Abstract
Background: Cardiac calmodulinopathy, characterized by a life-threatening arrhythmia and sudden death in the young, is extremely rare and caused by genes encoding calmodulin, namely calmodulin 1 (CALM1), CALM2, and CALM3.
Methods and Results: We screened 195 symptomatic children (age 0–12 years) who were suspected of inherited arrhythmias for 48 candidate genes, using a next-generation sequencer. Ten probands were identified as carrying variants in any of CALM1–3 (5%; median age 5 years), who were initially diagnosed with long QT syndrome (LQTS; n=5), catecholaminergic polymorphic ventricular tachycardia (CPVT; n=3), and overlap syndrome (n=2). Two probands harbored a CALM1 variant and 8 probands harbored 6 CALM2 variants. There were 4 clinical phenotypes: (1) documented lethal arrhythmic events (LAEs): 4 carriers of N98S in CALM1 or CALM2; (2) suspected LAEs: CALM2 p.D96G and D132G carriers experienced syncope and transient cardiopulmonary arrest under emotional stimulation; (3) critical cardiac complication: CALM2 p.D96V and p.E141K carriers showed severe cardiac dysfunction with QTc prolongation; and (4) neurological and developmental disorders: 2 carriers of CALM2 p.E46K showed cardiac phenotypes of CPVT. Beta-blocker therapy was effective in all cases except cardiac dysfunction, especially in combination with flecainide (CPVT-like phenotype) and mexiletine (LQTS-like).
Conclusions: Calmodulinopathy patients presented severe cardiac features, and their onset of LAEs was earlier in life, requiring diagnosis and treatment at the earliest age possible.
Calmodulin (CaM), a ubiquitously expressed calcium signaling protein, is encoded by 3 different genes, namely calmodulin 1 (CALM1), CALM2, and CALM3, located on distinct chromosomal loci.1,2 CaM, as a cellular calcium sensor, modulates various proteins, such as ion channels,3 ryanodine receptor channels,4 and Ca2+/CaM-dependent protein kinase.5 In particular, CaM is the actual Ca2+
sensor for Ca2+-dependent inactivation (CDI) of L-type Ca2+
channels (LCC).3,6 Since 2012, variants of the genes encoding CaM have been reported to be associated with early-onset inherited primary arrhythmia syndrome (IPAS), characterized by severe forms of long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and/or idiopathic ventricular fibrillation (VF).7 The disease entity has been recently called “cardiac calmodulinopathy”. However, their phenotypes vary considerably depending on variant location (CALM1, CALM2, or CALM3), and remain unclear due to their rarity.8
As mentioned above, 2 major phenotypes of cardiac calmodulinopathy are LQTS and CPVT. LQTS, a representative IPAS, presents prolongation of QT intervals on a 12-lead electrocardiogram (ECG) and a high risk of sudden cardiac death due to ventricular arrhythmias, termed as torsade de pointes (TdP).9 The prevalence of LQTS is approximately 1 in 2,000,10 and more than 10 genes have been reported to be associated with LQTS.11 LQTS caused by CALM1, CALM2, and CALM3 is categorized as LQT14, LQT15, and LQT16, respectively.11,12 CPVT, another form of IPAS, presents exercise-related bidirectional or polymorphic ventricular tachyarrhythmias in patients with a normal resting ECG and no structural heart disease.13 There are 3 confirmed CPVT variants; CPVT1 is caused by heterozygous mutation of the ryanodine receptor 2 (RYR2) gene,14 whereas CPVT2 and CPVT3 are caused by biallelic loss-of-function variants in the calsequestrin 2 (CASQ2)15 and trans-2,3-enoyl-CoA reductase like (TECRL) genes,16 respectively, although occasionally heterozygous CASQ2 variants have also been shown to cause CPVT2.15 In addition, calmodulin genes have been considered as important causative genes of CPVT.17
Recently, several calmodulin gene mutations were identified in our IPAS cohort that caused severe cardiac phenotypes. We have participated in the International Calmodulinopathy Registry,8 and several cases reported in the present study have been registered. However, because detailed clinical features, including ECGs, are not reported in the International Calmodulinopathy Registry, we sought to investigate the frequency and clinical characteristics of calmodulinopathy in our cohort of symptomatic Japanese IPAS children.
Methods
Study Cohorts
The study cohort comprised 195 symptomatic children with IPAS (age at diagnosis 0–12 years) from 195 unrelated families that were registered at Shiga University of Medical Science or Kyoto University Graduate School of Medicine between 1996 and 2022 for genetic analysis. The inclusion criteria was as follows: cardiopulmonary arrest (CPA), VF, sustained ventricular tachycardia (VT), high-degree atrioventricular block (AVB), personal history of syncope under emotional stress, seizures, or arrhythmic events. In addition, the child could not have any other gene variant related to LQTS/CPVT. The pathological conditions in these children were an LQTS phenotype in 43%, CPVT in 23%, LQTS+CPVT (overlapped) in 1%, idiopathic VF (including sudden infant death syndrome) in 12%, high-degree AVB in 4%, and others in 17% (e.g., sick sinus syndrome, seizures, palpitation). The cohort included 26 children who had lethal arrhythmic events (LAEs) when they were less than 6 years of age.
The diagnosis of LQTS was made by either of the following clinical features: (1) QTc prolongation on 12-lead ECG at rest, namely QTc ≥450 ms for males and ≥460 ms for females; and (2) QTc prolongation (≥480 ms) induced by an exercise stress test, although QTc at rest was borderline (within 440–450 ms).18 QT intervals were manually measured in Lead II or V5 and were corrected for the heart rate using Bazett’s formula (QTc).
