid deposition. If the biopsy shows amyloid deposits, a definitive diagnosis can be obtained by typing the amyloid precursor protein by immunohistochemical staining and mass spectrometry. In the pathological diagnosis of CA, the detection rate of amyloid deposits by myocardial biopsy is almost 100%, which is considered the gold standard for diagnosing this disorder. However, in some cases, especially in older patients, performing a myocardial biopsy involves an increased risk for complications. In CA, it is necessary to distinguish between the three typical types of amyloidosis, namely AL, ATTRwt, and ATTRv amyloidosis, and to introduce treatment specific to each disease at an early stage. To establish the diagnosis of AL amyloidosis, it is important to confirm the presence of amyloid deposits derived from immunoglobulin light chain in any organ. On the other hand, in the early stages of ATTRwt amyloidosis, amyloid deposits are difficult to detect in biopsies at sites other than the heart; thus, clinical diagnostic methods using diagnostic imaging such as pyrophosphate scintigraphy have been proposed in Europe and the US.1,160
8.1.1 Abdominal Fat Pad Biopsy
Abdominal fat pad biopsy is a safe and simple procedure; hence, it can be repeated over time for outpatient screening and follow-up. After the induction of local anesthesia in the abdominal fat pad around the umbilicus, the subcutaneous tissue is punctured using an 18G needle, which is reinserted several times while applying negative pressure to the syringe. When the needle is removed with the application of negative pressure, a small amount of adipose tissue is aspirated into the syringe. The collected tissue is crushed between two glass slides to prepare a smear or a formalin-fixed paraffin section, and pathological evaluation is performed by Congo red staining.197
The amyloid deposits around the adipocytes and small blood vessels contained in the adipose tissue (Figure 23). Abdominal fat pad biopsy can produce false-negative results because of the small amount of sample collected, but multiple biopsies may reduce this risk. The amount of tissue around the adipose tissue is usually small, and it is difficult to perform typing of the amyloid precursor proteins by immunohistochemical staining or mass spectrometry. The amyloid detection rate of abdominal liposuction biopsy is reported to be 79–84% for systemic AL amyloidosis,198–200
45–67% for ATTRv amyloidosis,190,200
and 14–15% for ATTRwt amyloidosis.190,200
In ATTRwt amyloidosis, the amount of amyloid deposited in adipose tissue is small and often sparse. If amyloid deposits are not detected using this method, deep and large skin tissue may be collected by surgical skin incision, as described below.201

8.1.2 Skin Biopsy
Skin biopsy by surgical incision or skin punch biopsy using a biopsy trepan is slightly more invasive than abdominal fat biopsy, but can preserve tissue morphology and collect a larger amount of tissue. Furthermore, in skin biopsy, the amyloid precursor protein can be identified by immunohistochemistry (IHC); hence, the possibility of false-negative results due to sampling errors can be reduced. After the induction of local anesthesia, the skin is cut into a spindle shape using a scalpel, and cross-sectional pathological images of the epidermis, dermis, and subcutaneous adipose tissue are obtained. In a punch biopsy, the epidermis, dermis, and subcutaneous adipose tissue are collected in a cylindrical shape using a biopsy trepan (inner diameter, 3–6 mm). Although the amount of sample collected is limited by the size of the trepan in skin biopsy, it is considered a simpler procedure that results in fewer scars than does abdominal fat pad biopsy. In ATTRwt amyloidosis, the utility of abdominal fat pad needle biopsy is limited because the amount of amyloid deposits is limited in the adipose tissue. Conversely, it has been reported that detection sensitivity can be improved by obtaining deep and large skin tissue via surgical incision.201,202
Moreover, in AL and ATTRv amyloidosis, the amyloid detection rate by skin biopsy is higher than that by abdominal fat pad biopsy.55,202
8.1.3 Lip Salivary Gland Biopsy
Lip salivary gland biopsy does not require sedation and is associated with few serious bleeding complications. Moreover, in lip salivary gland biopsy, the submucosal tissue can be reliably collected. As a general method, the mucosa side of the lower lip is locally anesthetized and incised, and a small amount of salivary gland tissue is collected. Next, after achieving hemostasis, suturing is performed. Amyloid deposits are frequently detected in small blood vessels and around the salivary glands, and salivary gland biopsy can be performed to obtain the amount of amyloid deposits required to identify the amyloid precursor protein by immunohistochemical staining.203
The detection rate of lip salivary gland biopsy is 81–89% for systemic AL amyloidosis204,205
and 91–100% for ATTRv amyloidosis.206,207
However, no studies evaluating ATTRwt amyloidosis using lip salivary gland biopsy have been conducted.
8.1.4 Gastrointestinal Biopsy
Gastric/duodenal biopsy using an endoscope is frequently performed to establish a diagnosis of systemic amyloidosis. In the gastrointestinal tract, amyloid deposits are detected on the muscularis mucosa and submucosal blood vessels. Collecting amyloid deposits on only the mucosal surface does not allow the presence of amyloid deposits in the submucosal blood vessels to be evaluated, which produces false-negative results. Therefore, it is necessary to collect amyloid deposits in the submucosa thoroughly while paying careful attention to gastrointestinal perforation and bleeding. Specifically, in ATTRwt amyloidosis, the amyloid deposits, which are nondiffusely distributed, are frequently detected in the submucosal vascular wall only (Figure 24); hence, biopsies at multiple sites are required. In the upper gastrointestinal tract, the detection rate of amyloid is higher in the order of the duodenum (part 2, bulbs) and stomach (vestibule).208,209
Using gastrointestinal biopsy tissue, the amyloid precursor protein can be identified by immunohistochemical staining or mass spectrometry. The amyloid detection rate in gastrointestinal tract biopsy is reported to be 72–95% for systemic AL amyloidosis210,211
and 86–100% for ATTRv amyloidosis.212,213
In a study comprising a few cases of ATTRwt amyloidosis, amyloid was detected in three of the eight cases (38%).26

8.1.5 Bone Marrow Biopsy
In AL amyloidosis, bone marrow biopsy is performed to evaluate the presence of abnormal plasma cells. In AL amyloidosis, bone marrow biopsy can detect 57–60% of amyloid deposits;214,215
therefore, bone marrow biopsy with Congo red staining is recommended when assessing the bone marrow. Since the amount of amyloid deposits in bone marrow biopsy tissues is often significantly small, it is frequently difficult to identify the amyloid precursor protein by IHC or mass spectrometry. Positive rates of amyloid deposition in ATTRv and ATTRwt amyloidosis in bone marrow biopsies are reported to be 41% and 30%, respectively.190
8.1.6 Renal Biopsy
In patients experiencing significant renal failure, such as proteinuria and nephrotic syndrome, renal biopsy frequently leads to the establishment of a diagnosis, especially in AL and AA amyloidosis. Amyloid deposits are usually detected in the glomeruli, interstitium, and vascular wall. Since renal biopsy is invasive, amyloid deposition can be detected in other biopsy sites, including the abdominal fat pad, skin, lip salivary gland, and gastrointestinal tract, in patients suspected to have systemic amyloidosis. ATTRv amyloidosis also presents with renal failure due to renal amyloidosis and nephrotic syndrome at the end stage, but renal biopsy is rarely performed because diagnosis has already been established by biopsy from other sites. In ATTRwt amyloidosis, no or only a small amount of amyloid is deposited in the kidney.
8.1.7 Nerve Biopsy
Nerve biopsy is performed to identify the cause of peripheral neuropathy of unknown cause. Regarding the biopsy site, it is common to select a sural nerve that does not contain a motor nerve component. ATTRv amyloidosis frequently develops with peripheral neuropathy, and the detection of amyloid deposits on nerve biopsy occasionally leads to the diagnosis of this disease. Amyloid deposits are predominantly proximal in peripheral nerves such as the dorsal root ganglia and autonomic ganglia;216
therefore, sural nerve biopsy might fail to detect amyloid deposits in the early stage of the disease.217
Although AL amyloidosis frequently presents with a variety of sensory, motor, and autonomic dysfunctions,218
invasive nerve biopsy is rarely performed if the disease is suspected.
8.1.8 Tendon/ligament Tissue Biopsy
In ATTR amyloidosis, amyloids are frequently found in various tendon and ligament tissues, including the lateral carpal ligament and the ligamentum flavum.20,22,219–221
Specifically, in bilateral carpal tunnel syndrome in older individuals, the rate of detection of TTR amyloid deposition is high when biopsy is performed during lateral carpal ligament release surgery.20,219
TTR amyloid deposition in the tendons and ligaments is considered to occur prior to cardiac lesions; therefore, if TTR amyloid deposition is observed in these tissues, it is necessary to monitor the appearance of the cardiac lesions carefully. On the other hand, in the early stages of ATTRwt amyloidosis, TTR amyloid deposition is frequently localized to the tendon and ligament tissues; therefore, it should be noted that tendon and ligament amyloid deposition cannot necessarily establish the pathological diagnosis of CA.
8.1.9 Other Organs
High levels of ALP or liver damage due to amyloid deposition may be observed in AL amyloidosis, but liver biopsy for the diagnosis of this disease is considered to result in a high risk of bleeding, and thus, is not generally recommended.
Table 23
shows detection rate of amyloid at screening biopsy sites other than the heart in CA.
Table 24
shows the recommendations and evidence for biopsy other than the heart in CA.
Table 23.
Detection Rate of Amyloid at Screening Biopsy Sites in CA
*When performing deeper and wider biopsies than usual. **Examination in a few cases.
Table 24.
Recommendations and Evidence for Biopsy (Other Than the Heart) in Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Perform abdominal fat pad biopsy as a means of detecting amyloid
other than myocardial biopsy when cardiac amyloidosis is
suspected |
I |
C |
B |
IVa |
Perform lip salivary gland biopsy*, skin biopsy**, and gastrointestinal
tract biopsy** as a means of detecting and typing other than
myocardial biopsy when cardiac amyloidosis is suspected |
I |
C |
B |
IVa* |
| IVb** |
Repeat biopsies from other sites (abdominal fat, skin, digestive
tract, labial salivary glands) when single biopsy failed to detect amyloid
deposits |
IIa |
C |
B |
IVa |
Perform abdominal fat pad biopsy, skin biopsy, gastrointestinal
biopsy, and labial salivary gland biopsy as a means of assessing
the severity and prognosis of cardiac amyloidosis |
III |
C |
B |
VI |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
8.1.10 Important Points in Biopsy From Organs Other Than the Heart
Abdominal fat pad biopsy is considered to be the simplest procedure, and to be beneficial as a screening test because it is minimally invasive and has a relatively high amyloid detection rate. When CA is clinically suspected, abdominal fat pad biopsy, along with M protein, urinary Bence Jones protein, free light-chain protein, and pyrophosphate scintigraphy are considered beneficial. On the other hand, typing the amyloid precursor protein by abdominal fat pad biopsy is considered difficult because of the small amount of tissue collected. The detection rates of lip salivary gland, stomach/duodenum, and skin biopsy are sufficiently high in AL and ATTRv amyloidosis, and these biopsy sites are considered beneficial when amyloid precursor protein typing (immunohistochemical staining and tissue mass spectrometry) is required. It should be noted that the amyloid detection rate in screening biopsies is not necessarily 100% at any site, and if biopsy from one site fails to detect amyloid deposition, then biopsy from another site or repeat biopsy is recommended. Conversely, in ATTRwt amyloidosis, amyloid detection rates at biopsy sites other than the heart are relatively insufficient. Therefore, biopsies from multiple sites are frequently required for the histological detection of amyloid, and in some cases, the detection of amyloid deposits in biopsy specimens from sites other than the heart can be difficult.
