Abstract
Epidemiological evidence of increased risks of cancer in heart failure (HF) patients and HF in cancer patients has suggested close relationships between the pathogenesis of both diseases. Indeed, HF and cancer share common risk factors, including aging and unhealthy lifestyles, and underlying mechanisms, including activation of the sympathetic nervous system and renin-angiotensin-aldosterone system, chronic inflammation, and clonal hematopoiesis of indeterminate potential. Mechanistically, HF accelerates cancer development and progression via secreted factors, so-called cardiokines, and epigenetic remodeling of bone marrow cells into an immunosuppressive phenotype. Reciprocally, cancer promotes HF via cachexia-related wasting and metabolic remodeling in the heart, and possibly via cancer-derived extracellular vesicles influencing myocardial structure and function. The novel concept of the “heart-cancer axis” will help in our understanding of the shared and reciprocal relationships between HF and cancer, and provide innovative diagnostic and therapeutic approaches for both diseases.
Recent progress and advances in cancer therapy have markedly improved the outcomes of cancer treatment.1 However, an increasing number of cancer drugs have significant and deleterious effects on cardiovascular (CV) homeostasis. CV toxicity of cancer chemotherapy can be morbid, and even fatal, and adversely force discontinuation of optimal cancer therapy. Therefore, cancer therapy-related CV adverse events have greater impact on the prognosis and quality of life in cancer patients and survivors than imagined.2 Although CV diseases and cancer have been regarded as disparate disease entities, the management of the cancer patients with CV comorbidities and complications require close communication between cardiologists and oncologists, and a multilayered framework of cardio-oncology/onco-cardiology services involving healthcare services, hospitals, medical institutions, and academic societies.3
Furthermore, recent studies have unveiled that CV health is significantly influenced not only by cancer therapy, but also by cancer per se.4–6 There is also accumulating evidence that failing hearts promote cancer progression.4–6 Therefore, direct and reciprocal interactions are indicated to exist between the heart and cancer.4–6 In addition, heart failure (HF) and cancer share common risk factors and pathophysiological mechanisms, including inflammation, metabolic remodeling, and neurohumoral activation.4–6 In this review, we present an emerging concept of the “heart-cancer axis” (Figure 1) and discuss shared risk factors, shared mechanisms, and reciprocal interactions between HF and cancer.
Epidemiological Evidence for a Relationship Between HF and Cancer
Recent epidemiological studies have indicated increased risks of cancer in HF patients and HF in cancer patients.4–6 In a community-based cohort study enrolling 596 pairs of HF patients and controls in Olmsted County in the US, HF patients had a higher risk of developing cancer (hazard ratio [HR] 1.68; 95% confidence interval [CI] 1.13–2.50) compared with controls.7 In another prospective cohort study conducted in Olmsted County, patients who developed HF after myocardial infarction (MI) had a higher risk of subsequent cancer (HR 2.16; 95% CI 1.39–3.35) than those without HF after MI.8 A large Danish cohort study of a total of 9,307 HF patients showed an increased risk of cancer with an incidence rate ratio (IRR) of 1.24 (95% CI 1.15–1.33) compared with the background population.9 The risk was increased for all major types of cancer except prostate cancer.9 A retrospective single-center cohort study enrolling 5,238 hospitalized patients with HF in Japan also demonstrated a higher incidence of cancer in HF patients than in controls (0.99% vs. 0.59%; P<0.001).10 Although conflicting results were reported in a cohort study enrolling male physicians,11 several nationwide or regional cohort studies using an administrative database12–16 and a meta-analysis17 support an increased risk of the incidence of cancer in HF patients.
Comorbid cancer is present in 15–22% of patients with HF with preserved ejection fraction (HFpEF)18–20 and in 13–19% of patients with HF with reduced ejection fraction (HFrEF).18,20 Cancer is a leading cause of non-CV death in HF patients.4,21 Although the prevalence of cancer as a comorbidity in randomized controlled trials (RCTs) is extremely low because active or recent cancer is included in the exclusion criteria,22 cancer has been the most frequently reported cause of non-CV death in RCTs enrolling patients with HFpEF23–26 or HFrEF.26–28 These studies suggest a significant impact of HF on the incidence and outcome of cancer.