CPVT was defined as the presence of bidirectional VT, polymorphic VT, or VF, which were documented through an exercise stress test and/or an ambulatory ECG in the absence of QT prolongation. The characteristic QRS morphologies were defined as a change in QRS axis every other beat, with 2 (bidirectional) or more types of patterns (polymorphic) during more than 4 consecutive beats.19
All patients and/or their guardians gave written informed consent in accordance with the guidelines approved by each institutional review board.
Genetic Testing
Genomic DNA was extracted from peripheral blood leukocytes. We have conducted genetic analyses for at least 48 genes listed in the Supplementary Table, including 14 confirmed arrhythmia-related genes, using the HaloPlex HS custom panel (Agilent Technologies, Santa Clara, CA, USA) and a benchtop-type next-generation sequencing machine (MiSeq; Illumina, San Diego, CA, USA). Data analyses were performed using SureCall software (Agilent Technologies). The variants were then confirmed using Sanger sequencing. Parents of patients with CALM1/2 variants received genetic analysis for the target variants. Annotation of CALM1 and CALM2 variants was based on accession numbers NM_006888.6 and NM_001743.6, respectively.
The variants in CALM1 and CALM2 were classified based on their pathogenicity according to the ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), VarSome (https://varsome.com/), and the American College of Medical Genetics and Genomics standards and guidelines.20 In all the probands with CALM1 and CALM2 variants, there were no pathogenic/likely pathogenic variants found, except those of the CALM genes.
Results
We identified a 7 variants in 10 of 195 symptomatic IPAS children (5%): 1 CALM1 variant in 2 probands and 6 CALM2 variants in 8 probands (Table). All of these, except 1, were de novo variants. The family members had no arrhythmic events or cardiac dysfunction. Figure 1 shows the topology of the CaM protein: 6 of 7 variants, indicated by different colors, are located in the middle of either EF hand III or IV. In contrast, p.E46K is located in the linker between EF hand I and II. Numbers in blue in Figure 1 correspond to each proband (#1–#10 in the Table), and different colors indicate the category of phenotypes. The names of the original amino acids and their numbers are indicated on the top of each symbol, and the mutated amino acids are indicated by colored symbols.
Table.
Children Carrying Mutations of a Calmodulin-Related Gene
| Phenotype |
Proband
no. |
Age of
onset
(years) |
Sex |
Gene |
Amino
acid |
Diagnosis |
HR
(beats/
min) |
QTc
(ms) |
Symptom |
CHD |
DD |
Drug
therapy |
ICD |
Documented
LAEs |
1 |
5 |
M |
CALM1 |
p.N98S |
CPVT/
LQTS |
98 |
467 |
VF |
None |
±* |
Atenolol |
+ |
| |
|
|
|
|
|
|
|
|
|
|
Flecainide |
|
| |
|
|
|
|
|
|
|
|
|
|
Mexiletine |
|
| 2 |
3 |
F |
CALM1 |
p.N98S |
CPVT |
92 |
420 |
VF |
None |
None |
Nadolol |
+ |
| 3 |
4 |
M |
CALM2 |
p.N98S |
LQTS |
86 |
635 |
VF |
None |
None |
Propranolol |
− |
| 4 |
5 |
M |
CALM2 |
p.N98S |
CPVT/
LQTS |
70 |
453 |
VF |
None |
None |
Propranolol |
− |
Suspected
LAEs |
5 |
5 |
M |
CALM2 |
p.D96G |
LQTS |
59 |
471 |
Syncope |
None |
None |
Bisoprolol |
− |
| 6 |
0 |
F |
CALM2 |
p.D132G** |
LQTS |
102 |
523 |
CPA |
None |
None |
Carteolol |
− |
| |
|
|
|
|
|
|
|
|
|
|
Mexiletine |
|
Critical cardiac
complication |
7 |
0 |
M |
CALM2 |
p.D96V |
LQTS |
98 |
689 |
Advanced
AVB,
deceased |
VSD,
ASD |
NA |
Propranolol |
− |
| 8 |
0 |
M |
CALM2 |
p.E141K |
LQTS |
61 |
788 |
AVB, low
LVEF,
deceased |
None |
NA |
− |
− |
Developmental
disorder |
9 |
5 |
M |
CALM2 |
p.E46K |
CPVT |
66 |
440 |
Syncope,
polymorphic |
PDA |
+ |
Carvedilol |
− |
| |
|
|
|
|
|
|
|
|
|
|
Flecainide |
|
| 10 |
9 |
M |
CALM2 |
p.E46K |
CPVT |
50 |
400 |
Bidirectional
VT,
bradycardia |
PDA |
+ |
Nadolol |
− |
| |
|
|
|
|
|
|
|
|
|
|
Flecainide |
|
*This proband had autism. **All of them except for CALM2 p.D132G were de novo variants (CALM2 p.D132G was unknown because her parents refused genetic testing). ASD, atrial septal defect; AVB, atrioventricular block; CALM1, calmodulin 1; CALM2, calmodulin 2; CHD, congenital heart disease; CPA, cardiopulmonary arrest; CPVT, catecholaminergic polymorphic ventricular tachycardia; DD, developmental disorder; HR, heart rate; ICD, implantable cardioverter defibrillator; LAEs, lethal arrhythmic events; LQTS, long QT syndrome; LVEF, left ventricular ejection fraction; NA, not affected; PDA, patent ductus arteriosus; QTc, corrected QT; VF, ventricular fibrillation; VSD, ventricular septal defect; VT, ventricular tachycardia.

The clinical diagnoses of the 10 children with CALM variants were LQTS in 5, CPVT in 3, and an overlap of both LQTS and CPVT in 2. Their ages at diagnosis ranged between 0 and 9 years, with a median of 5 years.