8.2 Endomyocardial Biopsy
CA is essentially diagnosed based on the pathological findings that amyloid deposits are recognized in the myocardial tissues on autopsy or EMB. Amyloid is pathologically defined as substances that turn red with Congo-red staining and show green birefringence through the polarized light microscope, known as the apple green sign. Amyloid deposits can be detected by EMB in almost all patients with CA, leading to the idea that “sampling error” need not be a concern during the process of investigating amyloid by EMB.
8.2.1 Light Microscopic Findings
It is important to suspect CA on investigating endomyocardial tissues conventionally stained by hematoxylin-eosin and Masson-trichrome. It may be difficult to differentiate amyloid from interstitial fibrosis by hematoxylin-eosin staining because both tissues can be observed as pale-pink vitreous amorphous (Figure 25A). On the other hand, Masson-trichrome staining can easily lead to precise differentiation: amyloid seems to be bluish-grey, whereas fibrosis looks clear blue (Figure 25B). Although direct fast scarlet 4BS or Dylon staining can be used in clinical practice, in principle, amyloid is defined using Congo-red staining (Figure 25C) and the polarized light microscope (Figure 25D).222
It is useful to ensure no co-staining of the internal elastic membrane to avoid false-positive results during these diagnostic processes.
8.2.2 Electron Microscopy Findings
Amyloid fibrils are observed as felt-like unbranched structures, 8–12 nm in width, in a disarrayed configuration (Figure 26). Even when amyloid deposits are not seen on light microscopy, electron microscopy can rarely show them. When evaluated by electron microscopy, histological tissues from EMB must be fixed by glutaraldehyde. When considering CA clinically as one of the differential diagnoses, part of the biopsied samples should be fixed immediately for electron microscopy.223
8.2.3 Immunohistochemical Analysis
Following the detection of amyloid, immunohistochemical analysis should be performed using specific antibodies to identify amyloid subtype.224
Above all, subtyping for transthyretin (Figure 25E), κ chain, or λ-chain is especially important in CA. However, specific antibodies have not been fully developed and widely spread. So it is not rare to get definite subtyping by the stained findings currently.
9. Pathology/Mass Spectrometry
9.1 Histopathological Differential Diagnosis of CA
Amyloid typing is essential in determining the treatment plan in patients with CA. After confirming amyloid deposition by Congo red staining, IHC is usually recommended. Since AL, AA, ATTR, and Aβ2-microglobulin amyloidosis are common in Japan, IHC is performed as the first step using antibodies recognizing these amyloid proteins (Class I, Grade A). However, amyloid proteins in AL amyloidosis may often be unrecognized by commercially available antibodies against κ- or λ-light chain because they are mainly composed of variable region fragments of immunoglobulin light chain.224
Therefore, it is recommended to use antibodies developed for diagnosis in AL amyloidosis225
(Class IIa, Grade B). At present, amyloidosis diagnostic services using those special antibodies are provided by the Amyloid Research Committee of the MHLW in Japan (http://amyloidosis-research-committee.jp/consultation/).
In ATTR amyloidosis, it is impossible to distinguish ATTRwt from ATTRv amyloidosis using only IHC; therefore,
TTR
gene analysis is necessary. In Japan, the most common
TTR
variant is V30M (p.V50M), which causes severe peripheral and autonomic neuropathy as well as cardiac involvement. However, some
TTR
variants, such as V122I and T60A, which cause severe CA, have also been detected in Japanese patients.226,227
In patients with these variants, the clinical features typically include late-onset cardiac failure, and the neuropathic symptoms, except for carpal tunnel syndrome, are very mild, which is similar to ATTRwt amyloidosis.
9.2 Amyloid Typing Using Mass Spectrometry
9.2.1 Detection of Serum Variant
TTR
As the majority of
TTR
variants are one amino acid substitutions, such as V30M, the presence of variant
TTR
in serum can be confirmed by the detection of the difference of molecular weight in one amino acid substitution using mass spectrometry.228
In healthy controls and patients with ATTRwt amyloidosis, only a wild-type
TTR
peak is seen on mass spectrometry. On the other hand, both wild-type and variant
TTR
peaks are detected in patients with ATTRv amyloidosis. However, the peak of variant
TTR
becomes undetectable in the case of small differences in the molecular weight, such as in E61K and E89Q.
9.2.2 Amyloid Typing Using Mass Spectrometry
When the type of amyloid protein is not confirmed by IHC, an investigation of amyloid protein is needed. With recent advances in mass spectrometry, the investigation of quite small amounts of amyloid protein is possible,229,230
and tiny biopsied samples, such as cardiac biopsy, can be analyzed. Even in formalin-fixed, paraffin-embedded samples, amyloid typing can be performed using LMD-LC-MS/MS.231
Protein analysis using LMD-LC-MS/MS is currently one of the most useful diagnostic procedures, and shows higher diagnostic accuracy than IHC.232
In LMD, deposited amyloid can be selectively isolated with a laser cutter (Figure 27A–F). Amyloid is composed of amyloid fibril protein and various amyloid-associated proteins, such as serum amyloid-P component; therefore, which protein is a main body of amyloid should be determined carefully. In Japan, amyloid protein typing using LMD is mainly carried out at Kumamoto University and Shinshu University, and diagnostic services can also be ordered online through the Amyloid Research Committee of the MHLW in Japan (http://amyloidosis-research-committee.jp/consultation/) (Table 25).

Table 25.
Levels of Recommendation and Evidence of Histopathological Diagnosis in Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| IHC after detecting amyloid deposits |
I |
B |
A |
IVb |
| Mass spectrometry for cases undiagnosed by IHC |
I |
B |
A |
IVb |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
10.1 Role of Genetic Testing for the Diagnosis of CA
Genetic testing in patients with CA is necessary for the definitive diagnosis of ATTRv and ATTRwt amyloidosis (Table 26). Genetic testing may also be performed for the purpose of presymptomatic genetic testing in at-risk relatives. The target gene for testing is
TTR.
Table 26.
Recommendations and Evidence Levels of Genetic Testing for the Definitive Diagnosis of ATTR-Type Cardiac Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| Detection of TTR variant |
I |
C |
A |
V |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
TTR
is composed of four exons and encodes 147 amino acids. Mature TTR protein consists of 127 amino-acids, as codons 1–20 encode a 20-amino-acid signal sequence. The most common pathogenic variant in Japan is c.148G>A (p.V50M). Historic protein numbering was based on the mature protein after cleavage of a 20-amino-acid signal sequence (e.g., p.Val50Met would be referred to as Val30Met). In this guideline, both historic and standard protein numbering as recommended by the Human Genomes Variation Society are employed.
In Japan, genetic testing should be performed in accordance with the “Guidelines for Genetic Tests and Diagnoses in Medical Practice” approved by the Japanese Association of Medical Sciences,233
and “genetic testing for the diagnoses of patients who have developed a disease” and “genetic testing for presymptomatic diagnosis” should be distinguished (Table 27).
Table 27.
Difference Between “Genetic Testing for the Diagnoses of Patients Who Have Developed a Disease” and “Genetic Testing for Presymptomatic Diagnosis”
| |
Purpose of the test |
Physician and medical
staff in charge of the test |
Cost of the test |
Genetic testing for patients
who have already
developed a disease |
Establishment of diagnosis
of ATTRv and ATTRwt
amyloidosis |
Attending doctor
(e.g., Cardiologist) |
Covered by public health
insurance including
genetic counseling fee |
Genetic testing for
presymptomatic diagnosis |
Prediction of disease
development of ATTRv
amyloidosis beforehand |
Genetic counseling team
• Clinical geneticists
• Genetic counselor
• Clinical psychologist |
Not covered by public
health insurance |
10.2 Genetic Testing for the Diagnoses of Patients Who Have Developed a Disease
Definitive diagnosis of patients with CA is essential to choose the optimal therapeutic strategy, and thus, genetic testing should be performed for patients who are suspected as having ATTR amyloidosis. However, sufficient consideration is necessary, as variants in the
TTR
gene have a profound impact on relatives. In particular, patients should be informed about the purpose and implication of the test, as well as the mode of inheritance, clinical course, prognosis, and therapy for the disease prior to testing. In addition, possible conditions after the results are obtained should be indicated. If necessary, the attending physician should arrange genetic counseling by experts so that patients can receive support for autonomous decision-making.
V122I (p.V142I) is a representative variant that shows a cardiac-dominant phenotype, and is found in 3.9% of the African-American population.234
Recently, a patient with this variant was also reported in Japan.226
In addition, several variants, including T60A (p.T80A), D38A (p.D58A), and S50I (p.S70I), have been associated with the cardiac-dominant phenotype in Japan. It is not rare that late-onset V30M patients show the cardiac-dominant phenotype. A considerable number of patients with late-onset ATTRv amyloidosis do not have a familial history of amyloidosis.42
Therefore, genetic testing is needed to differentiate ATTRwt from ATTRv amyloidosis, even in sporadic older patients.
In Japan,
TTR
gene analysis is covered by public health insurance (medical remuneration points: 3,880) in patients who are suspected as having ATTRv amyloidosis. A genetic counseling fee (medical remuneration points: 1,000) can be added when the results of genetic testing are disclosed at genetic counseling.
10.3 Genetic Testing for Presymptomatic Diagnosis
Patients with hereditary disorders often do not wish to disclose information about the disease to at-risk relatives. However, with early diagnosis, ATTRv amyloidosis is a treatable disease, and thus, the provision of information to at-risk relatives is highly important in terms of health management.235
Actually, it has been reported that the percentage of presymptomatic testing usage is higher in ATTRv amyloidosis than in untreatable diseases.236,237
It is desirable that the provision of information to at-risk relatives and presymptomatic testing be managed by the genetic counseling department. Presymptomatic testing of hereditary disorders should be managed in accordance with the “Guidelines for Genetic Tests and Diagnoses in Medical Practice” approved by the Japanese Association of Medical Sciences,233
and performed by an adequate genetic counseling team. As ATTRv amyloidosis is an adult-onset disease, it is necessary to confirm that 1) the client is an adult, 2) the client is willing to take the test, and 3) the client has sufficiently understood the information concerning the mode and risk of inheritance, clinical course, prognosis, and available therapeutic strategies of the disease, purposes and implications of the test, and possible conditions after the result is obtained.