There is a possibility that the association between HF and cancer is reciprocal. An increasing number of studies indicate that cancer history increases a risk of subsequent incidence of HF. A community-based retrospective cohort study of 36,236 adult-onset cancer patients surviving at least 2 years after diagnosis revealed a significantly increased risk of HF among survivors of breast cancer (IRR 1.35; 95% CI 1.19–1.54), lung and bronchus cancer (IRR 1.95; 95% CI 1.35–2.80), chronic lymphocytic leukemia (IRR 1.75; 95% CI 1.10–2.80), multiple myeloma (IRR 2.52; 95% CI 1.65–3.84), and non-Hodgkin lymphoma (IRR 2.01; 95% CI 1.51–2.67).29 In a population-based cohort study of 108,215 survivors of the 20 most common adult cancers who were alive 12 months after diagnosis, using multiple linked UK electronic health records databases, an increase in the risk of HF was also observed in patients with 10 cancers, including non-Hodgkin lymphoma (HR 1.94; 95% CI 1.66–2.25), leukemia (HR 1.77; 95% CI 1.50–2.09), multiple myeloma (HR 3.29; 95% CI 2.59–4.18), esophageal cancer (HR 1.96; 95% CI 1.46–2.64), lung cancer (HR 1.82; 95% CI 1.52–2.17), kidney cancer (HR 1.73; 95% CI 1.38–2.17), and ovarian cancer (HR 1.59; 95% CI 1.19–2.12).30 In the Prospective comparison of ARNI with ACEI to Determine Impact on Global Mortality and morbidity in Heart Failure (PARADIGM-HF) and Aliskiren Trial to Minimise Outcomes in Patients with Heart Failure (ATMOSPHERE) trials enrolling HFrEF patients and the Prospective comparison of ARNI with ARB Global Outcomes in Heart Failure with Preserved Ejection Fraction (PARAGON-HF) and Effects of Candesartan in Patients with Chronic Heart Failure and Preserved Ejection Fraction (CHARM-PRESERVED) trials enrolling HFpEF patients, incident cancer was diagnosed during the trials in 4.3% and 8.5% of HFrEF and HFpEF patients, respectively.31 Although the outcomes in HFpEF trials were similar with or without cancer history, HFrEF patients with cancer history had higher risks of HF hospitalization (HR 1.28; 95% CI 1.07–1.52; P<0.005) and non-CV death (HR 1.57; 95% CI 1.16–2.12; P<0.01) than those without.31 Indeed, the increased risk of HF after cancer diagnosis and treatment can be caused by the combined effects of cancer per se and cancer therapy-related cardiac toxicity. However, a recent study suggested that cardiac structure and function may be influenced by cancer per se prior to the initiation of chemotherapy.32 Interestingly, CV magnetic resonance-based evaluation of 381 patients with chemotherapy-naïve breast cancer or lymphoma and 102 healthy controls revealed significantly smaller chamber volumes and elevated strain and native myocardial T1 mapping in chemotherapy-naïve cancer patients, although left ventricular (LV) ejection fraction was similar.32
Shared Risk Factors Between HF and Cancer
The association between the development of HF and cancer appears to result, in part, from the risk factors common to CV diseases and cancer, including aging and unhealthy lifestyles, such as smoking, unhealthy dietary habits (e.g., more foods high in energy, sugar, fat, and salt and low in dietary fiber), sedentary behavior, high alcohol consumption, and environmental stress33,34 (Figure 1). In the prospective cohort studies originally aiming to investigate CV diseases, there was a consistent and significant inverse association between the number of the “Life’s Simple 7” CV health metrics (smoking, body mass index, physical activity, dietary habits, blood pressure, fasting glucose level, and total cholesterol level) defined by the American Heart Association35 and the risk of future cancer.36–39 An improvement in the CV health metrics over time and higher CV health metrics at baseline were related to a lower risk of cancer incidence,40 supporting the effectiveness of CV health promotion as a preventive strategy against future risk of both HF and cancer.