There were 4 major clinical categories: (1) documented LAEs (N98S; orange symbol, Figure 1), (2) suspected LAEs (yellow symbols, Figure 1), (3) critical cardiac complication (red symbols, Figure 1), and (4) complicated neurological disorders (E46K, green symbol, Figure 1; Table).
Documented LAEs (4 Probands)
Four preschoolers experienced LAEs without structural heart diseases. All of these children had a variant of p.N98S in either CALM1 or CALM2.
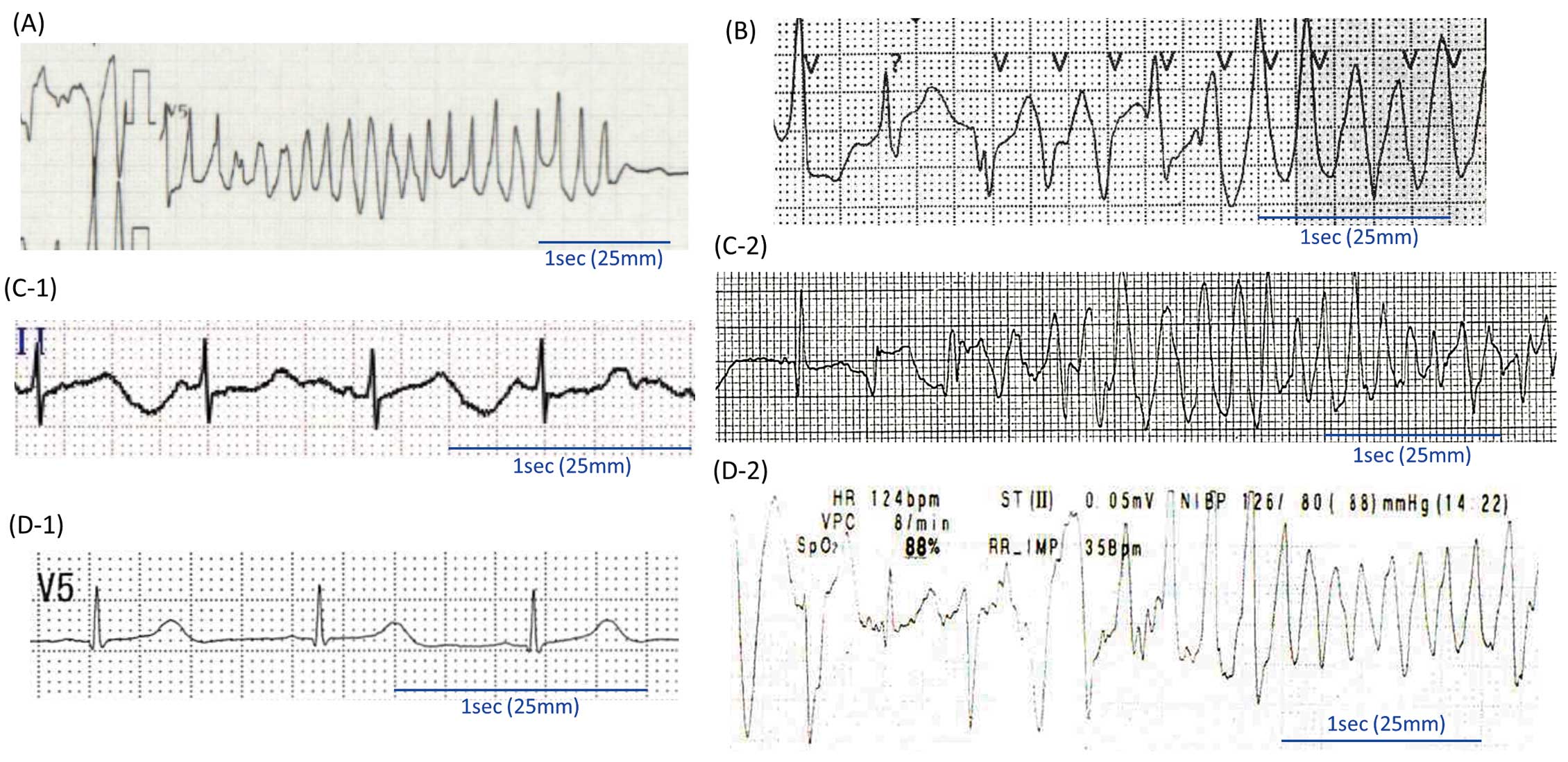
CALM1 p.N98S (2 Probands) A 5-year-old boy with autism (Proband #1; Table) experienced CPA while running and was resuscitated. Repetitive TdPs were documented in an emergency room (Figure 2A). His 12-lead ECG at rest showed distinct T wave alternans in the V4 lead (Supplementary Figure 1A). A Holter ECG recording documented polymorphic and bidirectional VTs during emotional stress (Supplementary Figure 1B). Even after the administration of oral atenolol (50 mg/day) and flecainide (80 mg/day), the patient experienced VT/VFs. Oral mexiletine (270 mg/day) was added, and an implantable cardioverter defibrillator (ICD) was implanted. Since then, there have been no VF events under the above medication.