ATTRv amyloidosis is an autosomal-dominant genetic disorder, and patients are usually heterozygous for a
TTR
pathogenic variant. Therefore, each asymptomatic child of a patient has a 50% risk of inheriting the
TTR
pathogenic variant.235
In early-onset families with the V30M variant in endemic areas, the penetration of the disease is approximately 100%. However, the penetration of the disease is not so high in late-onset families with the V30M variant in non-endemic areas, especially female carriers, who sometimes do not develop the disease throughout their lifetime. As the male:female ratio is approximately 4:1 in late-onset V30M patients in non-endemic areas,42
penetration is considered to be low in female patients. A limited number of patients with biallelic
TTR
pathogenic variants have been reported. In such a situation, all offspring of a proband will inherit the variant gene.235
When presymptomatic tests identify an asymptomatic variant carrier, the carrier should be followed on a periodic basis by the genetic counseling and clinical departments (e.g., Department of Cardiology, Department of Neurology), and psychological support and screening tests for the onset of amyloidosis should be provided.238
It should also be noted that presymptomatic testing is not covered by public health insurance in Japan.
Recommendations and evidence levels of genetic counseling for relatives of and patients with ATTRv amyloidosis are summarized in
Table 28.
Table 28.
Recommendations and Evidence Levels of Genetic Counseling for Patients and Relatives of ATTRv Amyloidosis
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| Genetic counseling for patients with ATTRv amyloidosis |
I |
C |
C1 |
V |
Genetic counseling for asymptomatic relatives of ATTRv
amyloidosis patients |
I |
C |
C1 |
V |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
Aortic stenosis combined with ATTR cardiac amyloidosis has attracted attention in recent years.239,240
Myocardial amyloid deposits are observed in 6% of patients aged ≥65 years with advanced aortic stenosis who have undergone aortic valve replacement.241
Another study reported that 99 mTc pyrophosphate scintigraphy detected myocardial amyloid deposits in 16% of patients who underwent transcatheter aortic valve implantation (TAVI) (mean age, 84±6 years, 68% male).29
The patients with positive results in 99 mTc pyrophosphate scintigraphy were of advanced age (mean age, 86.3±5.7 years). Positive results were more common among men than women (22% vs. 4%, respectively). Many patients with positive results demonstrated prolonged QRS duration; right bundle branch block; and low-flow low-gradient aortic stenosis. In addition, one study observed occult cardiac amyloidosis in 13.9% of advanced aortic stenosis patients prior to TAVI.242
In advanced aortic stenosis combined with ATTR cardiac amyloidosis, therapeutic intervention involves high risk. Some studies report that symptoms improve little despite treatment and prognosis is poor.241,243–245
However, occult amyloid deposition is commonly observed in elderly people.25
Different severities of cardiac amyloidosis may affect prognosis differently. In the examination of therapeutic interventions for aortic stenosis in the elderly, the coexistence of ATTR cardiac amyloidosis must always be considered. Treatment suitability should be determined on a case-by-case basis based on an accurate assessment of the severities of ATTR cardiac amyloidosis and aortic stenosis.
Both ATTR cardiac amyloidosis and advanced aortic stenosis result in left ventricular hypertrophy, hindering the suspicion of coexistent ATTR cardiac amyloidosis. However, ATTR cardiac amyloidosis can be suspected based on various red flags (carpal tunnel syndrome, etc.) or late enhancement or high extracellular volume in contrast-enhanced CT prior to TAVI.246
The causal relationship between aortic stenosis and ATTR cardiac amyloidosis is not well understood. In cases of surgically removed heart valves, amyloid in aortic stenosis was not only observed frequently (74%), it was observed more frequently than in aortic regurgitation or mitral valve stenosis.247
Aortic stenosis-induced valve degeneration, inflammation-induced atherosclerotic changes, and high shear stress are suggested to potentially result in the progression of aortic stenosis by promoting amyloid deposition in the aortic valve. However, cardiac load associated with aortic stenosis may lead to left ventricular remodeling and promote TTR amyloid deposition, resulting in ATTR cardiac amyloidosis.240
III. Treatment of Cardiac Amyloidosis (CA)
In general, heart diseases are roughly classified into pump failure and rhythm abnormality.
Figure 28
shows the overview of treatment for cardiac amyloidosis by stage of heart failure. The recommendations and evidence levels for drug therapy in patients with cardiac amyloidosis are presented in the following
Table 29.
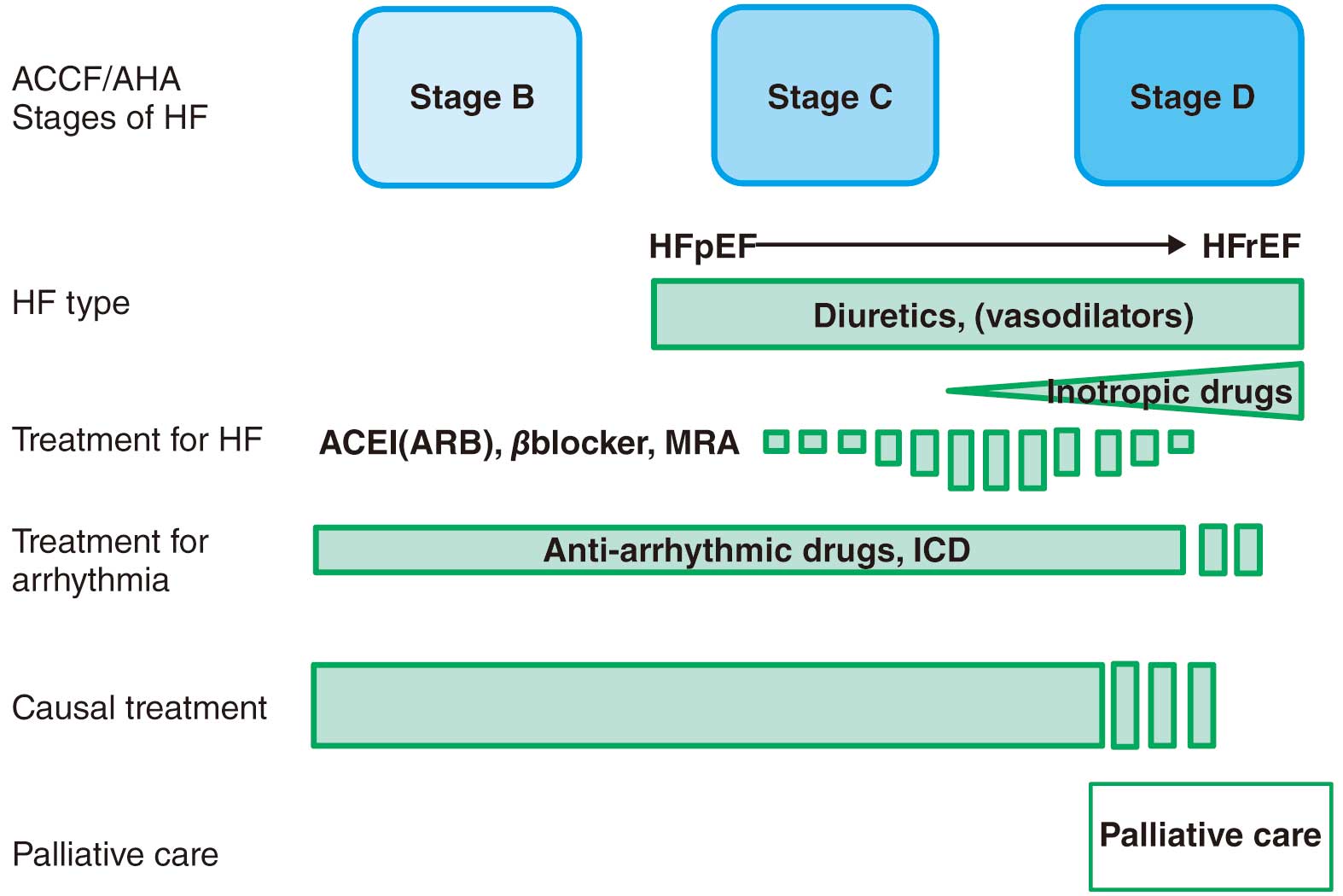
Table 29.
Recommendations and Levels of Evidence for Drug Therapy in Patients With CA
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| Diuretics for fluid retention |
I |
C |
C1 |
V |
| Nitrates or carperitide for pulmonary congestion in acute settings |
IIa |
C |
C1 |
V |
Catecholamines or phosphodiesterase inhibitors in pump failure
with low output |
IIa |
C |
C1 |
V |
Tolerated dosing of ACE inhibitors, β-blockers and aldosterone
antagonists to reduce clinical events |
IIb |
C |
C1 |
V |
| DOAC for AF |
I |
C |
B |
V |
| DOAC for AT with LV dysfunction |
IIa |
C |
C1 |
V |
| β-blocker for AF tachycardia |
II
(case by
case) |
C |
C1 |
V |
NonDPH Ca antagonist for ATTR amyloidosis with preserved
systolic function |
IIb
(case by
case) |
C |
C2 |
V |
NonDPH Ca antagonist for ATTR amyloidosis with impaired
systolic function |
III |
C |
C2 |
V |
| NonDPH Ca antagonist for AL amyroidosis |
III |
C |
C2 |
V |
| Digitalis for AF |
III |
C |
C2 |
V |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
1.1 Basic Policies for CA Treatment
As a clinical phenotype in CA patients, cardiologists encounter heart failure (HF) more often than arrhythmias.69
F treatment generally consists of two categories: one is to prolong lives through the prevention of disease progression, and the other is to relieve signs and symptoms while reducing the risk of death through improving hemodynamic deterioration. Furthermore, the former consists of two components: one is the intervention for long-lasting disease progression commonly seen in chronic HF, and the other is to treat the cause of HF. The basic policies for CA management are to deal with symptomatic complaints based on HF, together with the consideration of two therapeutic possibilities: general HF pharmacotherapy and causal treatment for CA.
1.2 Treatment to Prevent HF Progression
Few clinical studies have focused on HF management in CA patients. HF patients with left ventricular systolic dysfunction should receive pharmacological treatment generally recommended based on the clinical evidence, such as ACE inhibitors, angiotensin II receptor blockers, β-blockers, and aldosterone antagonists.5
However, there have been no trials so far to show their prognostic benefits in HF with preserved ejection fraction resembling the clinical phenotypes in most CA cases.5
Moreover, low cardiac output can be easily seen through the restrictive compromise in CA. The neurohumoral modulator agents described above not only have uncertain properties for improving the prognosis in diastolic dysfunction, but they may also induce adverse effects such as hypotension and renal impairment.249
Accordingly, causative therapy for amyloidosis has to play a major role to prevent disease progression in CA cases. It may be a wise policy to use neurohumoral modulators carefully in order to relieve HF symptoms partly through vasodilation and diuresis, as long as critical complications are not induced.