Shared Mechanisms Between HF and Cancer
HF and cancer share common pathways underlying the development and progression of diseases that include the sympathetic nervous system (SNS), renin-angiotensin-aldosterone system (RAAS), chronic inflammation, and clonal hematopoiesis of indeterminate potential (CHIP; Figure 1).5,6,33
SNS
Activation of the SNS increases circulating and local concentrations of catecholamines to maintain virtually all organ homeostasis against a wide variety of stress burdens, but excessive and prolonged SNS activation can adversely affect not only HF,41 but also cancer progression and metastasis.42 Epidemiological studies have indicated that exposure to β-adrenergic receptor blockers is associated with improved survival of patients with breast, prostate, lung, ovarian cancers, and malignant melanoma.42 Experimental animal studies have also demonstrated that chronic behavioral stress can promote the progression and metastasis of several cancers (breast, prostate, ovary, pancreas, neuroblastoma, malignant melanoma, and hematological malignancies), which can be attenuated by treatment with β-adrenergic receptor blockers.42 Mechanistically, SNS activation alters gene expression and cellular function in the tumor microenvironment, and thus promotes cancer progression and metastasis via a wide variety of processes, including inhibition of DNA damage repair and programmed cell death, oncogene activation, epithelial-mesenchymal transition, stimulation of inflammation and angiogenesis, and dysregulation of immune responses.42
RAAS
Activation of the RAAS plays a homeostatic role in the systemic circulation by regulating blood pressure and electrolyte and water balance, but it also promotes LV remodeling and HF,41,43 as well as cancer progression and metastasis.44,45 Genetic and epidemiological studies have provided evidence that some polymorphisms in RAAS genes are associated with a risk for developing breast cancer.44 Indeed, the RAAS components are expressed in tissues or cell lines of several cancers (breast, prostate, ovary, pancreas, and stomach),44 and cancer biology can be influenced by activation of the RAAS components. For example, it was reported that the angiotensin II receptor type 1 (AGTR1) gene was overexpressed in 10–20% of breast cancer patients, and that the angiotensin AT1
receptor blocker losartan attenuated tumor growth of AGTR1-overexpressing breast cancer cells implanted in the mammary fat pad of nude mice.46 RAAS activation can enhance the proliferation and survival of cancer cells, and further remodels the tumor microenvironment via modulation of inflammation, angiogenesis, and immune cell modulation.44,45,47
Chronic Inflammation
Chronic inflammation increases the risk of both HF and cancer.5,6,48 Sustained myocardial injury and overload triggers chronic inflammation in the heart, and some proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, and IL-18, have detrimental effects on LV structure and function, promoting adverse LV remodeling.49 In the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) trial, canakinumab (a monoclonal antibody against IL-1β) significantly reduced HF-related hospitalization and mortality in patients with prior MI and high-sensitivity C-reactive protein ≥2 mg/L,50 indicating a causal impact of inflammation on the pathogenesis of HF. Conversely, cancer cells are surrounded by stromal cells and inflammatory cells, forming a tumor microenvironment in which well-orchestrated reciprocal interactions promote tumorigenesis and malignant progression.51 Interestingly, in the CANTOS study, canakinumab treatment was associated with a significant reduction in lung cancer incidence and mortality,52 suggesting a pathogenic role of IL-1β in lung cancer.