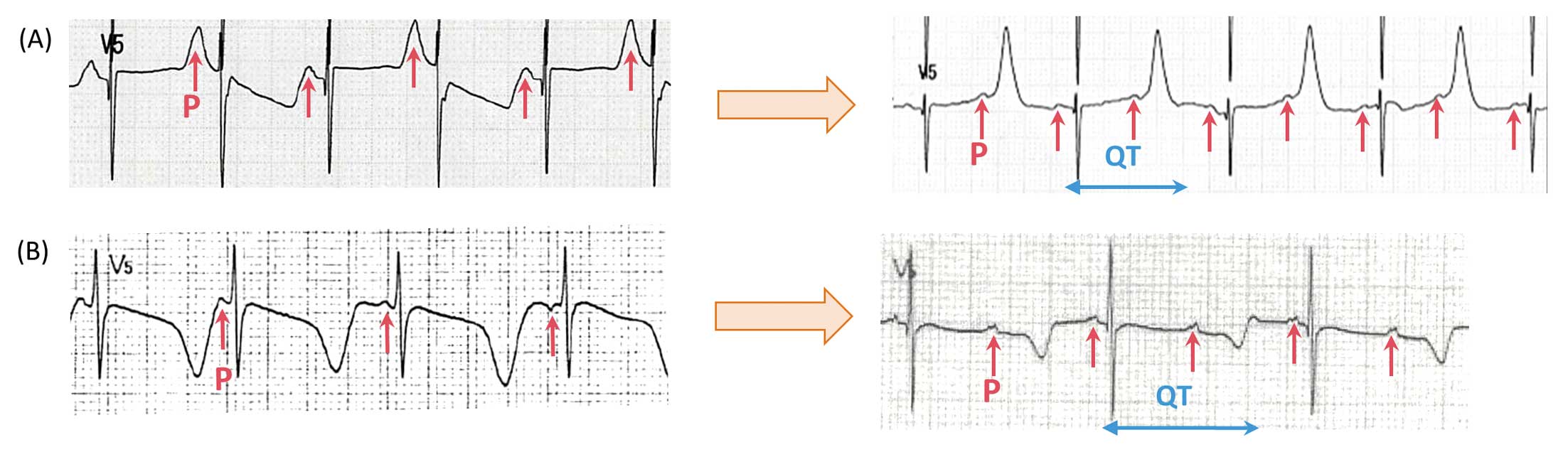
The second case was that of a 3-year-old girl with history of a normal delivery and developmental milestones (Proband #2; Table). The girl experienced CPA while running. An automated external defibrillator (AED) documented VF, and 3 AED deliveries were able to resuscitate her. Her QTc immediately after rescue was within the normal range (420 ms), but gradually prolonged to produce repetitive polymorphic and bidirectional VT and TdP (573 ms) after admission (Figure 2B). Landiolol (8 μg/kg/min) was effective at controlling LAEs during the acute phase, and was later replaced with oral nadolol 1.5 mg/kg/day. She received underwent ICD implantation following her first LAE episode. When she stopped taking nadolol, she had another VF attack that was successfully treated with ICD.
CALM2 p.N98S (2 Probands) A 4-year-old-boy (Proband #3; Table) had suddenly fallen while playing in the playground of a kindergarden, as reported previously.21 An AED documented VF, and an AED shock restored his heart rhythm to sinus rhythm (Figure 2C-1). During transfer to the hospital, his ECG showed transient VF spontaneously (Figure 2C-2), which required a defibrillation shock 4 times. A 12-lead ECG showed normal sinus rhythm with macroscopic T wave alternans and QT prolongation (635–736 ms; Supplementary Figure 2).21 Amiodarone infusion successfully suppressed VF and TdP. Echocardiography revealed normal cardiac function without congenital anomaly. The patient had been doing well under the initial therapy with oral propranolol (90 mg/day).
Another CALM2-N98S carrier was a 5-year-old boy with normal development (Proband #4; Table). Two elder siblings (an 11-year-old brother and a 9-year-old sister) were apparently healthy. His QTc was mildly prolonged on normal sinus rhythm (453 ms; Figure 2D-1; Supplementary Figure 3). At 5 years of age, he had a VF attack while running, and was resucitated with an AED. In the emergency room, polymorphic VTs recurred (Figure 2D-2). Oral propranolol (2 mg/kg/day) was then started and was effective in supressing LAE attacks.
Suspected LAEs (2 Probands)
CALM2 p.D96G The patient was a 5-year-old boy who had a syncopal attack while running (Proband #5; Table). Although his 12-lead ECG at rest showed mild QTc prolongation (471 ms: Figure 3A, Left), his QT interval was markedly prolonged during an exercise test (500 ms at recovery 3 min: Figure 3A, Right). The change in T wave morphology by exercise was similar to that seen in Type 1 LQTS (Supplementary Figure 4). This boy was treated with bisoprolol (0.5 mg/kg/day), which prevented the recurrence of syncopal attacks.
