Abstract
Improvements in the long-term survival of cancer patients have led to growing awareness of the clinical importance of cancer therapeutics-related cardiac dysfunction (CTRCD), which can have a considerable effect on the prognosis and quality of life of cancer patients and survivors. Under such circumstances, onco-cardiology/cardio-oncology has emerged as a new discipline, with the aim of best managing cardiovascular complications, including CTRCD. Despite the recent accumulation of epidemiological and clinical information regarding CTRCD, the molecular mechanisms underlying the pathogenesis of CTRCD by individual drugs remain to be determined. To achieve the goal of preventing cardiovascular complications in cancer patients and survivors, it is important to elucidate the pathogenic mechanisms and to establish diagnostic strategies with risk prediction and mechanism- and evidence-based therapies against CTRCD.
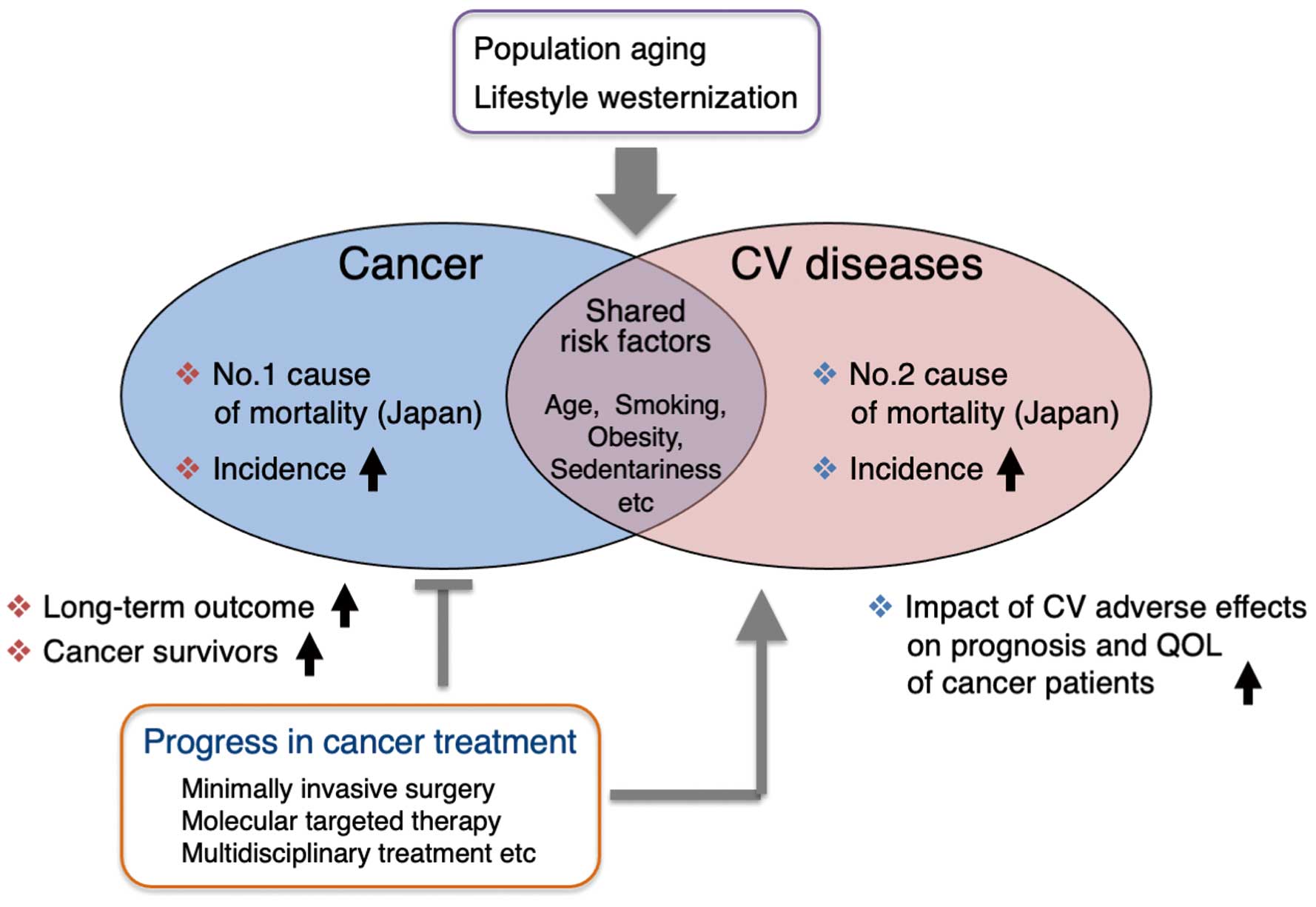
Cancer and cardiovascular disease remain the first and second leading causes of mortality in Japan, respectively.1
The incidence of most cancers and cardiovascular diseases increases with age, and the population of industrialized countries, including Japan, is progressively aging.1
According to Cancer Statistics in Japan (Cancer Information Service, National Cancer Center, Japan; 2020. Available from: http://ganjoho.jp/reg_stat/statistics/dl/index.html), the number of elderly patients with cancer is increasing in line with the rapid increase in the aging of the population, and elderly cancer patients are more likely to have cardiovascular comorbidities.2
In addition, some types of cancer and cardiovascular diseases share common risk factors, such as tobacco use, diet, obesity, and a sedentary lifestyle, which predisposes cancer patients to cardiovascular comorbidities.3
Conversely, advances in cancer therapy have markedly improved the long-term outcome of cancer patients, leading to an increase in the number of cancer survivors, but adverse cardiovascular events caused by cancer therapy have a greater effect on the prognosis and quality of life of cancer patients and survivors4
(Figure 1). Under such circumstances, it becomes increasingly important to manage the cardiovascular complications, which are related to both the cancer itself and the adverse effects of cancer therapies. Although oncology and cardiology have been separate medical fields for a long time, cardio-oncology or onco-cardiology emerges for solutions to address these unmet medical needs by developing interdisciplinary collaboration between oncology and cardiology specialists in clinical practice and medical research4,5
(Figure 2).