1.3 Treatment to Ameliorate HF Signs and Symptoms
Additional drugs should be given to patients with signs of congestion and low cardiac output. Although vasodilators and diuretics are generally used for the treatment of congestion, vasodilators can play a limited role because of the potential risk of hypotension occasionally seen in CA.248
Diuretic agents including loop diuretics have frequently been used to relieve congestion.249
However, intravascular volume depletion may cause hypotension in cases of restrictive hemodynamics, leading to diuretic resistance. Tolvaptan may be a therapeutic option in cases with loop-diuretic resistance presumably through aggressive fluid removal with preservation of intravascular volume by refilling from the extravascular space,250
even in cases with renal dysfunction and hyponatremia.
Inotropic drugs such as catecholamines and phosphodiesterase inhibitors are used for the treatment of low perfusion. These drugs have mainly been used temporarily in the acute phase, but oral inotropic agents may be unavoidable in cases with persistent low output during the chronic phase. Although not often recommended, oral inotropics may occasionally lead to relief of HF symptoms or avoid subsequent clinical events. Especially in patients in sinus rhythm, one must be careful not to reduce the heart rate excessively, because an increased heart rate has a compensatory role in a restrictive heart with low stroke volume.248
1.4 Palliative Care
Even with advances in therapy aimed at the cause, end-stage CA will continue to result in life-threatening conditions and a poor prognosis. Advance care planning is an important step to help individual patients to prepare for future changes in their disease condition. In addition, many CA patients suffering from general pain should be supported by healthcare professionals with multidisciplinary approaches.5
CA is a representative disease for considering palliative care (Table 30).
Table 30.
Recommendations and Levels of Evidence for Palliative Care for Patients With CA
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Conduct advance care planning (ACP) in which physicians share
decisions with the patient and his/her family members about
treatment and care before the patient becomes difficult to make his
or her own decisions |
I |
B |
B |
II |
Continued treatment to manage heart failure and complications and
relieve symptoms |
I |
C |
B |
II |
Multidisciplinary team-based frequent assessment for the patient’s
physical, mental and spiritual needs |
II |
C |
C1 |
VI |
(Adapted from Japanese Circulation Society and the Japanese Heart Failure Society Joint Working Group. 20185)
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
Patients with CA have various supraventricular (e.g., AF, atrial flutter, atrial tachycardia, conduction disturbance) and/or ventricular arrhythmia (e.g., ventricular tachycardia, ventricular fibrillation, pulseless electrical activity [PEA]). However, no evidence regarding the relationship between the prognosis of CA and these arrhythmias has been established because no international cohort or interventional studies have been conducted. Although a guideline is already available from the JCS, physicians should choose the therapy individually according of the characteristics of CA (e.g., etiology, general status, stage of amyloidosis, effectiveness of treatments, prognosis). Therefore, in this guideline, we have provided notes in regard to the use of previous guidelines for patients with CA.
2.1 Atrial Fibrillation, Atrial Flutter, and Atrial Tachycardia
In patients with CA, amyloid fibers deposit in the myocardial interstitial matrix. Diastolic dysfunction due to LV hypertrophy increases the LA pressure. The deposition of amyloid fibers in the atrium also results in conduction disturbances. These phenomena involve AF and/or other atrial arrhythmias. Sanchis et al. reported that 44% of 238 patients with CA already had AF at diagnosis (persistent AF: 60%; nonpersistent AF: 40%). AF was found in 26% of patients with AL and 60% with ATTR amyloidosis. Overall, in the observation period, AF was found in 64% of patients with CA.102
Pinney et al. also reported that 11% of patients with AL and 46% of those with ATTR amyloidosis had AF at diagnosis.70
The prognosis of patients with AF was worse than that of patients without AF.251
The prognosis with permanent AF did not differ from nonpermanent AF.102
2.2 Anticoagulant Therapy for AF
Edward et al. reported that transesophageal echocardiography revealed spontaneous echocardiogram contrast and atrial thrombi in 67% and 28% of patients with CA accompanied by AF, respectively; however, no differences were found between AL and other CAs.252
Furthermore, intracardiac mural thrombi were found in 35–50% of patients with CA without AF. AL amyloidosis, AF, LV diastolic dysfunction, tachycardia, and RV hypertrophy were independent risk factors for thoromboembolism.63,122
Therefore, anticoagulant therapy should be prescribed for all patients with CA accompanied by AF after an evaluation of hemorrhage risk. Patients with atrial tachycardia and systolic/diastolic dysfunction should also receive anticoagulant therapy. In patients who require tooth extraction or a surgical procedure, anticoagulant therapy should be discontinued in accordance with the 2020 JCS guideline.253
2.3 Heart Rate Control
Tachycardia due to AF (>130/min) can result in congestive heart failure, even if patients have no organic cardiac disorders. Therefore, heart failure occurs more frequently in patients with CA with diastolic dysfunction due to LV hypertrophy. Generally, the Race II Trial recommended mild heart rate control (<110/min) during AF to prevent heart failure and other cardiac events.254
Similarly, mild heart rate control was recommended for patients with CA. However, in patients with conduction disturbance, the dosage of medications to avoid AV block must be chosen carefully. Heart rate control is not needed in anemic patients. First of all, treatment for anemia should take precedence over heart rate control. In the general population, digitalis, β-blockers, verapamil, diltiazem, and amiodarone are available for heart rate control. However, digitalis is contraindicated in CA because the usual daily dose could cause life-threatening arrhythmia. Intravenous injection of landiolol hydrochloride (1–10 μg/kg/min) can reduce heart rate safely in patients with LV dysfunction (LVEF 25–50%). However, there is little evidence of its safety and effectiveness in patients with CA. Non-dihydropyridiane Ca blockers (verapamil and diltiazeme) have negative inotropic effects. These Ca blockers are contraindicated in patients with amyloidosis (especially AL amyloidosis) with LV dysfunction (Class III, level C). Bisoprolol fumarate and carvedilol can be prescribed to reduced heart rate in patients with heart failure; however, careful attention is needed when prescribing carvedilol to patients with autonomic dysfunction. Carvedilol containing α blockade could worsen postural hypotension.
2.4 Pulmonary Vein Isolation for AF
The indication of pulmonary vein isolation (PVI) for AF should be decided individually according to the 2018 JCS/JHRS Guideline on Non-Pharmacotherapy of Cardiac Arrhythmias.255
The etiology of CA, LA dilatation, LV hypertrophy, and other general statuses are important factors affecting prognosis after PVI. Recently, PVI for AF has been established as non-pharmacotherapy in the general population. The success rate in patients with paroxysmal AF is 80–90%. However, the effectiveness of PVI in patients with CA has not been thoroughly evaluated. Tan et al. reported that the success rate in patients with CA was lower than that in the general population (75% at 1 year and 60% at 3 years).256
Therefore, patients with paroxysmal AF without LA dilatation or LV hypertrophy may be candidates for PVI (Class IIb, level C). However, PVI is contraindicated for patients with AL amyloidosis, poor prognosis and severe LA dilatation, and LV hypertrophy (Class III, level C).
3. Device Therapy
3.1 AV Block
Patients with CA frequently have some sort of ECG abnormality.257
Penney et al. reported that AV conduction abnormalities were recognized in 43% of patients with AL and 58% of patients with ATTR amyloidosis.70
Pacemaker implantation is indicated for 20% of patients with CA. The indication of an implantable pacemaker for AV block can be discussed according to the 2018 JCS/JHRS Guideline on Non-Pharmacotherapy of Cardiac Arrhythmias.255
Amyloidosis is generally a progressive disease. Therefore, the time courses until advanced or complete AV block differ among patients. Moreover, some patients are accompanied by life-threatening ventricular arrhythmias. Thus, scheduled ECG or Holter ECG and implantable cardiac monitoring (ICM) should be performed for the early detection of these arrhythmias. Vincent et al. demonstrated that pacemaker implantation was indicated within 45 months in patients with CA with the following findings: first-degree AV block, Wenckbach rate <100 bpm, AH interval >70 ms, HV interval >55 ms, and bundle block). In addition, the preventive implantation of a pacemaker for such high risk patients has been shown to be effective for avoiding cardiac events.258
However, the usefulness of preventive pacemaker implantation has not been established (Class IIb, level C).
3.2 Sick Sinus Syndrome and AF Bradycardia
The indication for an implantable pacemaker for sick sinus syndrome and AF bradycardia can be discussed according to 2018 JCS/JHRS Guideline on Non-Pharmacotherapy of Cardiac Arrhythmias.255
AF is more frequently accompanied by AV block in patients with CA than in the general population. Asymptomatic AF bradycardia is not recommended for pacemaker implantation; however, AF bradycardia accompanied by heart failure could be a candidate for pacemaker implantation. Regardless of the presence of symptoms (e.g., syncope, convulsion, dizziness, dyspnea, easy fatigability) in patients with AF bradycardia, the relationship between the presence of symptoms and bradycardia need to be verified by scheduled ECG or Holter ECG and ICM.
3.3 Cardiac Resynchronized Therapy (CRT)
Generally, cardiac resynchronized therapy (CRT) for patients with heart failure has been recognized as an effective therapy for LV dysfunction associated with intraventricular conduction disturbance, especially left bundle branch block (LBBB). However, no evidence has been established for CA. To date, only case reports or small cohort studies have been published. Therefore, the evidence level is not available. The indication for CRT should be discussed individually. Candidate patients can be discussed according to the 2018 JCS/JHRS Guideline on Non-Pharmacotherapy of Cardiac Arrhythmias.255
CRT can be indicated for patients with LBBB and a relatively good prognosis (>1 year) (Class IIb, level C). CRT is contraindicated for patients with a poor prognosis (<1 year), a QRS interval less than 150 ms, and conduction disturbances other than LBBB (Class III, level C).
3.4 Implantable Cardioverter-Defibrillators (ICDs)
Previous reports have demonstrated the occurrence of sudden cardiac death in 10–30% of patients with CA.259,260
Especially in AL amyloidosis, the prognosis is extremely poor and PEA is occasionally documented. Therefore, reviews regarding the efficacy of ICDs do not recommend ICD implantation for patients with amyloidosis.261,262
In a Japanese cohort study (Nippon Storm), few cases of implanted ICDs for CA were reported.263
Kristen reported that many patients with CA eventually died as a result of ventricular fibrillation or PEA, and no significant difference in prognosis was found between patients with and without an implanted ICD.261
On the other hand, favorable results for ICD implantation were reported in patients with preserved cardiac function who received effective therapy.264
Therefore, indications for ICDs must be carefully discussed. The 2018 JCS/JHRS Guideline on Non-Pharmacotherapy of Cardiac Arrhythmias255
is limited as a reference regarding the indication for ICD in patients with CA. ICD implantation is not strongly recommended for prevention against sudden cardiac death in any patients (so called Class I indication). ICD implantation for patients with a poor prognosis (<1 year) is contraindicated (Class III, level C). A limited number of patients (e.g., those with mild hypertrophy and preserved systolic/diastolic function, those with a good prognosis after adequate therapy) could be candidates for ICD implantation (Class IIb, level C). In the future, the indications for ICD implantation may expand if cardiac involvement can be eliminated after early diagnosis and adequate therapy can be provided. These issues require further discussion.