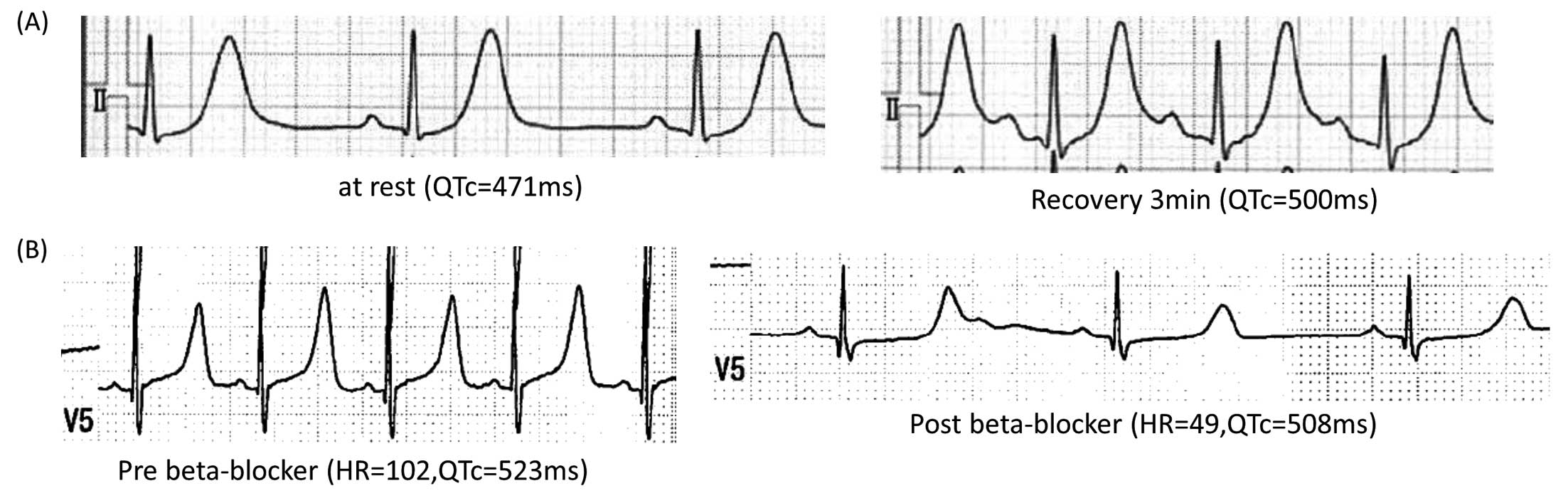
CALM2 p.D132G Another patient in this group was a woman who was 22 years old at the time of genetic diganosis of calmodulinopathy (Proband #6; Table). The patient had once became cyanotic and experienced transient respiratory arrest at the age of 9 month, when she was wailing. She was then successfully resuscitated by cardiopulmonary resuscitation. Upon arriving at hospital, her 12-lead ECG showed significant QTc prolongation (523 ms; Figure 3B, Left). At that time, the patient had been treated with carteolol (20 mg/day) and mexiletine (40 mg/day). The carteolol slowed her heart rate and effectively supressed arrhythmia events, but her QTc was not shortened (508 ms; Figure 3B, Right). Her T-wave showed late-appearance T waves, similar to those seen in LQT3 or LQT8.22–25 At a later age, before genetic testing, an exercise stress test induced no ventricular arrhythmias but did induce QTc prolongation (536 ms; recovery 4 min; data not shown).
Critical Cardiac Complication (2 Probands)
Two probands started to present severe cardiac phenotypes in their fetal period, and died within 2 years.
CALM2 p.D96V The patient was a 0-year-old boy without remarkable family history (Proband #7; Table). Before birth, it was identified that he had mild sinus bradycardia. He was born at 38 weeks gestation (3,221 g). His 12-lead ECG presented bizarre T wave alternans with marked QTc prolongation (671 ms; Figure 4A, Left) and 2:1 AVB, which developed on the Postnatal Day 5 (Figure 4A, Right).
Echocardiography detected significant heart anomalies: a large perimembranous ventricular septal defect, a large secundum atrial septal defect, and a left ventricular non-compaction (Supplementary Figure 5). Although his left ventricular ejection fraction (LVEF) was 60%, congestive heart failure worsened due to the combination of bradycardia and congestive heart failure due to a large amount of left to right shunts. Pacemaker implantation was performed on Day 10. He was administered continuous intravenous landiolol followed by oral bisoprolol.
At the age of 3 months, patch closure of the ventricular septal defect, direct closure of the atrial septal defect, and pulmonary artery debanding were performed. However, he died at 6 months of age due to severe heart failure.
CALM2 p.E141K The patient was a male newborn who was diagnosed with fetal hydrops and 2 : 1 AVB at 26 weeks gestation with markedly prolonged QTcs (atrial rate 110 beats/min, ventricular rate 55 beats/min, QTc 788 ms; Proband #8; Table). Echocardiography revealed that his LVEF was markedly decreased (<20%) without any anomaly of the cardiac anatomy. He was born preterm and with a low birth weight (1,800 g at 34 weeks gestation). An ECG at birth showed a profound bradycardia for age (Figure 4B, Left) and subsequently 2:1 AVB with QT prolongation (atrial rate 124 beats/min, ventricular rate 62 beats/min, and QTc 744 ms; Figure 4B, Right).
Although he received pacemaker implantation and multiple medical therapies for severe heart failure, he died at 1.5 years of age due to uncontrollable arrhythmia and refractory heart failure.
Phenotype With Neurological and Developmental Disorders (2 Probands)
Two CALM2-E46K carriers from unrelated families were first diagnosed with neurological and developmental disorders, patent ductus arteriosus (PDA), and CPVT.
One of these was a 5-year-old boy, who was born with a very low birth weight (1,098 g, 28 weeks gestation; Proband #9; Table). The patient underwent PDA closure surgery at 58 weeks of age. He experienced repetitive syncopal attacks while emotionally excited, as well as a sudden collapse while walking, with cyanosis, and was later rescued by AED. His ambulatory ECG showed a sinus bradycardia considering his age (65 beats/min; Figure 5A, Left) and sustained polymorphic and bidirectional VTs (Figure 5A, Right). Oral carvedilol (5 mg/day) and flecainide (100 mg/day) were prescribed, but the patient could not take medication by himself due to severe developmental disorders, including autism. His parents usually took care of him, but sometimes he failed to take the drugs as prescribed. All subsequent VT/VF events occurred in the absence of drugs.

The last patient was a 9-year-old boy (Proband #10; Table). This boy was born small for gestational age (2,444 g) with trivial PDA. He also had autism and epilepsy, and experienced syncope while running. Ambulatory ECG showed sinus bradycardia for his age (35–50 beats/min) and polymorphic VT (Figure 5B). QTc was not prolonged (440 ms). The patient received nadolol (1 mg/kg/day) at the diagnosis, and flecainide (100 mg/day) was added for frequent premature ventricular contractions from at age of 15 years.