Cardiovascular complications caused by cancer therapy include cancer therapeutics-related cardiac dysfunction (CTRCD), coronary artery disease, venous thromboembolism, arterial hypertension, arrhythmias, peripheral vascular disease and stroke, valvular heart disease, pericardial disease, and pulmonary hypertension5,6
(Figure 2). Of these, CTRCD is the most concerning because it is life-threatening and occasionally leads to an unwanted interruption of cancer therapy. Since the first discovery of cardiotoxic effects of anthracyclines in the 1970s, many researchers have reported various molecular mechanisms that potentially underlie anthracycline-induced cardiotoxicity.7
However, the culprit molecules or pathways remain to be determined, and we have little chance of preventing or mitigating the cardiotoxic effects of anthracycline using effective pharmacological interventions.8
Furthermore, more and more molecularly targeted anticancer drugs, including monoclonal antibodies against human epidermal growth factor receptor 2 (HER2) or vascular endothelial growth factor (VEGF), tyrosine kinase inhibitors (TKIs), and proteasome inhibitors, have been added to the list of drugs that may potentially cause cardiotoxicity. In this review, we update our current understanding of CTRCD from the viewpoint of onco-cardiology.
CTRCD From the Onco-Cardiology Viewpoint
When the term “cardio-oncology” was first mentioned by Daniela Cardinale in 1996,9
the dose-dependent cardiotoxicity of anthracyclines was already recognized, and the need for a comprehensive approach to anthracycline cardiotoxicity led to the birth of a new discipline, cardio-oncology (Figure 3).10
Thereafter, dedicated units for this medical discipline emerged in cancer centers in the US and Europe. The University of Texas MD Anderson Cancer Center established an “onco-cardiology unit” with equipment found in cardiac catheterization laboratories in 200011
(Figure 3). At present, cardio-oncology or onco-cardiology services are emerging in countries worldwide. Which term is more appropriate, “cardio-oncology” or “onco-cardiology”, is a puzzling problem to be reconciled. One may imagine that “cardio-oncology” means “oncology specialized for cardiac cancers”, whereas the term “onco-cardiology” better defines a “subspecialty of cardiology dedicated to cardiovascular management of cancer patients”.10
However, in reality, the term “cardio-oncology” is by far the most prevalent, as indicated by the number of citations in PubMed (Figure 3). The first “onco-cardiology unit” in Japan was established in Osaka Medical Center for Cancer and Cardiovascular Diseases, the predecessor of the Osaka International Cancer Institute, in 2010, and the term “onco-cardiology” has been used more often than “cardio-oncology” in Japan. Therefore, in this article, we use the term “onco-cardiology”.
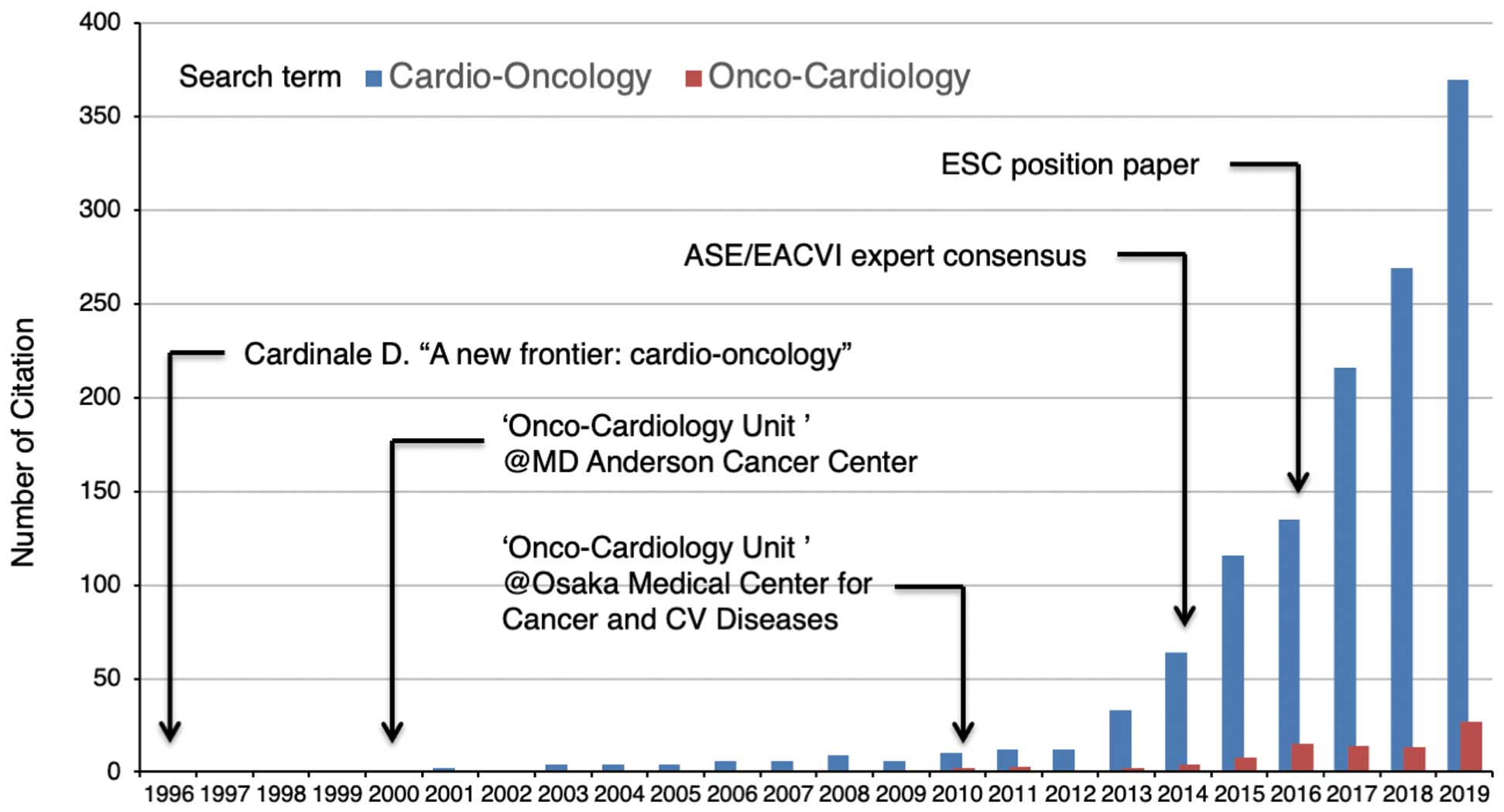
Growing demands for a mechanistic understanding of pathophysiology and better clinical practice have bolstered research activities in onco-cardiology, as revealed by a rapid increase in the number of publications in this field4
(Figure 3). It has become increasingly important to that specialists in oncology and cardiology exchange views, share common experiences, and provide an expert consensus. In 2014, the American Society of Echocardiography and the European Association of Cardiovascular Imaging published an expert consensus for multimodality imaging evaluation of CTRCD, defining it as a decrease in left ventricular (LV) ejection fraction (LVEF) of more than 10 percentage points, to a value below 53%, the lower limit of normal for 2-dimensional echocardiography12
(Figure 3). However, it remains contentious as to whether the proposed cut-off value of LVEF is relevant, because the institutional lower limit of LVEF ranges from 50% to 55%. In the position paper published in 2016 by the European Society of Cardiology, CTRCD was simply defined as a decrease in LVEF of more than 10 percentage points, to a value below the lower limit of normal6
(Figure 3). Recent studies revealed that LVEF is insufficient for the early detection of CTRCD, and that global longitudinal strain obtained by speckle tracking is a superior parameter for the accurate and early prediction of a subsequent decline in LVEF.13
Echocardiography remains the mainstay for the detection and evaluation of CTRCD; multimodality imaging techniques, including cardiac magnetic resonance imaging, are also helpful.6
In addition, cardiac biomarkers, such as B-type natriuretic peptide and cardiac troponin, are widely used for early detection and risk stratification of CTRCD.6
In accordance with the increasing need for a standardized approach to CTRCD, several guidelines have been developed.14–17
After the publication of a pivotal trial of trastuzumab for HER2-positive breast cancer reporting a considerable increase in the incidence of CTRCD,18
CTRCD was intuitively classified according to reversibility and structural abnormalities: Type 1 CTRCD is irreversible and dose-dependent with structural abnormalities, whereas Type 2 CTRCD is reversible and dose-independent without structural abnormalities.19