Clonal Hematopoiesis of Indeterminate Potential
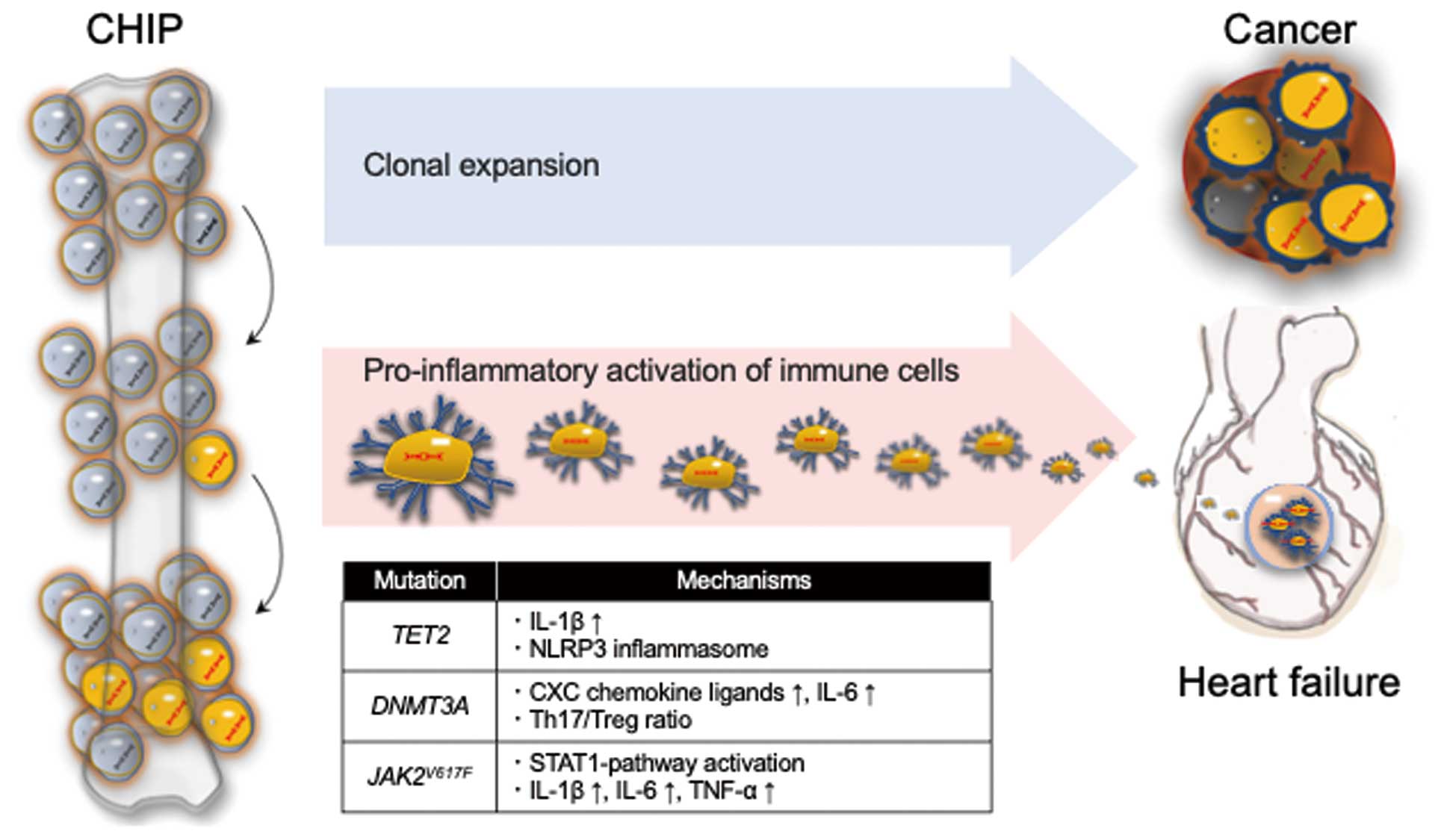
Hematopoietic stem cells (HSCs) undergo clonal expansion with age due to the acquisition of somatic mutations. Recent genetic analyses of large populations revealed that somatic mutations of the genes promoting clonal expansion of HSCs, such as DNA methyltransferase 3 α (DNMT3A), tet methylcytosine dioxygenase 2 (TET2), Janus kinase 2 (JAK2), ASXL transcriptional regulator 1 (ASXL1), tumor protein p53 (TP53), protein phosphatase, Mg2+/Mn2+
dependent 1D (PPM1D), and splicing factor 3b subunit 1 (SF3B1), were prevalent in 5–10% of individuals 70 years of age or older.53–55 CHIP is defined by the presence of somatic variants in genes implicated in hematological malignancies with a variant allele frequency of at least 2%, but without being affected by hematological malignancies or other clonal diseases.56 A previous study showed that the presence of CHIP was associated with an increased risk of all-cause mortality (HR 1.4; 95% CI 1.1–1.8).55 It also increased the risk of hematopoietic malignancies (HR 11.1; 95% CI 3.9–32.6), but only a few percentage of individuals developed hematological malignancies, and death from hematological malignancies did not account for the increased mortality.55 Notably, an increased risk of coronary artery disease (HR 2.0; 95% CI 1.2–3.4) and ischemic stroke (HR 2.6; 95% CI 1.4–4.8) possibly contributed to the increased risk of all-cause mortality in this population.55 Thereafter, increasing evidence suggests a link between CHIP and a broader spectrum of age-related CV diseases,56–58 as well as atherosclerotic CV disease.59,60
Evidence is accumulating that CHIP is associated with CV mortality in HFrEF and involved in the development of HFpEF.61,62 Mechanistically, HSCs harboring these somatic mutations give rise to proinflammatory immune cells that exacerbate the HF phenotypes (Figure 2). For example, competitive bone marrow transplantation with Tet2-deficient cells or myeloid cell-specific Tet2 gene disruption worsened LV remodeling after transverse aortic constriction (TAC) or ligation of left anterior descending artery in mice through macrophage proinflammatory activation involving the IL-1β/NLR family pyrin domain containing 3 (NLRP3) inflammasome.63 Similarly, clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9-mediated Dnmt3a gene disruption in HSCs exacerbated LV remodeling after angiotensin II infusion in mice, and Dnmt3a-disrupted macrophages showed enhanced proinflammatory activity with upregulation of CXC chemokine ligands and IL-6.64 A recent study of patients with aortic valve stenosis undergoing transcatheter aortic valve implantation revealed that those harboring somatic mutations in the DNMT3A or TET2 gene had an increased all-cause mortality after the successful implantation, and DNMT3A-mutation carriers showed a significant increase in the T helper 17 (Th17) cells/regulatory T (Treg) cells ratio, indicating a proinflammatory T-cell polarization.65 Myeloid-restricted JAK2V617F