Discussion
The present study reports the frequency and clinical characteristic of patients with variants in the calmodulin gene in our cohort of 195 symptomatic IPAS children whose genetic background is unclear. With gene panel sequencing, we identified 10 probands with 7 different CALM1 or CALM2 variants. This frequency of CALM variants (5%) was not too low and cannot be overlooked. In particular, in our IPAS child cohort there were 26 children who experienced LAEs before the age of 6 years. Thus, 35% of them were found to carry CALM variants (Table), indicating that calmodulinopathy is not a rare disease among LAE patients aged <6 years.
Interestingly, even though being located at the same residue N98, the variant N98S produced distinct phenotypes: CPVT by CALM1 and LQTS by CALM2. Carriers of p.N98S variant (Probands #1–#4; Table) experienced documented LAEs. The same variants have been reported previously.7,26,27CALM1 p.N98S attenuates calcium-binding affinity and exhibits an aberrant interaction with the RYR2 calmodulin-binding-domain peptide,7 thus producing a phenotype showing CPVT. Conversely, CALM2 p.N98S was previously reported in a case of LQTS.25 Using human induced pluripotent stem cell cell-derived cardiomyocytes (iPSC-CMs), we demonstrated that the CALM2 p.N98S significantly impairs inactivation of the LCC current, and thereby significantly prolongs the action potential duration.27 The variant mainly affected the CDI through the Ca2+-calmodulin pathway, and this contrasts with the notion that Timothy syndrome-related calcium voltage-gated channel subunit alpha1 C (CACNA1C) mutations impaired voltage-dependent inactivation of LCC, thereby prolonging action potential duration and the QT interval.28–30
CALM2 p.D96V (Proband #7) and CALM2 p.E141K (Proband #8) caused very critical cardiac phenotypes: not only arrhythmic events, but also severe cardiac dysfunction, including congenital anomalies. In addition, the phenotypes of these children resembled those of classical Timothy syndrome, which is caused by CACNA1C p.G406R/G402S.31 The CALM2 p.D96V variant that was first reported by Crotti et al in 201312 was found to slow the CDI of cardiac LCC by reducing Ca2+
affinity for the C-domain of CaM.
Boczek et al32 and Wren et al33 previously reported CALM1 and CALM3 variants at the E141 locus. We identified a variant at the same codon, but in CALM2. The clinical features of our CALM2 p.E141K carrier were comparable to those of the CALM3 p.E141K carrier,33 and were more severe than those of the CALM1 p.E141G carrier.32CALM1 p.E141G has been shown to not only delay the CDI, but also increase late sodium currents.32 Conversely, the CALM3 p.E141K variant reduces the binding affinity of the C-domain for Ca2+
and delays the CDI process, which is similar to the Ca2+
channel variants reported in Timothy syndrome.33,34 Although the p.E141K variant was located in CALM2 in our study (Proband 8; Figure 1), the underlying mechanism appeared similar to that of CALM3 p.E141K.33 Thus, CALM2 p.D96V and CALM2 p.E141K variants lead to a dysfunction of calcium channels, as observed in LQT8.
Phenotypic differences between CALM2 p.D96G and p.D96V carriers (Probands #5 and #7; Table) were notable even though the amino acid location was same. The CALM2 p.D96V carrier (Proband #7) showed more severe cardiac phenotypes and died at 6 months of age. Conversely, the CALM2 p.D96G carrier (Proband #5) presented only syncope, which was suspected as an LAE. In addition, CALM2 p.D132G at the third amino acid in EF hand IV showed similar phenotypes to those of the patient bearing the CALM2 p.D96G variant. Although they are located in other EF hands, the position and substitution of amino acids were the same (Figure 1). In a previous report about CALM2 p.D132H and CALM1 p.D132V,35 the phenotypes and physiological changes of the CALM2 p.D132H carrier were very similar to those of our CALM2 p.D96V carrier. Functional analysis of CALM1 p.D132V showed a significantly delayed CDI. However, it was found to be milder than that caused by CALM2 p.D132H, including cardiac phenotypes.
CALM2 p.E46K is located in the linker between EF hand I and II and was associated with neurological and developmental disorders. Because this position is outside of CaM’s calcium-binding sites (EF hands), the cardiac phenotypes may differ to those in carriers with mutations in the EF hands. Recently, our group reported functional analysis using iPSC-CMs, finding that mutant cells of CALM2 p.E46K exhibited increased RyR2-binding affinity, and a facilitative effect on RyR2.36 Then, the phenotype in our 2 p.E46K carriers resembled that of CPVT. In addition, surprisingly, both of these 2 p.E46K carriers had PDA, but the mechanism responsible is unclear.
Our mutations seem to be de novo variants, except for the 1 patient whose parents rejected genetic testing, because all the family members of our probands were healthy. However, if there are many children with calmodulinopathy (but their parents without cardiac symptoms), we should be suspect the mosaicism, such as that reported previously regarding CALM3-D130G.37
Overall, the clinical features of calmodulinopathy vary considerably in a variant-specific manner (Figure 1). That is, the phenotypes of carriers of the same variant resembled each other considerably, such as in the 4 patients with CALM1 or 2 p.N98S and 2 patients with CALM2 p.E46K. There appeared to be 3 major determinants of severity in calmodulinopathy: (1) the location of the variant; (2) the amino acid substituted; and (3) the subset of calmodulin gene (CALM1, CALM2, and CALM3).