Examples of drugs causing Type 1 and Type 2 CTRCD are anthracyclines and trastuzumab, respectively, although both are likely to induce cardiotoxicity through a combined action. However, this simple classification has been challenged because a substantial proportion of anthracycline-induced cardiotoxicity is reversible, especially when early detection and prompt therapy of cardiotoxicity is feasible.20
Given that the number of anticancer drugs for which CTRCD is a concern is growing, it becomes increasingly important to understand the molecular and cellular basis of CTRCD for each class of anticancer drugs (Figure 4).

Anthracyclines
Anthracyclines are cytotoxic agents with potent antitumor effects that have commonly been used for the treatment of a variety of solid tumors and hematological malignancies.6–8,19
However, the clinical use of anthracyclines is limited because of concerns about their cardiotoxicity, which is dose-dependent21
and can occur acutely or chronically months to years later.6,19
Overall, the incidence of doxorubicin-related CTRCD at cumulative doses of 400, 550 and 700 mg/m2
has been reported to be 3–5%, 7–26%, and 18–48%, respectively.6,21
On the basis of clinical studies reporting an increased risk of CTRCD with a cumulative doxorubicin dose of at least 250 mg/m2, the Clinical Practice Guideline from the American Society of Clinical Oncology (ASCO) recommended that patients with high-dose anthracycline treatment (doxorubicin ≥250 mg/m2, epirubicin ≥600 mg/m2) should be considered at an increased risk of developing CTRCD.15
Although the total cumulative dose is a major risk factor, there is individual variability in susceptibility to CTRCD due to the presence of other risk factors, including older age (>65 years), pediatric age (<18 years), concomitant chemotherapy using trastuzumab, prior or concurrent mediastinal radiation therapy, cardiac comorbidities, and hypertension.6
Although the precise mechanism underlying anthracycline-induced CTRCD has not been fully elucidated,7,8
reactive oxygen species (ROS) are the central determinant of cardiotoxicity, as well as antitumor effects22
(Figure 4). The quinone moiety of doxorubicin forms a semiquinone free radical after 1-electron reduction and, under oxic conditions, redox cycling gives rise to persistent formation of ROS, which eventually damage cellular membranes, mitochondria, and nuclear DNA.22
In addition, doxorubicin accumulates in the mitochondria and increases mitochondrial iron levels by reducing the expression of ATP-binding cassette (ABC) transporter ABCB8, a transporter facilitating mitochondrial iron export, in cardiomyocytes.23
Consequently, the doxorubicin-iron complex in mitochondria further stimulates free radical formation via ferrous/ferric redox cycling,22
and further exacerbates cell damage.23
Dexrazoxane, an iron chelator, is a drug used to mitigate anthracycline-induced cardiotoxicity approved in the US and Europe8
that has superior effects in reducing mitochondrial iron levels over deferoxamine, an iron chelator used therapeutically in patients with iron overload.23
The important role of mitochondrial iron accumulation in doxorubicin cardiotoxicity is supported by experimental results showing that dexrazoxane prevented doxorubicin-induced cardiac dysfunction in mice whereas deferoxamine did not.23
Anthracyclines also induce DNA damage by free radical-independent mechanisms. The anthracycline molecule intercalates into double-stranded DNA and inhibits not only DNA and RNA synthesis, but also DNA unwinding mediated by topoisomerase II (Top2).7
Top2 unwinds the double-helical DNA by a sequential catalytic cycle of nicking, controlled rotation, and religation.24
Doxorubicin interferes with Top2 binding to DNA at higher concentrations (>10 µM), and induces DNA double-strand breaks (DSBs) by disrupting the cycle of Top2-DNA interactions (Top2 poisoning) and blocking DNA religation at lower concentrations (<1 µM),24
which accounts for the potent cytotoxic activity against cancer cells. Human cells express 2 distinct isoforms of Top2: Top2α and Top2β. Top2α is highly expressed in proliferating cells in normal tissues and cancer, whereas Top2β is expressed in quiescent cells, such as postnatal cardiomyocytes.24,25
Recently, interaction with Top2β has emerged as a key mechanism underlying anthracycline cardiotoxicity.7,8,25
Cardiomyocyte-specific deletion of the
Top2b
gene in mice prevented doxorubicin-induced DNA DSBs in cardiomyocytes, and improved LV systolic function and survival.25
Top2β-dependent DNA DSBs induce activation of the p53 pathway and downregulation of proliferator-activated receptor-γ coactivator 1α and 1β, leading to aberrant mitochondrial biogenesis and function, decreased ATP production, increased ROS generation, and eventually apoptotic cell death of cardiomyocytes.25
Anthracycline cardiotoxicity is a multifactorial process that is incredibly complex and involves a number of changes in the structure and function of intracellular molecules and organelles7,8,19