4. Treatment for AL Amyloidosis
The aim of treatment for AL amyloidosis is to decrease FLC, which is a major cause of toxic amyloid fibrils. The National Comprehensive Cancer Network (NCCN) guideline recommends the combination of cyclophosphamide (CPA), bortezomib (BOR), and dexamethasone (DEX) (CyBorD) as treatment for both transplant-eligible and transplant-ineligible patients.265
This combination is also recommended in the United Kingdom (UK) guideline.266
Unfortunately, BOR has not been approved for primary AL amyloidosis in Japan (as of March 2020). The UK guideline notes that treatment-related toxicity is increased when BOR is used for patients with AL amyloidosis as compared with MM, and dose reduction is recommended. No randomized, clinical trial has shown the efficacy and safety of BOR-including regimens to date.
In Japan, standard treatment for AL amyloidosis is the combination of oral melphalan (MEL) and dexamethasone (DEX) (MEL/DEX), and high-dose melphalan and stem-cell transplantation (HDM/ASCT) may lead to a higher response rate and long-term survival for eligible patients.267–273
There are few data on the superiority of HDM/ASCT compared with other treatment modalities. A randomized trial reported in 2007 failed to show the superiority of HDM/ASCT over oral MEL/DEX.272
The lack of evaluation of cardiac biomarkers may lead to high treatment-related mortality (TRM) in patients receiving HDM/ASCT.
The indications for HDM/ASCT have been reported from the Mayo Clinic274
and the UK amyloidosis treatment trial.275
A recent review from the Mayo Clinic provides simple criteria276
(Table 31).
Table 31.
Guidelines for Patient Selection for Stem Cell Transplantation (Mayo Clinic)
| Suggested requrement |
| “Physiologic” age < 70 |
| Creatinine clearance > 30 mL/min |
| cTnT < 0.06 ng/mL |
| Systolic blood pressure ≥ 90 mmHg |
| NYHA Class I/II |
(Adapted from Sher T, et al. 2016276) ©2016 American Society for Blood and Marrow Transplantation. Licensed under the CC BY-NC-ND 4.0.
MEL/DEX seems to lack power and have low efficacy, but it is recommended for HDM/ASCT-ineligible patients because of low treatment toxicity. Palladini reported that better survival is expected in hematological responders.277
Although the efficacy of BOR and thalidomide (THAL) has been reported from the USA and Europe, these drugs are not approved in Japan as of March 2020. The combination of CPA, BOR, and DEX is reported as CyBorD from the Mayo Clinic278
and as CVD from the National Amyloidosis Center, London.279
Both reports showed efficacy and safety, and such combination treatment will be a powerful strategy if eligible patients are carefully chosen. Venner reported the result of a matched pair analysis showing better response with CVD than CTD.280
Approval of BOR and THAL for primary AL amyloidosis is expected.
5. Disease-Modifying Therapy for ATTRv Amyloidosis (see to
CQ3)
The clinical efficacy of disease-modifying therapies including liver transplantation, transthyretin (TTR) tetramer stabilizers, and gene therapies has been proven for hereditary ATTR (ATTRv) amyloidosis.
Figure 29
shows the process of ATTR amyloid fibril formation and the mechanism of action of disease-modifying therapies approved in Japan.
5.1 Liver Transplantation
Since most serum TTR is produced by the liver, liver transplantation was first performed in a patient with ATTRv amyloidosis in 1990. After liver transplantation, the serum concentration of TTR decreased rapidly281,282
(Figure 29). Since then, liver transplantation has been performed for this disease at many hospitals worldwide, and useful clinical data are accumulating. According to data from the Familial Amyloidotic Polyneuropathy World Transplant Registry and Domino Liver Transplant Registry,283
2,236 liver transplantations have been performed to date with 5-year and 20-year survival rates of 77%284
and 55%,285
respectively. Prognosis after liver transplantation is affected by TTR genotype, and it was reported that the 5-year survival rate was 82% in V30M (p.V50M) patients, whereas that in non-V30M patients was 59%.286
Among V30M patients, the prognosis of early-onset patients is excellent,51
although that of elderly male patients is relatively poor.287,288
The main cause of death after liver transplantation is cardiac-related death.283,284
Liver transplantation decreases the amount of amyloid deposition in abdominal fat tissue289
and improves peripheral and autonomic nerve function in early-onset V30M patients.290
However, turnover of amyloid is different among organs, and progression of amyloid deposition is often observed in the heart after liver transplantation.291–293
In addition, it should be noted that ocular amyloidosis and leptomeningeal/cerebral vessel amyloidosis are not relieved by liver transplantation due to variant TTR synthesis by the retinal pigment epithelium and choroid plexus.294–296
The situation surrounding the use of liver transplantation for ATTRv amyloidosis in Japan is different from that in other areas of the world. Cadaveric liver tissue is mainly used for transplantation outside Japan, whereas liver from live donors is mainly used in Japan due to a lack of brain dead donors. It takes considerable time to obtain live donor liver transplantation after registration with the Japan Organ Transplant Network because the urgency of transplantation for ATTRv amyloidosis is relatively low compared with other diseases. Requisites for live donors include healthy adults ≤65 years, relatives ≤ sixth-degree or spouse’s relatives ≤ third-degree, blood type possible for blood transfusion, and not having a pathogenic TTR variant.
5.2 TTR Tetramer Stabilizer
TTR exists as a tetramer in the human body, but dissociation of the tetramer to a monomer is necessary to form amyloid fibrils (Figure 29). It has been shown that the TTR tetramer structure is destabilized in ATTRv patients due to TTR gene variation.47
Based on these findings, research on small molecules that bind to and stabilize TTR tetramer advanced, and tafamidis was developed (Figure 29). Tafamidis has been shown to inhibit progression of peripheral neuropathy and decrease all-cause death and cardiovascular-related hospitalizations.
The first randomized, controlled trial of tafamidis (Fx-005 study)297
enrolled 125 ATTRv patients with V30M variant and administered tafamidis (tafamidis meglumine 20 mg/day) or placebo for 18 months. The primary endpoints were treatment response (with response defined as <2-point increase in the Neuropathy Impairment Score in the Lower Limbs [NIS-LL]) and change from baseline in total quality of life as measured by the Norfolk Quality of Life–Diabetic Neuropathy total score. Fx-005 study demonstrated that significantly more tafamidis patients than placebo patients were NIS-LL responders (60.0% vs. 38.1%, respectively), and tafamidis patients had better preserved total quality of life (0.1 vs. 8.9, respectively) in the efficacy-evaluable (EE) population. Based on these findings, tafamidis was approved in 2013 in Japan for the inhibition of peripheral neuropathy in ATTRv amyloidosis. Concerning ATTR-type cardiac amyloidosis (ATTRv and ATTRwt), a second randomized, controlled trial of tafamidis (ATTR-ACT study)3
was conducted and demonstrated that tafamidis significantly inhibited the primary endpoints (i.e., all-cause death and cardiovascular-related hospitalizations) compared with placebo. Based on these findings, an additional indication for tafamidis, ATTR-type cardiac amyloidosis (both ATTRwt and ATTRv), was approved in March, 2013 in Japan (refer to “
Chapter III, 6. Treatment for ATTRwt Amyloidosis
”).
Similar to tafamidis, diflunisal, a well-known nonsteroidal anti-inflammatory drug, also has a TTR tetramer stabilization effect and was proven to inhibit progression of peripheral neuropathy in a randomized, controlled trial.298
However, diflunisal is not currently available in Japan and is not approved for the treatment of ATTRv amyloidosis.
5.3 Nucleic Acid Medicine
ATTRv amyloidosis is considered to be a good target for gene silencing therapy because ATTRv amyloidosis is a typical gain of toxic function disease, and TTR knockout mice do not show any abnormal phenotype.299
In addition, delivery of a nucleic acid drug is relatively easy, since most TTR is produced by the liver.
Under these circumstances, patisiran, a small interfering RNA (siRNA) therapeutic agent targeting TTR mRNA, was developed, and the result of a randomized, controlled trial (APOLLO study) was reported in 2018.300
The primary endpoint of the APOLLO study was change from baseline of the modified Neuropathy Impairment Score+7 (mNIS+7), and 225 ATTRv amyloidosis patients were randomly assigned to receive patisiran 0.3 mg/kg or placebo by intravenous injection every 3 weeks for 18 months. Patisiran reduced the serum TTR level by approximately 80% from baseline, and significantly improved the mNIS+7 compared with placebo (changes from baseline in the patisiran group and in the placebo group were −6.0 and 28.0, respectively). In addition, the analysis of the cardiac subpopulation, patients with left ventricular wall thickness ≥13 mm and no history of hypertension or aortic valve disease, showed that patisiran significantly inhibited deterioration of left ventricular wall thickness, global longitudinal strain, cardiac output, end-diastolic volume, and NT-proBNP.300–302
Regarding the safety, although the incidence of infusion-related reaction was observed in about 20% of the patients with patisiran, this treatment was well tolerated.Based on the result of the APOLLO study, patisiran was approved and became available in September, 2019 in Japan for TTR-type amyloid neuropathy (indication described in the package insert).
Inotersen, an antisense oligonucleotide therapeutic agent targeting TTR mRNA, was also proven to inhibit mNIS+7 and improve the Norfolk Quality of Life-Diabetic Neuropathy score compared with placebo in a randomized, controlled trial of ATTRv amyloidosis.303
However, serious adverse events including glomerulonephritis and thrombocytopenia were reported in the inotersen group, and all patients need to receive enhanced monitoring for these events. Due to the serious adverse events described above, a phase III clinical trial of inotersen was cancelled in Japan; therefore, inotersen is not approved in Japan (Table 32).
Table 32.
Recommendations and Evidence Levels of Disease Modifying Therapies for ATTRv Neuropathy and Cardiomyopathy*
| |
COR |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
| Liver transplantation |
I |
C |
B |
IVb |
| Tafamidis for neuropathy** |
I |
B |
B |
II |
| Patisiran for neuropathy |
I |
B |
B |
II |
Tafamidsis for patients with NYHA Class I/II and satisfy patient
requirements in the statement of the Japanese Circulation Society** |
IIa |
B |
B |
II |
Tafamidsis for patients with NYHA Class III and satisfy patient
requirements in the statement of the Japanese Circulation Society** |
IIb |
B |
B |
II |
Tafamidis should be administered with a dosage of 20 mg for neuropathy and 80 mg for cardiomyopathy (dosage of tafamidis can be reduced in the case of intolerance to 80 mg).