Beta-blocker therapy was reported to be effective in treating LAEs caused by calmodulinopathy and LQTS. Regarding our critical cases, β-blocker therapy was successful in suppressing LAEs; however, severe heart failure was complicated and uncontrollable. As an additional drug therapy, flecainide and mexiletine were useful in our patients. In 3 of our patients who showed CPVT phenotypes, flecainide suppressed their ventricular arrhythmias; in addition, the effect of flecainide was demonstrated in a functional analysis.35 Conversely, additional mexiletine was used for LQTS phenotypes. Thus, although β-blocker therapy can be used broadly for calmodulinopathy, additional drug therapy should be chosen in accordance with the phenotype. To the best of our knowledge, the onset of calmodulinopathy in most cases is in childhood. However, several patients had been prescribed anti-arrhythmic drugs under suspicion of LQTS/CPVT, without identification of pathogenic gene variants. As long as taking appropriate drug therapy, the outcome of calmodulinopathy in adults would be favorable.
As we recently reported,38 in Japan, a school-based ECG screening system is effective for identifying children who are at risk of LAEs. Currently, all children undergo an ECG recording before entering primary school (age 6 years). However, in order to detect calmodulinopathy, the age of ECG screening should be earlier than 6 years based upon our findings.
In conclusion, cardiac calmodulinopathy presents serious and potentially lethal phenotypes in very early stages of life and therefore requires diagnosis and treatment at the earliest age possible.
Acknowledgments
The authors are grateful to all the cardiologists and pediatricians in Japan for clinical information.
Sources of Funding
This work was supported, in part, by MEXT KAKENHI (Grant no. 19K08555 and 22K08179 [to M.F.] and 18K07875 [to S.O.]) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, as well as a grant from the Ministry of Health, Labor and Welfare of Japan for Clinical Research on Intractable Disease (H29-055 [to M.H., S.O.]).
Disclosures
The authors declare that there is no conflict of interest in this work.
IRB Information
This study was approved by the Institutional Review Board of Shiga University of Medical Science (Reference no. G2011-128). The study complied with the Declaration of Helsinki and written informed consent was obtained from all patients or their guardians.
Data Availability
The data underlying this article will be shared upon reasonable request to the corresponding author.
Supplementary Files
Please find supplementary file(s);
https://doi.org/10.1253/circj.CJ-23-0195
References
- 1.
Wawrzynczak EJ, Perham RN. Isolation and nucleotide sequence of a cDNA encoding human calmodulin. Biochem Int 1984; 9: 177–185.
- 2.
Fischer R, Koller M, Flura M, Mathews S, Strehler-Page MA, Krebs J, et al. Multiple divergent mRNAs code for a single human calmodulin. J Biol Chem 1988; 263: 17055–17062.
- 3.
Budde T, Meuth S, Pape HC. Calcium-dependent inactivation of neuronal calcium channels. Nat Rev Neurosci 2002; 3: 873–883, doi:10.1038/nrn959.
- 4.
Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science 2004; 304: 432–435, doi:10.1126/science.1093490.
- 5.
Sorensen AB, Søndergaard MT, Overgaard MT. Calmodulin in a heartbeat. FEBS J 2013; 280: 5511–5532, doi:10.1111/febs.12337.
- 6.
Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 1999; 22: 549–558, doi:10.1016/s0896-6273(00)80709-6.
- 7.
Nyegaard M, Overgaard MT, Søndergaard MT, Vranas M, Behr ER, Hildebrandt LL, et al. Mutations in calmodulin cause ventricular tachycardia and sudden cardiac death. Am J Hum Genet 2012; 91: 703–712, doi:10.1016/j.ajhg.2012.08.015.
- 8.
Crotti L, Spazzolini C, Tester DJ, Ghidoni A, Baruteau AE, Beckmann BM, et al. Calmodulin mutations and life-threatening cardiac arrhythmias: Insights from the International Calmodulinopathy Registry. Eur Heart J 2019; 40: 2964–2975, doi:10.1093/eurheartj/ehz311.
- 9.
Moss AJ, Schwartz PJ, Crampton RS, Tzivoni D, Locati EH, MacCluer J, et al. The long QT syndrome: Prospective longitudinal study of 328 families. Circulation 1991; 84: 1136–1144.
- 10.
Schwartz PJ, Stramba-Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, et al. Prevalence of the congenital long-QT syndrome. Circulation 2009; 120: 1761–1767, doi:10.1161/CIRCULATIONAHA.109.863209.
- 11.
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm 2013; 10: e75–e106, doi:10.1016/j.hrthm.2013.05.014.
- 12.
Crotti L, Johnson CN, Graf E, De Ferrari GM, Cuneo BF, Ovadia M, et al. Calmodulin mutations associated with recurrent cardiac arrest in infants. Circulation 2013; 127: 1009–1017, doi:10.1161/CIRCULATIONAHA.112.001216.
- 13.
Aizawa Y, Komura S, Okada S, Chinushi M, Morita H, Ohe T. Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT). Int Heart J 2006; 47: 381–389, doi:10.1536/ihj.47.381.
- 14.
Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, et al. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation 2001; 103: 196–200, doi:10.1161/01.cir.103.2.196.
- 15.
Lahat H, Pras E, Olender T, Avidan N, Ben-Asher E, Man O, et al. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am J Hum Genet 2001; 69: 1378–1384, doi:10.1086/324565.
- 16.
Devalla HD, Gélinas R, Aburawi EH, Beqqali A, Goyette P, Freund C, et al. TECRL, a new life-threatening inherited arrhythmia gene associated with overlapping clinical features of both LQTS and CPVT. EMBO Mol Med 2016; 8: 1390–1408, doi:10.15252/emmm.201505719.
- 17.