(Figure 4). For example, other mechanisms contributing to anthracycline cardiotoxicity include mitochondrial dysfunction, dysregulation of Ca2+
homeostasis, dysregulation of the ubiquitin protease system, disturbance of autophagy fluxes, endoplasmic reticulum stress, cardiomyocyte cell death, and endothelial dysfunction. Many molecular pathways and interactions contribute to anthracycline-induced cardiomyocyte injury or dysfunction, and various potential therapeutic targets for cardioprotection have emerged.8
Although many strategies to activate endogenous prosurvival pathways hold promise in animal models of anthracycline-induced CTRCD, the obstacle that needs to be overcome before these strategies can be used clinically is ensuring these approaches protect the heart without compromising the antitumor efficacy of the anthracyclines. Further investigations are needed to explore the overarching mechanism of anthracycline cardiotoxicity taking into consideration phenotypic diversity and pathophysiological heterogeneity.
HER2 Inhibitors
The ErbB receptor family of tyrosine kinases has 4 members (epidermal growth factor receptor [EGFR], HER2, HER3, and HER4) and plays an important role in the control of fundamental cellular responses, including cell metabolism and survival as well as cell proliferation and differentiation.26
Ligand binding to ErbB receptors stimulates different signaling networks.26
Amplification or overexpression of the
HER2
gene is observed in 25–30% of breast cancers, and was associated with worse prognosis under conventional treatment.26
A pivotal randomized trial of trastuzumab, a humanized monoclonal antibody against HER2, in combination with standard chemotherapy showed clinical benefit in patients with metastatic HER2-positive breast cancer, but a high incidence of LV dysfunction was observed among patients who had received trastuzumab and anthracycline-based therapy simultaneously.18
Heart failure in New York Heart Association (NYHA) Class III or IV occurred in 27% of patients given trastuzumab plus anthracycline/cyclophosphamide, compared with only 8% of patients given anthracycline/cyclophosphamide alone.18
Unlike anthracycline cardiotoxicity, trastuzumab cardiotoxicity is largely reversible and dose-independent without structural abnormalities, and typically occurs during or early after treatment.19
The concomitant use of anthracyclines should be avoided because major adjuvant trials indicated that the administration of trastuzumab simultaneously or shortly after anthracyclines led to a higher incidence of CTRCD.19
Other risk factors for trastuzumab cardiotoxicity are the prior use of anthracyclines, lower baseline LVEF, arterial hypertension, and older age (>60 years).6,27
The Clinical Practice Guideline from the ASCO suggests that patients receiving low-dose anthracycline treatment (doxorubicin <250 mg/m2, epirubicin <600 mg/m2) followed by trastuzumab and those receiving trastuzumab alone with the presence of any risk factor (more than 2 cardiovascular risk factors including smoking, hypertension, diabetes, dyslipidemia, and obesity, older age [≥60 years], and compromised cardiac function) are at an increased risk of developing CTRCD.15
The mechanism underlying trastuzumab-induced CTRCD is not completely understood26,27
(Figure 4). In mice, cardiomyocyte-specific deletion of the erb-b2 receptor tyrosine kinase 2 (Erbb2) gene (the mouse homolog of HER2) led to LV dysfunction and increased sensitivity to anthracycline cardiotoxicity.28,29
Cardiomyocyte apoptosis was increased in the hearts of cardiomyocyte-specific
Erbb2-deficient mice, and overexpression of anti-apoptotic Bcl-xL partially rescued the heart failure phenotype.28
Consistent with these findings, stimulation with neuregulin-1, a HER2 ligand released from endothelial cells, inhibited myofibrillar degeneration and apoptosis, and promoted survival in cultured cardiomyocytes.30
These observations suggest that trastuzumab-induced CTRCD occurs as an adverse consequence of interfering with the homeostatic and cytoprotective effects of HER2 on cardiomyocytes.26,27
Therefore, trastuzumab cardiotoxicity could be pronounced when the heart is stressed by hemodynamic overload or cytotoxic damage by the concomitant or prior use of anthracyclines.6,19,27
Following the success of trastuzumab, lapatinib, pertuzumab, and ado-trastuzumab emtansine (T-DM1) were approved for worldwide HER2-directed therapies. Lapatinib is a TKI of HER2 and EGFR, and pertuzumab is a humanized monoclonal antibody against HER2 that blocks dimerization of HER2 with other ErbB receptors. T-DM1 consists of trastuzumab covalently linked to the cytotoxic agent DM1. Interestingly, the cardiac safety of pertuzumab plus trastuzumab was similar to that of trastuzumab alone,27
and the incidence of CTRCD was low in patients treated with lapatinib or T-DM1.27
The specific mode of action of individual HER2 inhibitors may account for the differences in clinical manifestations of cardiotoxicity, and further investigations are needed to understand the precise mechanisms of class- and drug-specific cardiotoxicities of HER2 inhibitors.