mutation induced signal transducer and activator of transcription 1 (STAT1) activation, upregulation of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α in myeloid cells, and promoted LV remodeling after TAC or MI in mice.66
HF Accelerates Cancer Development
Recent experimental studies using animal models have suggested a causal role of HF in cancer development and progression, supporting the epidemiological evidence for the impact of HF on the incidence and outcome of cancer. For example, post-MI HF increased the number and size of intestinal tumors in Apcmin
mice, which are prone to develop intestinal adenomas67 (where min [multiple intestinal neoplasia] is a mutant allele of the adenomatous polyposis coli [Apc] locus). Heterotopic transplantation of failing hearts after MI into Apcmin
mice with a normal native heart did not impair normal systemic circulation, but increased intestinal tumor growth, suggesting that tumor growth was enhanced independently of hemodynamic impairment but by cardiac-excreted factors.67 Indeed, serpin 3A, among several proteins elevated in the blood of HF patients, was capable of promoting the proliferation of cultured human colon cancer cells.67 Early LV remodeling after TAC also enhanced the tumor growth of implanted breast cancer cells and tumor metastasis of cancer cells directly injected into the tail vein.68 In that experiment, periostin expression was elevated in both the heart and serum of TAC-operated mice, and the increase in the proliferation of cultured breast cancer cells induced by the serum of TAC-operated mice was abrogated by depletion of periostin from the serum, suggesting periostin as a putative HF-associated factor promoting tumor growth.68 Similarly, LV hypertrophy induced by transgenic expression of activating transcription factor 3 (ATF3) promoted tumor growth and metastasis of implanted breast cancer cells in mice, possibly via multiple secreted factors.69 These animal experiments provide mechanistic insights into the role of HF-originated secreted factors, so-called cardiokines, in the accelerated cancer development in HF patients (Figure 1). In addition, HF drives cancer progression via a cancer-promoting landscape of immune cells. MI by left anterior descending artery ligation accelerated the tumor growth of implanted breast cancer cells in mice, which was dependent on increased supply and recruitment of Ly6Chigh
monocytes to tumors.70 Mechanistically, MI induced Ly6Chigh
monocytes in the bone marrow reservoir to an immunosuppressive phenotype via sustained epigenetic reprogramming, leading to accelerated tumor growth70 (Figure 1).