*Refer to Table 33 for therapeutic options in each patient. **Refer to section of “ATTRwt amyloidosis” concerning indication of tafamidis for ATTRv cardiac amyloidosis.
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
5.4 Therapeutic Options in ATTRv Amyloidosis
ATTRv amyloidosis is a lethal disease if disease-modifying therapies are not given. Therefore, either liver transplantation or pharmacotherapy (tafamidis or patisiran) should be performed. Therapeutic options in each patient should be decided based on characteristics of each therapy (Table 33) and the patient’s condition, including TTR genotype, severity of amyloidosis, general condition, and social environment, such as occupation.
Table 33.
Characteristics of Disease Modifying Therapies for ATTRv Amyloidosis
Disease modifying
therapy |
Merits (efficacy, safety, etc.) |
Points of attention |
| Liver transplantation |
Lon-term evidence of efficacy in patients with early-
onset V30M variants (peripheral nerve damage is
stopped or slightly improved if transplantation is
performed in early stage of the disease) |
Invasiveness
Shortage of donor
Requirement for immunosuppressive drugs after
transplantation
Transplant-related complications (biliary obstruction,
infection, rejection, etc.)
Limitation of efficacy in non-V30M patients
Progression of cardiac amyloidosis after transplantation
due to wild-type TTR deposition
Progression of ocular and leptomeningeal/cerebral
vessel amyloidosis after transplantation |
| Tafamidis |
Inhibition of progression of peripheral neuropathy in
V30M patients
Reduce frequency of all cause of death and
cardiovascular-related hospitalizations
Inhibition of deterioration of cardiac failure symptoms
Excellent safety profile
Good compliance as being a once a day oral drug |
Can not stop progression of pheripheral neuropathy
and cardiac failure completely
No evidence concerning inhibition of progression of
peripheral neuropathy in non-V30M patients (tafamidis
covered by public health insurance for both V30M and
non-V30M patients in Japan)
No evidence concerning ocular and leptomeningeal/
cerebral vessel amyloidosis
High medical cost |
| Patisiran |
Arrest or slight improvement of peripheral neuropathy
Inhibition of deterioration of echocardiographic
parameters and NT-proBNP |
Lack of long term evidence (efficacy, safety)
Intravenous injection every 3 weeks (require
approximately 3 h including premedications)
No evidence concerning ocular and leptomeningeal/
cerebral vessel amyloidosis
High medical cost |
Liver transplantation was the only therapeutic option for ATTRv amyloidosis for a long time and was still a first-line therapy for early-onset V30M patients even after approval of tafamidis.75
However, pharmacotherapy (tafamidis or patisiran) has become first-line therapy for all ATTRv amyloidosis patients because patisiran showed an excellent therapeutic effect on peripheral neuropathy, comparable with liver transplantation in the APOLLO study (Table 33). In particular, the indications for liver transplantation are limited because cardiac amyloidosis progresses even after liver transplantation. Since a limited amount of evidence has been accumulated to date, combination therapy with patisiran and tafamidis should be considered only in patients who do not respond to monotherapy with each drug.
6. Treatment for ATTRwt Amyloidosis (see also CQ3)
6.1 TTR Stabilizers
The tetrameric structure of TTR is thought to be destabilized by gene mutations or aging, and dissociates into monomers, causing misfolding and polymerization. TTR stabilizers suppress amyloid fibril formation by binding at the thyroxine-binding site of TTR to stabilize the tetrameric structure.
In a phase 2/3 study in 2012,297
tafamidis was well tolerated in patients with ATTRv amyloidosis with peripheral neuropathy for 18 months, and it suppressed the progression of peripheral nerve symptoms. In 2013, approval for tafamidis was granted in Japan.
The ATTR-ACT,3
a phase III international, multicenter, double-blind, randomized clinical study, was conducted to investigate the efficacy, safety, and tolerability of an oral daily dose of 20 mg or 80 mg tafamidis meglumine compared with placebo over a 30-month period in 441 patients with ATTR-CM (including both ATTRwt and ATTRv amyloidosis). In the primary analysis, a significant reduction in the hierarchical combination of both all-cause mortality and the frequency of cardiovascular-related hospitalizations was observed in the tafamidis groups (264 cases) compared with the placebo group (177 cases). Tafamidis significantly reduced all-cause mortality (tafamidis group: 29.5% [78/264 cases]; placebo group: 42.9% [76/177 cases]) and cardiovascular event-related hospitalization frequency compared with the placebo group (0.48/year in the tafamidis group; 0.70/year in the placebo group) (Figure 30).3

In the secondary study end points, a decline in functional capacity as measured by 6-min walking test distance was alleviated in the tafamidis group, and a decline in QOL as evaluated by the Kansas City Cardiomyopathy Questionnaire-Overall Summary score was also improved compared with the placebo group at 30 months. Tafamidis was also well tolerated, with an observed safety profile comparable to that of placebo.
In prespecified subgroups, tafamidis was effective for both ATTRwt and ATTRv amyloidosis. No significant difference in the primary end point between the high- and low-dose groups was found, but all-cause mortality was significantly higher in the high-dose group (Figure 31).3
However, it took more than 1.5 years from the start of treatment to observe a decrease in mortality due to tafamidis, and it could not be confirmed to be effective by subgroup analysis in patients with NYHA class III at baseline with cardiovascular-related hospitalization, suggesting the importance of early administration.
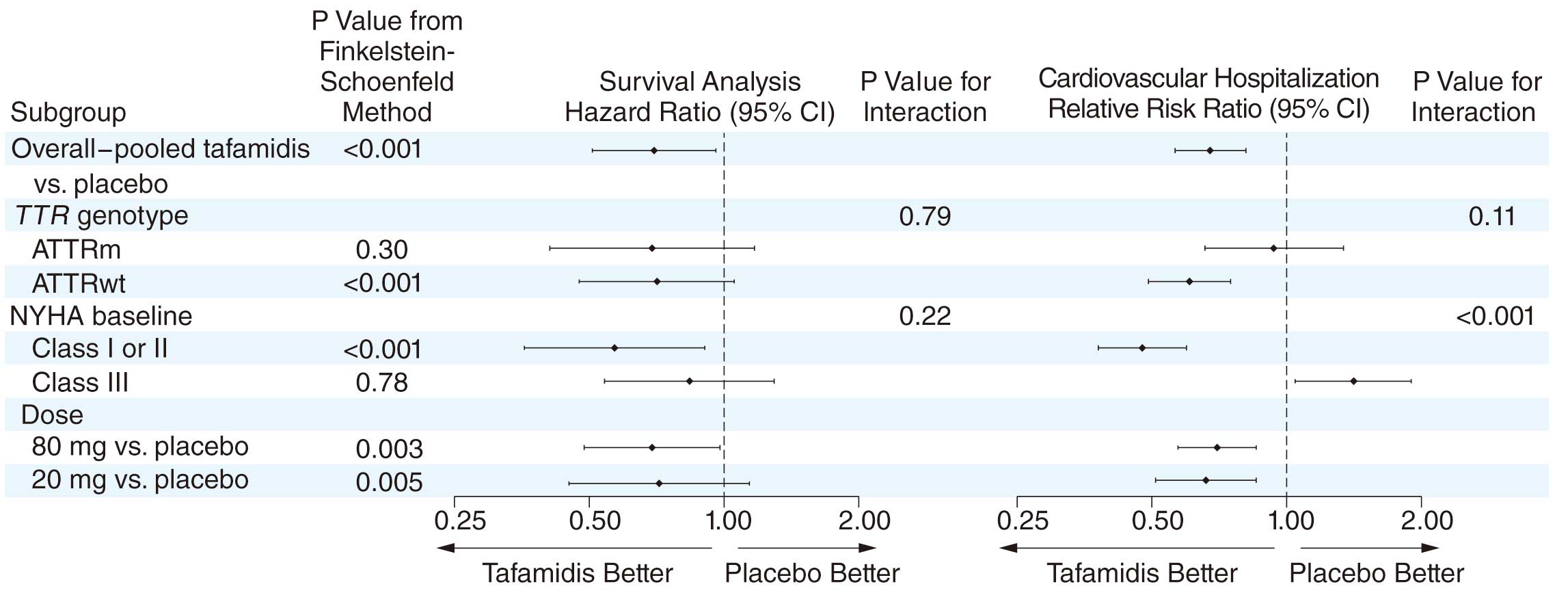
Following the results of the ATTR-ACT, ATTR-CM (including both ATTRwt and ATTRv amyloidosis) was added to the indications for tafamidis in March 2019. Although the efficacy of tafamidis has been demonstrated in a patient group enrolled in the ATTR-ACT, the efficacy in those excluded from this study (Table 34), such as those with NYHA class IV, remains unknown. The MHLW has set the requirements for prescribing tafamidis, and the JCS has established the certification for prescribing facilities and physicians to promote the appropriate use of this drug.
Table 35
shows the recommendations and levels of evidence for tafamidis for ATTRwt CA.
Table 34.
Main Patient Groups Excluded in ATTR-ACT Study
| · Heart failure not caused by ATTR-CM |
| · NYHA Class IV |
| · AL amyloidosis |
| · Implanted with any cardiac device |
| · Estimated glomerular filtration rate (eGFR) ≤25 mL/min/1.73 m2 |
| · Liver transaminase more than twice the upper normal limit |
| · Notable undernutrition patients: Modified body mass index (serum albumin (g/L)×BMI) <600 |
| · Patients taking oral nonsteroidal anti-inflammatory drug, doxycycline, calcium antagonist, digitalis |
(Source: Prepared based on Maurer MS, et al. 20183)
Table 35.
Classes of Recommendation and Levels of Evidence for Tafamidis in the Treatment of ATTRwt Cardiac Amyloidosis
| |
COR* |
LOE |
GOR
(MINDS) |
LOE
(MINDS) |
Tafamidis for ATTRwt cardiac amyloidosis patients with NYHA
Class I / II who meet patient requirements in the JCS statement |
IIa |
B |
B |
II |
Tafamidis for ATTRwt cardiac amyloidosis patients with NYHA
Class III who meet patient requirements in the JCS statement |
IIb |
B |
B |
II |
COR, class of recommendation; LOE, level of evidence; GOR (MINDS), grade of recommendation (MINDS); LOE (MINDS), level of evidence (MINDS).
*The JCS statement is subject to change, so always refer to the latest information (see CQ3–1).