Angiogenesis Inhibitors
Invasive tumor growth depends on the formation of new blood vessels (angiogenesis) to accommodate the increased demands for the delivery of oxygen and nutrients and removal of catabolites.31
Judah Folkman’s foresight of “anti-angiogenesis”32
as a novel strategy for cancer treatment led to the identification of VEGF as a key mediator of angiogenesis with considerable anticancer therapeutic potential.33
The first angiogenesis inhibitor bevacizumab, a humanized monoclonal antibody against VEGF, was approved for the treatment of metastatic colorectal cancer.34
Thereafter, other angiogenesis inhibitors were developed, including: sunitinib, sorafenib, vandetanib, axitinib, pazopanib, regorafenib, and lenvatinib, TKIs targeting the signaling cascade of VEGF and other growth factors, such as platelet-derived growth factor (PDGF) and fibroblast growth factor; ramucirumab, an anti-VEGF receptor (VEGFR) 2 monoclonal antibody; and aflibercept, a soluble VEGF receptor decoy that has a high binding affinity for VEGF-A, VEGF-B, and placental growth factor. Despite providing considerable benefits in the treatment of advanced solid cancers, angiogenesis inhibitors can cause adverse cardiovascular toxicities, including CTRCD, hypertension, and thromboembolism.6,35
The incidence of CTRCD associated with bevacizumab and TKIs has been reported to be 1.6–4% and 3–19%, respectively, although the rate varies widely depending on the drug, treatment protocol, and diagnostic criteria.6
In most cases, CTRCD occurs early, and is reversible under careful cardiovascular management.6,35
The mechanism underlying angiogenesis inhibitor-induced CTRCD is multifactorial, and is not completely understood35
(Figure 4). The administration of clinically relevant doses of TKIs unexpectedly induced a broad spectrum of changes in transcriptome and kinome profiles in mouse heart.36
Similarly, treatment with TKIs caused cytotoxicity not only in human induced pluripotent stem cell (hiPSC)-derived endothelial cells, but also in hiPSC-derived cardiomyocytes (hiPSC-CMs) and fibroblasts,37
indicating a direct toxic effect of these TKIs on cardiomyocytes and other non-endothelial cells. Importantly, in response to hemodynamic overload, the myocardium undergoes hypertrophic growth and simultaneously promotes VEGF-mediated angiogenesis to meet the demands for a blood supply that is sufficient to sustain the increase in myocardial mass and performance.38
Inhibition of cardiac angiogenesis by administration of a soluble form of fms-like tyrosine kinase 1, a decoy for VEGF, exacerbated LV dysfunction in mice with pressure overload.39
The mismatch resulting from insufficient angiogenesis may be one of the critical mechanisms underlying the pathogenesis of CTRCD associated with angiogenesis inhibitors. Angiogenesis inhibitors also cause arterial hypertension via many molecular mechanisms,6,35
and an increase in afterload potentially contributes to the development of CTRCD.
Proteasome Inhibitors
The advent of proteasome inhibitors (bortezomib, carfilzomib, and ixazomib) and immunomodulatory drugs has greatly improved the overall survival of patients with multiple myeloma (MM).40
Proteasome inhibitors selectively inhibit the 26S proteasome that is responsible for proteasomal degradation of ubiquitinated proteins, and exerts a cytotoxic effect on myeloma cells by leading to the accumulation of misfolded proteins and reversing cell cycle and apoptotic regulatory pathways, thereby triggering apoptosis.41
A pivotal randomized trial of carfilzomib in combination with lenalidomide and dexamethasone reported significantly improved progression-free survival in patients with relapsed MM, but the incidence of CTRCD was higher in patients receiving carfilzomib (all grades: 6.4%; Grade 3 or higher: 3.8%) than in those receiving lenalidomide and dexamethasone alone (all grades: 4.1%; Grade 3 or higher: 1.8%).42
In a randomized trial of carfilzomib and dexamethasone vs. bortezomib and dexamethasone for relapsed or refractory MM, CTRCD of Grade 3 or higher occurred more frequently in the carfilzomib group (1.7%) than in the bortezomib group (0.4%), although progression-free survival was significantly longer in the carfilzomib group.43
Cardiovascular adverse events, such as CTRCD and hypertension, are more severe with carfilzomib than bortezomib, possibly due to the potent and irreversible activity of carfilzomib on proteasome inhibition.40,44
Most cardiovascular adverse events occur within the first 3 months of therapy,44
and are reversible and manageable.45
Although the frequency of discontinuation or death due to cardiotoxicity is low,45
careful cardiovascular management is recommended for patients receiving proteasome inhibitors, because a high proportion of MM patients are elderly and comorbid with cardiovascular diseases and risk factors.