Cancer Promotes HF
Cachexia occurs in 50–80% of patients with advanced cancer, and accounts for 20% of cancer-related deaths.71 Cancer-associated cachexia is characterized by weight loss, anorexia, and systemic inflammation, which lead to wasting and functional disturbance in multiple organs, including the heart.71 In individuals who died of cancer, it was reported that heart weight was significantly lower in those with than without cachexia.72,73 Cardiac wasting with LV wall thinning elevates ventricular wall stress, according to Laplace’s law, and possibly increases the susceptibility to HF under hemodynamic overload74 (Figure 1). An experimental study revealed that intraperitoneal injection of AH-130 hepatoma cells in rats resulted in a progressive decline in body weight and heart weight, which was associated with a significant deterioration in LV function.75 Notably, bisoprolol and spironolactone, but not imidapril, significantly attenuated cardiac wasting and dysfunction in this hepatoma cancer cachexia model.75 In another study using mice with subcutaneous implantation of c26 colon adenocarcinoma cells, losartan prevented tumor-related wasting and LV dysfunction, as well as tumor progression.76
Cancer cells can promote metabolic remodeling in the heart77 (Figure 1). Mutations in genes encoding isocitrate dehydrogenase 1 and 2 (IDH1/2) result in accumulation of D2-hydroxyglutaric acid (D2-HG), contributing to various cancers, such as acute myeloid leukemia, cholangiocarcinoma, chondrosarcoma, and glioma.78 Induction of a germline IDH2 mutation resulted in a dilated cardiomyopathy-like phenotype in mice, with ultrastructural changes such as cardiomyocyte apoptosis, mitochondrial damage, and glycogen accumulation.79 Subcutaneous implantation of U87 glioma cells bearing an IDH2 mutation in nude mice increased serum D2-HG concentrations, and induced cardiac hypertrophy accompanied by an increase in cardiomyocyte apoptosis.79 In the isolated rat heart, D2-HG induced LV dysfunction through inhibition of α-ketoglutarate dehydrogenase activity, a key regulatory enzyme of cellular energy, which impairs oxidative phosphorylation and promotes compensatory epigenetic modifications.80 These results indicate a potential role of cancer-derived D2-HG in developing metabolic and functional abnormalities in the heart as a paraneoplastic condition.
Cancer cells secrete more extracellular vesicles (EVs) than non-cancerous cells. These cancer-derived EVs carry regulatory molecules such as oncoproteins, oncopeptides, microRNAs (miRNAs), mRNAs, and long non-coding RNAs, and mediate intercellular communications between cancer cells and neighboring cells or distant cells.81 EVs are implicated not only in cancer development and progression, but also in pathological process of various diseases, including HF.81 In particular, recent studies have suggested the involvement of a number of miRNAs in myocardial hypertrophy, inflammation, and cell death, and circulating miRNAs are emerging as a useful biomarker for the diagnosis and prognostic prediction of HF.82 It is quite conceivable that miRNAs in cancer-derived EVs can influence the cardiac phenotype (Figure 1). However, future studies will be required to identify the specific miRNAs contained in cancer-derived EVs that bridge the gap in how cancer promotes HF.
Conclusions
Recent epidemiological, clinical, and mechanistic evidence has supported close and reciprocal relationships between HF and cancer. HF and cancer have several lifestyle risk factors in common. In Japan, cancer and CV disease have been the first and second leading causes of death,3 respectively, and thus, adherence to healthy lifestyle behaviors is of utmost importance to prevent the development of these 2 major killer diseases and to extend healthy life expectancy. Importantly, a better understanding of the shared and reciprocal mechanisms between HF and cancer will shed light on the unsolved issues in their complicated pathogenesis, and further advance the innovative diagnostic and therapeutic approach for both diseases.
Disclosures
I.K. and H.A. are members of Circulation Journal’s Editorial Team. H.A. has received remuneration for lectures from Daiichi Sankyo Co., Ltd., Bayer Yakuhin, Ltd., Novatis Pharma K. K., Pfizer Japan Inc., Bristol Myers Squibb Company, Ohtsuka Pharmaceutical Co., Ltd., and Viatris Inc. J.I. has received remuneration for lecture from AstraZeneka K.K. and scholarship funds from Pfizer Japan Inc. The remaining authors have no conflicts of interest to disclose.