*Only the ATTR-ACT has been reported as a randomized controlled trial for ATTR. Therefore, the recommendations and evidence levels associated with tafamidis greatly depend on the results of this trial, and the following points need to be noted.
· NYHA Class IV patients were excluded from the study, and thus, the efficacy and safety of tafamidis in such patients have not been confirmed.
· The efficacy and safety of tafamidis in the population excluded from the ATTR-ACT (Table 34) have not been confirmed.
· In the ATTR-ACT trial, it took >1 year after administration to confirm the effectiveness of tafamidis. Therefore, in cases with a life expectancy not expected to be >1 year, the administration of tafamidis should be carefully considered.
6.2 Nucleic Acid Medicine
Regarding the neurological symptoms of patients with ATTRv amyloidosis who have peripheral neuropathy, the efficacy of two oligonucleotide-based drugs that knock down the expression of TTR — patisiran, a siRNA drug, and inotersen, an antisense oligo drug — have been confirmed in phase III trials.300,303
Patisiran was approved in Japan in June 2019 (for details, see “
Chapter III, Section 5. Treatment for ATTRv Amyloidosis
”). Since the efficacy of these nucleic acid drugs for ATTR has not been sufficiently evaluated, a phase III trial of patisiran, APOLLO-B, and a clinical trial of the next-generation inotersen, AKTEA-TTR-LRX, are underway for ATTR, including ATTRwt amyloidosis.
IV. CA Diagnostic Algorithm
The diagnostic algorithm for CA is shown in
Figure 32.1,75,304–309
[CQ1] What is the role of 99 mTc-PYP scintigraphy (bone scintigraphy) in clinical practice for CA?
[Response]
99 mTc-PYP scintigraphy is fairly effective for detecting both ATTRv and ATTRwt CA1,2,173
(see “
Chapter II, Section 6. Nuclear Imaging
”). When 99 mTc-PYP scintigraphy yields a positive result, ATTRv and ATTRwt CA must be differentiated with genetic testing.
In the visual assessment of 99 mTc-PYP scintigraphy, AL amyloidosis sometimes presents as Grade 2, thereby requiring hematology testing to rule out monoclonal immunoglobulin (M protein). If 99 mTc-PYP scintigraphy yields a positive result and AL amyloidosis is excluded, the diagnostic specificity and positive predictive value for ATTR are reportedly 100%.1
Physiological uptake into blood pools may result in false-positive results. Therefore, SPECT should be performed to assess myocardial uptake accurately.
An important consideration in the use of 99 mTc-PYP scintigraphy is selecting suitable cases (see in
Chapters II
and
IV). 99 mTc-PYP scintigraphy positivity can be assessed using the Kumamoto criteria:99
high-sensitivity cardiac troponin T ≥0.03 ng/mL, LV posterior wall thickness ≥13.6 mm, and QRS width ≥120 ms. For patients who meet two and three of these criteria, 99 mTc-PYP scintigraphy positivity rates are 63% and 96%, respectively. Thus, the Kumamoto criteria may be useful when selecting patients to undergo 99 mTc-PYP scintigraphy.
In real-world clinical practice, the suitability of 99 mTc-PYP scintigraphy must be considered on a case-by-case basis.
Table 36
shows the clinical scenarios and recommendation levels for 99 mTc-PYP scintigraphy based on existing academic evidence.160,308,310
Table 36.
Clinical Scenarios and Recommendation Levels for
99 mTc-PYP Scintigraphy
| Clinical scenario |
Recommendation
level |
| Suspicion of ATTR cardiac amyloidosis |
Recommended |
| Cardiac hypertrophy and previous bilateral carpal tunnel syndrome |
Recommended |
| Cardiac amyloidosis suspected in echocardiography, CMR, or cardiac CT |
Recommended |
| Low-flow, low-gradient aortic stenosis in a patient aged ≥60 years |
Recommended |
Persistently mildly elevated high-sensitivity cardiac troponin T in a patient aged ≥70
years |
Considered |
Heart failure of unidentified cause with left ventricular hypertrophy in a patient aged
≥60 years |
Considered |
Atrioventricular block, bundle branch block, or atrial fibrillation with cardiac hypertrophy
in a patient aged ≥60 years |
Considered |
| Symptoms of polyneuropathy or autonomic neuropathy |
Considered |
| Monitoring of progress for a patient diagnosed with ATTR cardiac amyloidosis |
Not recommended |
| Suspicion of AL amyloidosis |
Not recommended |
AL, amyloid light-chain; ATTR, amyloid transthyretin; CMR, cardiac magnetic resonance imaging; CT, computed tomography.
1. Biopsy From Other Than the Heart
Generally, pathological information can be obtained from the tissues or organs related to symptoms in patients with systemic amyloidosis. Abdominal wall fat, skin, salivary gland of the lip, and the intestinal tract are selected as non-invasive biopsy sites.26,190,198–201,204,205,207–215
The detection rate of amyloid protein from various tissues/organs varies depending on the type of amyloid precursor protein. If amyloid protein is detected from non-cardiac organs in suspected amyloidosis patients with clinical and imaging findings, it is acceptable to make a diagnosis of cardiac amyloidosis.
1.1 Biopsy From Other Than the Heart in AL Amyloidosis
In patients with systemic AL amyloidosis, abdominal fat pad, lip salivary gland, and bone marrow biopsies are commonly performed. The amyloid protein detection rate at each biopsy site is shown in
Table 37. In suspected AL amyloidosis with evidence of monoclonal proteins, bone marrow biopsy is often performed to search for abnormal plasma cells. Bone marrow biopsy is not the first choice for screening, because the detection rate of amyloid deposits by bone marrow biopsy is relatively low in AL amyloidosis. It is recommended that bone marrow specimens be assessed by Congo red staining for detection of amyloid protein. Although the amyloid detection rate of liver and spleen biopsies is high, they should be avoided because they are highly invasive procedures inducing bleeding complications.
Table 37.
Biopsy Sites in Amyloidosis
Author
(reference number) |
Subjects |
Biopsy site |
Detection rate |
| Garcia Y198 |
97 suspected amyloidosis undergoing
abdominal fat pad excisional biopsy |
Abdominal fat excisional
tissue |
AL 79%, ATTR 12% |
| van Gameren II199 |
120 amyloidosis (38 AA, 70 AL, 12 ATTR) |
Abdominal fat aspiration
tissue |
AA 92%, AL 94%, ATTR 83% |
| Quarta CC200 |
600 cardiac amyloidosis (216 AL, 113 ATTRv,
271 ATTRwt) |
Abdominal fat aspiration
tissue |
AL 84%, ATTRv 45%, ATTRwt
15% |
| Suzuki T204 |
34 patients with evidence of monoclonal
proteins and symptoms related amyloidosis |
Skin, bone marrow, and
Labial salivary gland |
Skin 72%, Bone marrow 77%,
Labial salivary gland 89% |
| Hachulla E205 |
59 cases undergoing labial salivary gland
biopsy (30 patients with AA or AL amyloidosis) |
Labial salivary gland |
Labial salivary gland 86% |
| Swan N214 |
100 AL amyloidosis |
Bone marrow |
Bone marrow 60% |
| Kimmich C215 |
168 AL amyloidosis |
Bone marrow, abdominal
fat aspiration tissue |
Bone marrow 57%, abdominal
fat 96% |
| Fine NM190 |
286 ATTR amyloidosis with positive extra-
cardiac biopsy (186 ATTRv and 100 ATTRwt) |
Myocardium, abdominal fat
aspiration tissue |
Myocardium 100%, abdominal
fat (ATTRv 67%, ATTRwt 14%) |
| Lechapt-Zalcman E207 |
32 polyneuropathy with unknown etiology |
Labial salivary gland |
ATTRv 100% |
| Ikeda S201 |
11 ATTRwt |
Abdominal wall skin |
Skin 73% |
1.2 Biopsy From Other Than the Heart in ATTR
In transthyretin-related (ATTR) amyloidosis, biopsies are often performed from the abdominal fat pad, labial salivary gland, and gastrointestinal tract. As shown in
Table 37, the amyloid protein detection rates of biopsies in patients with wild-type transthyretin-related (ATTRwt) amyloidosis were relatively low, such as 14–15% for the abdominal fat pad200,214
and 38% for the intestinal tract.26
It has been demonstrated that the detection rate of abdominal wall skin biopsy was 73%.201
Skin biopsy can yield improvement of the sensitivity for diagnosis of ATTRwt amyloidosis. Since there are some localized cases of ATTR amyloidosis with limited amyloid deposition in tendon and ligament tissues, diagnosis of systemic ATTR amyloidosis based on pathological findings of tendon and ligament tissues alone should be avoided.
*Usefulness of bone scintigraphy and endomyocardial biopsy in ATTR
It has been reported that the detection rate of amyloid protein by EMB was almost 100% in patients with ATTR amyloidosis.190
The frequency of sampling errors from myocardium is extremely low. In particular, the amyloid protein detection rate of biopsies other than the heart is not high at the initial stage of ATTRwt amyloidosis. Bone scintigraphy leads to the diagnosis of ATTRwt cardiomyopathy being made reliably with EMB.1,160
2. Endomyocardial Biopsy
EMB can show amyloid fibrils in clinically suspected patients with hypertensive heart disease or HCM on echocardiography. Therefore, it is important to pathologically differentiate cardiac amyloidosis from other hypertrophic cardiomyopathies. In high-risk patients with conduction disturbances and advanced age, biopsy is performed to detect amyloid protein from the abdominal fat pad, skin, lip salivary gland, and intestinal tract. A non-invasive abdominal fat biopsy is often performed in such cases. However, the amyloid detection rate of abdominal liposuction biopsy is not high in patients with ATTRwt amyloidosis.200,214
Other biopsy sites, especially myocardium in reference to bone scintigraphy, are selected. Patients with myocardial uptake on bone scintigraphy and no FLC and confirmation of pathological amyloid protein cannot be definitely diagnosed as having ATTR amyloidosis, but such cases are regarded as probable ATTR amyloidosis (see “
Chapter I, Section 5. Diagnostic Criteria”).
[CQ3] What patients should receive tafamidis for CA?
1. Statement by the Japanese Circulation Society (JCS)
Tafamidis (Vyndaqel®) received approval in Japan for suppressing the progression of peripheral neuropathy caused by ATTRv amyloidosis in September 2013. Following the results of the ATTR-ACT,3
in March 2019, ATTR CA was added to the indications for this drug.
According to the precautions related to the efficacy of tafamidis, the Health Care Division of the MHLW states, “When applying this drug, refer to the latest guidelines and confirm that the diagnosis of ATTR has been determined.” It was recommended that tafamidis should be used in patients judged to be eligible by a physician skilled in diagnosing and treating ATTR.
In response to the recommendations of the MHLW, the JCS has begun to prepare guidelines for CA, and has issued a statement that stipulates the requirements of facilities and physicians for the introduction of tafamidis (shown below), in addition to the patient requirements from the MHWL (Table 38).311,312
Table 38.