The mechanism underlying proteasome inhibitor-induced CTRCD is multifactorial, and is not precisely understood40,46
(Figure 4). The ubiquitin-proteasome system plays a critical role in cellular homeostasis and integrity in the cardiovascular system, and both the myocardium and vasculature are damaged by proteasome inhibition. The cytotoxic effect can differ according to the nature of the proteasome inhibitor, the dosage and duration of treatment, and the cellular context-dependent levels of oxidative stress and misfolded proteins.40,46
Endothelial toxicity can potentiate coronary vasospasm, as well as hypertension and venous thromboembolism.40,46
A recent study demonstrated that administration of carfilzomib induced LV dysfunction in mice by increasing protein phosphatase 2A activity and subsequent inhibition of AMP-activated protein kinase α (AMPKα) phosphorylation and autophagy-related proteins.47
Interestingly, metformin, the first-line drug for the treatment of diabetes, is an AMPKα activator and autophagy inducer, and coadministration of metformin prevented carfilzomib-induced CTRCD without interfering with proteasome inhibition, highlighting metformin as a potential cardioprotective agent against carfilzomib cardiotoxicity.47
Genetic Architecture of CTRCD
Clinical risk factors for CTRCD have been proposed, but a wide range of phenotypic viability between individuals indicates a significant effect of genetic predisposition on the development of CTRCD.6,15
Genetic predisposition to CTRCD has been studied by 2 different approaches, namely using pharmacogenomics and patient-specific hiPSC-CMs.
Several genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPs) that occur more or less frequently in patients with anthracycline cardiotoxicity, including: rs1786814 in CUGBP Elav-like family member 4 (CELF4
; sarcomere structure and function); rs2229774 in retinoic acid receptor γ (RARG
;
TOP2B
expression); rs7853758 in solute carrier family 28 member 3 (SLC28A3
; drug transport); rs17863783 in uridine 5′-diphospho-glucuronosyltransferase family 1 member A6 (UGT1A6
; drug metabolism); and rs28714259, an intergenic variant.48
When combined with clinical risk factors, genetic risk profiling using these SNPs effectively predicted the risk of anthracycline cardiotoxicity.49
Further identification of genetic risk factors will not only improve the accuracy of pharmacogenomic risk prediction, but also yield novel insights into the molecular pathophysiology.
A GWAS of 280 patients treated for childhood cancer identified a non-synonymous variant in the
RARG
gene that was highly associated with anthracycline cardiotoxicity.50
RARG protein binds to the
TOP2B
promoter, and represses
TOP2B
gene expression. Interestingly, the RARG variant protein did not repress
TOP2B
expression as effectively as wild-type RARG protein. This finding suggests that the
RARG
variant is associated with higher
TOP2B
expression levels, which leads to increased susceptibility to anthracycline cardiotoxicity.50
A recent study exploring rare variants in dilated cardiomyopathy genes demonstrated that truncating variants in the titin (TTN) gene contributed to the susceptibility of developing CTRCD.51
The
TTN
gene encodes the giant protein titin, which is a molecular bidirectional spring essential for the structural integrity and function of sarcomeres,52
and
TTN-truncating variants are more frequently observed in patients with CTRCD (7.5%),51
as well as in patients with secondary cardiomyopathies, such as peripartum cardiomyopathy (10%)53
and alcoholic cardiomyopathy (9.9%).54
These data suggest that the cardiomyopathic phenotype may be accelerated by environmental factors, including cancer chemotherapy as “second hits” in genetically susceptible patients (e.g., those harboring
TTN-truncating variants).
hiPSC-CMs represent a novel and evolving technology that has been used to model cardiovascular diseases in vitro. hiPSC-CMs possess patient-specific genetic variations, and patient-specific responses to anticancer drugs, and allow for the in vitro evaluation of the safety of anticancer drugs for a particular patient.55
Patient-specific hiPSC-CMs recapitulate the prediction to doxorubicin-induced cardiotoxicity of individual patients at the cellular level. hiPSC-CMs derived from breast cancer patients who experienced doxorubicin cardiotoxicity were consistently more sensitive to doxorubicin-induced cytotoxicity than those from patients without doxorubicin cardiotoxicity, as evidenced by cellular viability or caspase activation.56
Similarly, hiPSC-CMs derived from patients who developed CTRCD after trastuzumab treatment were more sensitive to trastuzumab-induced decreases in contraction velocity and deformation, but not to cellular toxicity, than hiPSC-CMs from patients without cardiotoxicity.57
The use of hiPSC-CMS for in vitro modeling of CTRCD is promising, but is currently at the preliminary stages in the clinical setting for the prediction of the risk of CTRCD in individual patients on the basis of the cardiotoxicity of patient-specific hiPSC-CMs. This is because the cellular properties of hiPSC-CMs remain variable, and these variations may alter the results of phenotyping and affect reproducibility.58
Conclusions
This article provides an overview of our current knowledge of the clinical presentation and pathophysiology of CTRCD induced by anthracyclines, HER2 inhibitors, angiogenesis inhibitors, and proteasome inhibitors, which are the representative classes of anticancer drugs demanding diagnostic and therapeutic management from the onco-cardiology viewpoint. Anticancer drugs often target molecules essential to cardiovascular homeostasis, and off-target effects can also induce a toxic phenotype. With the rapid ongoing progress in the research and development of cancer therapeutics, the number of anticancer drugs with potential cardiotoxic effects will increase. The accumulation of clinical data, in particular on the newer anticancer drugs, is urgently needed to establish evidence-based practice for the diagnosis, prevention, and treatment of CTRCD. In addition, basic cardiovascular research has a major role in defining the pathogenic mechanisms, as well as establishing diagnostic strategies with risk prediction and therapeutic strategies for CTRCD. To solve the unmet needs in onco-cardiology, interdisciplinary collaboration is required to integrate the many different perspectives of clinicians, basic researchers, regulatory scientists, medical assistants, and even patients.