Walsh R, Adler A, Amin AS, Abiusi E, Care M, Bikker H, et al. Evaluation of gene validity for CPVT and short QT syndrome in sudden arrhythmic death. Eur Heart J 2022; 43: 1500–1510, doi:10.1093/eurheartj/ehab687.
- 18.
Schwartz PJ. Idiopathic long QT syndrome: Progress and questions. Am Heart J 1985; 109: 399–411, doi:0002-8703(85)90626-X.
- 19.
Napolitano C, Priori SG. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm 2007; 4: 675–678, doi:10.1016/j.hrthm.2006.12.048.
- 20.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015; 17: 405–424, doi:10.1038/gim.2015.30.
- 21.
Fujita S, Nakagawa R, Futatani T, Igarashi N, Fuchigami T, Saito S, et al. Long QT syndrome with a de novo CALM2 mutation in a 4-year-old boy. Pediatr Int 2019; 61: 852–858, doi:10.1111/ped.13959.
- 22.
Malfatto G, Beria G, Sala S, Bonazzi O, Schwartz PJ. Quantitative analysis of T wave abnormalities and their prognostic implications in the idiopathic long QT syndrome. J Am Coll Cardiol 1994; 23: 296–301.
- 23.
Moss AJ, Zareba W, Benhorin J, Locati EH, Hall WJ, Robinson JL, et al. ECG T-wave patterns in genetically distinct forms of the hereditary long QT syndrome. Circulation 1995; 92: 2929–2934.
- 24.
Harada M, Suzuki H, Ohno S, Ozawa J, Saitoh A, Horie M. Dynamic QT changes in long QT syndrome type 8. Circ J 2019; 83: 1614, doi:10.1253/circj.CJ-18-0984.
- 25.
Fukuyama M, Ohno S, Ozawa J, Kato K, Makiyama T, Nakagawa Y, et al. High prevalence of late-appearing T-wave in patients with long QT syndrome type 8. Circ J 2020; 84: 559–568, doi:10.1253/circj.CJ-19-1101.
- 26.
Makita N, Yagihara N, Crotti L, Johnson CN, Beckmann BM, Roh MS, et al. Novel calmodulin mutations associated with congenital arrhythmia susceptibility. Circ Cardiovasc Genet 2014; 7: 466–474, doi:10.1161/CIRCGENETICS.113.000459.
- 27.
Yamamoto Y, Makiyama T, Harita T, Sasaki K, Wuriyanghai Y, Hayano M, et al. Allele-specific ablation rescues electrophysiological abnormalities in a human iPS cell model of long-QT syndrome with a CALM2 mutation. Hum Mol Genet 2017; 26: 1670–1677, doi:10.1093/hmg/ddx073.
- 28.
Boczek NJ, Miller EM, Ye D, Nesterenko VV, Tester DJ, Antzelevitch C, et al. Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 2015; 12: 211–219, doi:10.1016/j.hrthm.2014.09.051.
- 29.
Fukuyama M, Wang Q, Kato K, Ohno S, Ding WG, Toyoda F, et al. Long QT syndrome type 8: Novel CACNA1C mutations causing QT prolongation and variant phenotypes. Europace 2014; 16: 1828–1837, doi:10.1093/europace/euu063.
- 30.
Ozawa J, Ohno S, Melgari D, Wang Q, Fukuyama M, Toyoda F, et al. Increased CaV1.2 late current by a CACNA1C p.R412M variant causes an atypical Timothy syndrome without syndactyly. Sci Rep 2022; 12: 18984, doi:10.1038/s41598-022-23512-2.
- 31.
Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, et al. Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci USA 2005; 102: 8089–8096, doi:10.1073/pnas.0502506102.
- 32.
Boczek NJ, Gomez-Hurtado N, Ye D, Calvert ML, Tester DJ, Kryshtal D, et al. Spectrum and prevalence of CALM1-, CALM2-, and CALM3-encoded calmodulin variants in long QT syndrome and functional characterization of a novel long QT syndrome-associated calmodulin missense variant, E141G. Circ Cardiovasc Genet 2016; 9: 136–146, doi:10.1161/CIRCGENETICS.115.001323.
- 33.
Wren LM, Jiménez-Jáimez J, Al-Ghamdi S, Al-Aama JY, Bdeir A, Al-Hassnan ZN, et al. Genetic mosaicism in calmodulinopathy. Circ Genom Precis Med 2019; 12: 375–385, doi:10.1161/CIRCGEN.119.002581.
- 34.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 2004; 119: 19–31, doi:10.1016/j.cell.2004.09.011.
- 35.
Pipilas DC, Johnson CN, Webster G, Schlaepfer J, Fellmann F, Sekarski N, et al. Novel calmodulin mutations associated with congenital long QT syndrome affect calcium current in human cardiomyocytes. Heart Rhythm 2016; 13: 2012–2019, doi:10.1016/j.hrthm.2016.06.038.
- 36.
Gao J, Makiyama T, Yamamoto Y, Kobayashi T, Aoki H, Maurissen TL, et al. Novel calmodulin variant p.E46K associated with severe catecholaminergic polymorphic ventricular tachycardia produces robust arrhythmogenicity in human induced pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol 2023; 16: e011387, doi:10.1161/CIRCEP.122.011387.
- 37.
Bhuiyan ZA, Bdier A, Al-Aama JY, Abramova T, George AL. Discordance between germline and blood mosaicism in calmodulinopathy. Circ Genom Precis Med 2022; 15: e003695, doi:10.1161/CIRCGEN.121.003695.
- 38.
Fukuyama M, Horie M, Aoki H, Ozawa J, Kato K, Sawayama Y, et al. School-based routine screenings of electrocardiograms for the diagnosis of long QT syndrome. Europace 2022; 24: 1496–1503, doi:10.1093/europace/euab320.