A Statement for the Appropriate Administration of Tafamidis in Patients With Transthyretin Cardiac Amyloidosis
| 1) Patient requirements |
| ① ATTRwt cardiac amyloidosis |
| a. A medical history of hospitalization for heart failure or a heart failure condition requiring treatment with a diuretic for improvement |
| b. Presence of amyloid deposits in biopsy tissue (cardiac or non-cardiac) |
| c. Transthyretin precursor protein identification by immunohistochemistry |
| d. Evidence of cardiac involvement on echocardiography with an end-diastolic interventricular septal wall thickness of >12 mm |
| ② ATTRv amyloidosis |
| a. A cardiomyopathy symptom and a TTR gene mutation associated with cardiomyopathy |
| b. A medical history of hospitalization for heart failure or a heart failure condition requiring treatment with a diuretic for improvement |
| c. Evidence of cardiac involvement on echocardiography with an end-diastolic interventricular septal wall thickness of >12 mm |
| d. Presence of amyloid deposits in biopsy tissue (cardiac or noncardiac) |
| 2) Facility requirements |
As a general rule, only facilities that meet all of the following requirements ① to ⑥ are permitted to start administration of tafamidis
meglumine: |
| ① Cardiologist training facility certified by the JCS |
| ② Radiologist general training institute certified by the Japan Radiological Society |
| ③ Pathologist training facility certified by the Japan Society of Pathology |
| ④ Hematologist Training Facility certified by the Japanese Society of Hematology |
| ⑤ Neurologist educational facilities certified by the Japanese Society of Neurology |
| ⑥ Institution that treats more than 15 cases of myocardial biopsy annually |
| 3) Physician requirements |
| Physicians who fulfill the following requirements ① or ② and commit to ③: |
① Physicians who have used tafamidis meglumine for ATTRv amyloidosis before the approval date of the indication expansion for
ATTR-CM |
② Physicians who have identified at least three cases of ATTR by identifying the TTR precursor protein by immunohistological staining or
mass spectrometry from a biopsy tissue sample obtained at their own institution or by asking another institution for identification |
| ③ Registration of all administration cases |
Continuous therapy for patients who have been introduced to the treatment can be prescribed at a cooperating hospital, considering
the convenience of patients, but the facilities that, and physicians who, have introduced tafamidis meglumine are responsible for
maintaining follow-up of the clinical course. |
(Excerpted from Endo J, et al. 2019311)
2. What patients with ATTR amyloidosis should take tafamidis?
A:
ATTR patients with NYHA functional class I or II are eligible for active treatment using tafamidis. In addition, it is necessary to determine the indication for treatment in consideration of general conditions such as comorbidities and frailty.
Class of Recommendation: IIa
Level of Evidence: B
Grade of Recommendation (MINDS): B
Level of Evidence (MINDS): II
Commentary
A tafamidis phase III trial (ATTR-ACT) showed that patients with ATTR in the tafamidis oral administration group had better survival compared with controls.3
In particular, patients with NYHA functional class I or II showed significant improvements in both all-cause mortality and cardiovascular-related hospitalizations.
ATTR amyloidosis is a progressive disease that shows poor prognosis, although the prognosis of the ATTR type is not as bad as that of the AL type.313,314
In patients with ATTR amyloidosis, there are also several reports that there is no difference between ATTRwt and ATTRv amyloidosis in terms of prognosis.77,313
In ATTRv amyloidosis, it is necessary to consider the patient’s general condition because of neuropathy. In ATTRwt amyloidosis, cardiac events are the most common cause of death.31,315
There are several reports, mainly from overseas, on the prognostic factors for patients with ATTR, but in those reports, the number of target patients is only about 100. An accurate prognostic prediction method has not yet been established. The following prognostic factors have been raised from previous reports: age, NYHA functional class, biomarkers (e.g., BNP/NT-proBNP, troponin, renal function, uric acid), echocardiographic indices (e.g., LVEF, relative wall thickness, strain), and CMR (e.g., LGE, ECV).31,33,54,70,92,93,148,153,157,314,316–319
In ATTRv amyloidosis, MIBG scintigraphy was reported to be useful for prediction.176
AF, a common electrocardiographic finding in patients with ATTR amyloidosis, has been reported to be associated with the onset of heart failure, but not clearly associated with survival.102,103,105
There are few prognostic data for Japanese patients with ATTR, and further large scale investigations are needed.101,320
With regard to predicting survival in heart failure (not limited to CA), many risk prediction models using multiple clinical indicators have been reported.321–325
However, whether these models are useful for predicting events in patients with ATTR amyloidosis remains unclear. In general, for patients with heart failure, the most consistently reported predictors were age, NYHA functional class, renal function, low blood pressure, low body mass index, malnutrition, and exercise capacity.326,327
In addition, in aged patients with heart failure, the presence of comorbidities is often the primary determinant of prognosis, rather than heart failure itself. Although it is reported that cardiac events are the most frequent mode of death in patients with ATTR, the indication for treatment using determined in comorbidities (e.g., renal dysfunction, cerebrovascular disorder, COPD, anemia), general condition, and nutritional status, including frailty and sarcopenia.
3. What are echocardiographic findings to recommend the use of tafamidis?
• There are no data about echocardiographic findings to determine whether tafamidis should be used.
• The effects of tafamidis are expected in patients with ATTR amyloidosis in the early stage. Therefore, it is important to diagnose ATTR amyloidosis at an earlier stage using specific cardiac echocardiographic findings.
A placebo-controlled randomized trial revealed that tafamidis improved prognosis (all-cause mortality and cardiovascular events) in patients with ATTR amyloidosis and inhibited the reduction of frailty and QOL.3
However, the survival rate (end point: all-cause mortality) at 1.5 years after administration was almost the same between patients who received tafamidis and those who received placebo, and gradually became different after 1.5 years. Therefore, although tafamidis appears to inhibit the progression of the disease stage, it does not seem to improve the pathology of ATTR amyloidosis. In patients with NYHA I or II, the risk of all-cause mortality and/or hospitalization due to cardiovascular events was reduced by tafamidis, although it did not improve in patients with NYHA III. Judging from these results, tafamidis appears to be effective for inhibiting disease progression in patients with ATTR amyloidosis with NYHA I or II. Therefore, ATTR amyloidosis needs to be diagnosed and treatment with tafamidis needs to be started before patients show obvious symptoms of heart failure with more than NYHA III.
To our knowledge, no studies have investigated echocardiographic findings for predicting prognostic improvement with tafamidis. Therefore, we conclude that there are no data about echocardiographic findings to determine whether tafamidis should be used. However, considering the effects of tafamidis described above, it is important to identify patients with ATTR amyloidosis at an early stage by echocardiography. To confirm these findings, we reviewed previous studies about echocardiographic findings as prognostic predictors and specific findings on echocardiography seen at an early stage of ATTR amyloidosis.
Regarding echocardiographic findings as prognostic predictors in patients with ATTR amyloidosis, LVEF was the most reported. Grogan et al.33
reported that LVEF <50% was an independent prognostic predictor in patients with ATTRwt CA (hazard ratio [HR]: 1.85, 95% confidence interval [CI]: 1.12–3.06). Moreover, in patients with ATTRwt CA, Connors et al.31
and Hanson et al.319
reported that reduced LVEF was a prognostic predictor. LVEF has ever been the most evidence-available echocardiographic finding as a prognostic predictor. Therefore, it is important to diagnose ATTR amyloidosis and start tafamidis before LVEF begins to reduce. On the other hand, previous studies have reported that myocardial contraction fraction (MCF), which is the ratio of LV mass to LV stroke volume, is useful for predicting prognosis in patients with ATTR amyloidosis. Rubin et al.318
reported that MCF <25% (HR: 8.5, 95% CI: 4.8–14.9) is more useful as a prognostic predictor than LVEF <50% (HR: 2.8, 95% CI: 1.8–4.4) in patients with ATTR amyloidosis. The usefulness of MCF was also shown in patients with AL amyloidosis;328
thus, MCF as well as LVEF should be considered as prognostic predictors for patients with amyloidosis.
Moreover, some studies have reported that tricuspid annular plane systolic excursion (TAPSE) and mitral annular plane systolic excursion (MAPSE) are more useful prognostic predictors than LVEF, MCF, and LV global strain.93,329
TAPSE was significantly lower in patients with than in those without RV LGE on CMR (RV LGE-positive: TAPSE 12 [9–15] mm, RV LGE-negative: TAPSE 20 [18–25] mm). RV LGE reflected RV fibrosis in patients with CA and was related to prognosis.329
Tafamidis should be started before these echocardiographic findings are detected. This treatment strategy could contribute to improved prognosis in patients with ATTR amyloidosis.
As clues for the early diagnosis of ATTR amyloidosis, apical sparing (apical-to-basal strain ration >2:1), in which LV motion at the apex is well maintained compared with basal motion, and relatively high LVEF values (EF-to-strain ratio >4) were specific findings in CA.309,330
Moreover, it has been reported that differences in strain values between LV apex and basal increased by using exercise stress echocardiography, and this difference in strain values was related to symptoms in patients with CA (e.g., NYHA class, metabolic equivalents at peak exercise, maximum V˙O2).331
Apical sparing is observed both in patients with ATTR amyloidosis and in those with AL amyloidosis. However, the strain value at the LV apex in patients with ATTR amyloidosis has been shown to be lower than that in patients with AL amyloidosis.332
The earlier diagnosis of ATTR amyloidosis by using these echocardiographic findings may lead to earlier administration of tafamidis.
[CQ4] What is the role of genetic testing in clinical practice for cardiac amyloidosis?
In clinical practice, genetic testing is essential for the definitive diagnosis of ATTRv and ATTRwt amyloidosis in patients with cardiac amyloidosis. Genetic testing may be performed for the purpose of presymptomatic testing of relatives. The target gene for the test is
TTR. If a pathogenic variant is not identified, ATTRv amyloidosis can be completely ruled out. Situations necessary for the
TTR
gene test are as follows:
1. Differential diagnosis of ATTRv and ATTRwt amyloidosis in a patient diagnosed as having ATTR amyloidosis by biopsy or complementary tests such as 99 mTc-pyrophosphate scintigraphy.
2. Definitive or differential diagnosis of ATTRv amyloidosis in a cardiomyopathy patient with neuropathy and/or a family history (genetic test may be performed before biopsy in this situation).
3. Definitive diagnosis of ATTRv amyloidosis in symptomatic relatives of an ATTRv amyloidosis patient.
4. Presymptomatic diagnosis in asymptomatic relatives of an ATTRv amyloidosis patient (genetic counseling by specialists is necessary before testing in presymptomatic testing; presymptomatic testing is not covered by public health insurance in Japan).
