Disclosures
H.A. has received trust research/joint research funding from Ono Pharmaceutical, scholarship funding from Mitsubishi-Tanabe Pharma, and remuneration for lectures from Daiichi Sankyo and Bayer Yakuhin. The remaining authors have no conflicts of interest to declare.
References
- 1.
Ministry of Health Labour and Welfare, Japan. Vital statistics. 2019. https://www.mhlw.go.jp/english/database/db-hw/vs01.html (accessed June 25, 2020).
- 2.
Okura Y, Ozaki K, Tanaka H, Takenouchi T, Sato N, Minamino T. The Impending epidemic of cardiovascular diseases in patients with cancer in Japan. Circ J 2019; 83: 2191–2202.
- 3.
Mehta LS, Watson KE, Barac A, Beckie TM, Bittner V, Cruz-Flores S, et al. Cardiovascular disease and breast cancer: Where these entities intersect: A scientific statement from the American Heart Association. Circulation 2018; 137: e30–e66.
- 4.
Barac A, Murtagh G, Carver JR, Chen MH, Freeman AM, Herrmann J, et al. Cardiovascular health of patients with cancer and cancer survivors: A roadmap to the next level. Am J Coll Cardiol 2015; 65: 2739–2746.
- 5.
Minami M, Matsumoto S, Horiuchi H. Cardiovascular side-effects of modern cancer therapy. Circ J 2010; 74: 1779–1786.
- 6.
Zamorano JL, Lancellotti P, Rodriguez Munoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for Cancer Treatments and Cardiovascular Toxicity of the European Society of Cardiology (ESC). Eur Heart J 2016; 37: 2768–2801.
- 7.
Renu K, Abilash VG, Tirupathi Pichiah PB, Arunachalam S. Molecular mechanism of doxorubicin-induced cardiomyopathy: An update. Eur J Pharmacol 2018; 818: 241–253.
- 8.
Raber I, Asnani A. Cardioprotection in cancer therapy: Novel insights with anthracyclines. Cardiovasc Res 2019; 115: 915–921.
- 9.
Cardinale D. A new frontier: Cardio-oncology. Cardiologia 1996; 41: 887–891 (in Italian).
- 10.
Okwuosa TM, Prabhu N, Patel H, Kuzel T, Venugopal P, Williams KA, et al. The cardiologist and the cancer patient: Challenges to cardio-oncology (or onco-cardiology) and call to action. Am J Coll Cardiol 2018; 72: 228–232.
- 11.
Yeh ET. Onco-cardiology: The time has come. Tex Heart Inst J 2011; 38: 246–247.
- 12.
Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: A report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr 2014; 27: 911–939.
- 13.
Negishi K, Negishi T, Haluska BA, Hare JL, Plana JC, Marwick TH. Use of speckle strain to assess left ventricular responses to cardiotoxic chemotherapy and cardioprotection. Eur Heart J Cardiovasc Imaging 2014; 15: 324–331.
- 14.
Armenian SH, Hudson MM, Mulder RL, Chen MH, Constine LS, Dwyer M, et al. Recommendations for cardiomyopathy surveillance for survivors of childhood cancer: A report from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol 2015; 16: e123–e136.
- 15.
Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol 2017; 35: 893–911.
- 16.
Denlinger CS, Sanft T, Baker KS, Broderick G, Demark-Wahnefried W, Friedman DL, et al. Survivorship, version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2018; 16: 1216–1247.
- 17.
Curigliano G, Lenihan D, Fradley M, Ganatra S, Barac A, Blaes A, et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Ann Oncol 2020; 31: 171–190.
- 18.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001; 344: 783–792.
- 19.
Ewer MS, Ewer SM. Cardiotoxicity of anticancer treatments. Nat Rev Cardiol 2015; 12: 547–558.
- 20.
Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation 2015; 131: 1981–1988.
- 21.
Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003; 97: 2869–2879.
- 22.
Keizer HG, Pinedo HM, Schuurhuis GJ, Joenje H. Doxorubicin (adriamycin): A critical review of free radical-dependent mechanisms of cytotoxicity. Pharmacol Ther 1990; 47: 219–231.
- 23.
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest 2014; 124: 617–630.
- 24.
Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem Biol 2010; 17: 421–433.
- 25.
Zhang S, Liu X, Bawa-Khalfe T, Lu LS, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med 2012; 18: 1639–1642.
- 26.
Cote GM, Sawyer DB, Chabner BA. ERBB2 inhibition and heart failure. N Engl J Med 2012; 367: 2150–2153.
- 27.
Florido R, Smith KL, Cuomo KK, Russell SD. Cardiotoxicity from human epidermal growth factor receptor-2 (HER2) targeted therapies. J Am Heart Assoc 2017; 6: pii:e006915.
- 28.