References
- 1.
Islami F, Ward EM, Sung H, Cronin KA, Tangka FKL, Sherman RL, et al. Annual report to the nation on the status of cancer, part 1: National cancer statistics. J Natl Cancer Inst 2021; 113: 1648–1669.
- 2.
Kadowaki H, Akazawa H, Ishida J, Komuro I. Cancer therapeutics-related cardiac dysfunction: Insights from bench and bedside of onco-cardiology. Circ J 2020; 84: 1446–1453.
- 3.
Oka T, Akazawa H, Sase K, Hatake K, Komuro I. Cardio-oncology in Japan: The rapidly rising sun. JACC CardioOncol 2020; 2: 815–818.
- 4.
de Boer RA, Meijers WC, van der Meer P, van Veldhuisen DJ. Cancer and heart disease: Associations and relations. Eur J Heart Fail 2019; 21: 1515–1525.
- 5.
de Boer RA, Hulot JS, Tocchetti CG, Aboumsallem JP, Ameri P, Anker SD, et al. Common mechanistic pathways in cancer and heart failure. A scientific roadmap on behalf of the Translational Research Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail 2020; 22: 2272–2289.
- 6.
de Wit S, Glen C, de Boer RA, Lang NN. Mechanisms shared between cancer, heart failure, and targeted anti-cancer therapies. Cardiovasc Res 2023; 118: 3451–3466.
- 7.
Hasin T, Gerber Y, McNallan SM, Weston SA, Kushwaha SS, Nelson TJ, et al. Patients with heart failure have an increased risk of incident cancer. J Am Coll Cardiol 2013; 62: 881–886.
- 8.
Hasin T, Gerber Y, Weston SA, Jiang R, Killian JM, Manemann SM, et al. Heart failure after myocardial infarction is associated with increased risk of cancer. J Am Coll Cardiol 2016; 68: 265–271.
- 9.
Banke A, Schou M, Videbaek L, Moller JE, Torp-Pedersen C, Gustafsson F, et al. Incidence of cancer in patients with chronic heart failure: A long-term follow-up study. Eur J Heart Fail 2016; 18: 260–266.
- 10.
Sakamoto M, Hasegawa T, Asakura M, Kanzaki H, Takahama H, Amaki M, et al. Does the pathophysiology of heart failure prime the incidence of cancer? Hypertens Res 2017; 40: 831–836.
- 11.
Selvaraj S, Bhatt DL, Claggett B, Djousse L, Shah SJ, Chen J, et al. Lack of association between heart failure and incident cancer. J Am Coll Cardiol 2018; 71: 1501–1510.
- 12.
Schwartz B, Schou M, Gislason GH, Kober L, Torp-Pedersen C, Andersson C. Prevalence and incidence of various cancer subtypes in patients with heart failure vs matched controls. Int J Cardiol 2020; 316: 209–213.
- 13.
Kwak S, Kwon S, Lee SY, Yang S, Lee HJ, Lee H, et al. Differential risk of incident cancer in patients with heart failure: A nationwide population-based cohort study. J Cardiol 2021; 77: 231–238.
- 14.
Roderburg C, Loosen SH, Jahn JK, Gansbacher J, Luedde T, Kostev K, et al. Heart failure is associated with an increased incidence of cancer diagnoses. ESC Heart Fail 2021; 8: 3628–3633.
- 15.
Bertero E, Robusto F, Rulli E, D’Ettorre A, Bisceglia L, Staszewsky L, et al. Cancer incidence and mortality according to pre-existing heart failure in a community-based cohort. JACC CardioOncol 2022; 4: 98–109.
- 16.
Paterson DI, Wiebe N, Cheung WY, Mackey JR, Pituskin E, Reiman A, et al. Incident cardiovascular disease among adults with cancer: A population-based cohort study. JACC CardioOncol 2022; 4: 85–94.
- 17.