Crone SA, Zhao YY, Fan L, Gu Y, Minamisawa S, Liu Y, et al. ErbB2 is essential in the prevention of dilated cardiomyopathy. Nat Med 2002; 8: 459–465.
- 29.
Ozcelik C, Erdmann B, Pilz B, Wettschureck N, Britsch S, Hubner N, et al. Conditional mutation of the ErbB2 (HER2) receptor in cardiomyocytes leads to dilated cardiomyopathy. Proc Natl Acad Sci USA 2002; 99: 8880–8885.
- 30.
Jay SM, Murthy AC, Hawkins JF, Wortzel JR, Steinhauser ML, Alvarez LM, et al. An engineered bivalent neuregulin protects against doxorubicin-induced cardiotoxicity with reduced proneoplastic potential. Circulation 2013; 128: 152–161.
- 31.
Apte RS, Chen DS, Ferrara N. VEGF in signaling and disease: Beyond discovery and development. Cell 2019; 176: 1248–1264.
- 32.
Zetter BR. The scientific contributions of M. Judah Folkman to cancer research. Nat Rev Cancer 2008; 8: 647–654.
- 33.
Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discov 2016; 15: 385–403.
- 34.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–2342.
- 35.
Touyz RM, Herrmann J. Cardiotoxicity with vascular endothelial growth factor inhibitor therapy. NPJ Precis Oncol 2018; 2: 13.
- 36.
Stuhlmiller TJ, Zawistowski JS, Chen X, Sciaky N, Angus SP, Hicks ST, et al. Kinome and transcriptome profiling reveal broad and distinct activities of erlotinib, sunitinib, and sorafenib in the mouse heart and suggest cardiotoxicity from combined signal transducer and activator of transcription and epidermal growth factor receptor inhibition. J Am Heart Assoc 2017; 6: pii:e006635.
- 37.
Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, et al. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med 2017; 9: pii:eaaf2584.
- 38.
Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ Res 2014; 114: 565–571.
- 39.
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007; 446: 444–448.
- 40.
Li W, Garcia D, Cornell RF, Gailani D, Laubach J, Maglio ME, et al. Cardiovascular and thrombotic complications of novel multiple myeloma therapies: A review. JAMA Oncol 2017; 3: 980–988.
- 41.
Adams J. The proteasome: A suitable antineoplastic target. Nat Rev Cancer 2004; 4: 349–360.
- 42.
Stewart AK, Rajkumar SV, Dimopoulos MA, Masszi T, Spicka I, Oriol A, et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med 2015; 372: 142–152.
- 43.
Dimopoulos MA, Moreau P, Palumbo A, Joshua D, Pour L, Hajek R, et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): A randomised, phase 3, open-label, multicentre study. Lancet Oncol 2016; 17: 27–38.
- 44.
Cornell RF, Ky B, Weiss BM, Dahm CN, Gupta DK, Du L, et al. Prospective study of cardiac events during proteasome inhibitor therapy for relapsed multiple myeloma. J Clin Oncol 2019; 37: 1946–1955.
- 45.
Waxman AJ, Clasen S, Hwang WT, Garfall A, Vogl DT, Carver J, et al. Carfilzomib-associated cardiovascular adverse events: A systematic review and meta-analysis. JAMA Oncol 2018; 4: e174519.
- 46.
Gavazzoni M, Vizzardi E, Gorga E, Bonadei I, Rossi L, Belotti A, et al. Mechanism of cardiovascular toxicity by proteasome inhibitors: New paradigm derived from clinical and pre-clinical evidence. Eur J Pharmacol 2018; 828: 80–88.
- 47.
Efentakis P, Kremastiotis G, Varela A, Nikolaou PE, Papanagnou ED, Davos CH, et al. Molecular mechanisms of carfilzomib-induced cardiotoxicity in mice and the emerging cardioprotective role of metformin. Blood 2019; 133: 710–723.
- 48.
Linschoten M, Teske AJ, Cramer MJ, van der Wall E, Asselbergs FW. Chemotherapy-related cardiac dysfunction: A systematic review of genetic variants modulating individual risk. Circ Genom Precis Med 2018; 11: e001753.
- 49.
Visscher H, Ross CJ, Rassekh SR, Barhdadi A, Dube MP, Al-Saloos H, et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J Clin Oncol 2012; 30: 1422–1428.
- 50.
Aminkeng F, Bhavsar AP, Visscher H, Rassekh SR, Li Y, Lee JW, et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat Genet 2015; 47: 1079–1084.
- 51.
Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, Lunde IG, Wakimoto H, Smith AM, et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation 2019; 140: 31–41.
- 52.
Ware JS, Cook SA. Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nat Rev Cardiol 2018; 15: 241–252.
- 53.
Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, et al. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med 2016; 374: 233–241.
- 54.
Ware JS, Amor-Salamanca A, Tayal U, Govind R, Serrano I, Salazar-Mendiguchia J, et al. Genetic etiology for alcohol-induced cardiac toxicity. Am J Coll Cardiol 2018; 71: 2293–2302.
- 55.
Stack JP, Moslehi J, Sayed N, Wu JC. Cancer therapy-induced cardiomyopathy: Can human induced pluripotent stem cell modelling help prevent it? Eur Heart J 2019; 40: 1764–1770.
- 56.
Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, et al. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med 2016; 22: 547–556.
- 57.
Kitani T, Ong SG, Lam CK, Rhee JW, Zhang JZ, Oikonomopoulos A, et al. Human-induced pluripotent stem cell model of trastuzumab-induced cardiac dysfunction in patients with breast cancer. Circulation 2019; 139: 2451–2465.
- 58.
Yang X, Pabon L, Murry CE. Engineering adolescence: Maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res 2014; 114: 511–523.