Camilli M, Chiabrando JG, Lombardi M, Del Buono MG, Montone RA, Lombardo A, et al. Cancer incidence and mortality in patients diagnosed with heart failure: Results from an updated systematic review and meta-analysis. Cardiooncology 2023; 9: 8.
- 18.
Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA, et al. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol 2012; 59: 998–1005.
- 19.
Lund LH, Donal E, Oger E, Hage C, Persson H, Haugen-Lofman I, et al. Association between cardiovascular vs. non-cardiovascular co-morbidities and outcomes in heart failure with preserved ejection fraction. Eur J Heart Fail 2014; 16: 992–1001.
- 20.
Kitai T, Miyakoshi C, Morimoto T, Yaku H, Murai R, Kaji S, et al. Mode of death among Japanese adults with heart failure with preserved, midrange, and reduced ejection fraction. JAMA Netw Open 2020; 3: e204296.
- 21.
Hamaguchi S, Kinugawa S, Sobirin MA, Goto D, Tsuchihashi-Makaya M, Yamada S, et al. Mode of death in patients with heart failure and reduced vs. preserved ejection fraction: Report from the registry of hospitalized heart failure patients. Circ J 2012; 76: 1662–1669.
- 22.
Khan MS, Samman Tahhan A, Vaduganathan M, Greene SJ, Alrohaibani A, Anker SD, et al. Trends in prevalence of comorbidities in heart failure clinical trials. Eur J Heart Fail 2020; 22: 1032–1042.
- 23.
Zile MR, Gaasch WH, Anand IS, Haass M, Little WC, Miller AB, et al. Mode of death in patients with heart failure and a preserved ejection fraction: Results from the Irbesartan in Heart Failure With Preserved Ejection Fraction Study (I-Preserve) trial. Circulation 2010; 121: 1393–1405.
- 24.
Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 2014; 370: 1383–1392.
- 25.
Vaduganathan M, Patel RB, Michel A, Shah SJ, Senni M, Gheorghiade M, et al. Mode of death in heart failure with preserved ejection fraction. J Am Coll Cardiol 2017; 69: 556–569.
- 26.
Vergaro G, Ghionzoli N, Innocenti L, Taddei C, Giannoni A, Valleggi A, et al. Noncardiac versus cardiac mortality in heart failure with preserved, midrange, and reduced ejection fraction. J Am Heart Assoc 2019; 8: e013441.
- 27.
McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 2014; 371: 993–1004.
- 28.
Tini G, Bertero E, Signori A, Sormani MP, Maack C, De Boer RA, et al. Cancer mortality in trials of heart failure with reduced ejection fraction: A systematic review and meta-analysis. J Am Heart Assoc 2020; 9: e016309.
- 29.
Armenian SH, Xu L, Ky B, Sun C, Farol LT, Pal SK, et al. Cardiovascular disease among survivors of adult-onset cancer: A community-based retrospective cohort study. J Clin Oncol 2016; 34: 1122–1130.
- 30.
Strongman H, Gadd S, Matthews A, Mansfield KE, Stanway S, Lyon AR, et al. Medium and long-term risks of specific cardiovascular diseases in survivors of 20 adult cancers: A population-based cohort study using multiple linked UK electronic health records databases. Lancet 2019; 394: 1041–1054.
- 31.
Dobbin SJH, Shen L, Petrie MC, Packer M, Solomon SD, McMurray JJV, et al. Characteristics and outcomes of patients with a history of cancer recruited to heart failure trials. Eur J Heart Fail 2023; 25: 488–496.
- 32.
Labib D, Satriano A, Dykstra S, Hansen R, Mikami Y, Guzzardi DG, et al. Effect of active cancer on the cardiac phenotype: A cardiac magnetic resonance imaging-based study of myocardial tissue health and deformation in patients with chemotherapy-naive cancer. J Am Heart Assoc 2021; 10: e019811.
- 33.
Koene RJ, Prizment AE, Blaes A, Konety SH. Shared risk factors in cardiovascular disease and cancer. Circulation 2016; 133: 1104–1114.
- 34.
Meijers WC, de Boer RA. Common risk factors for heart failure and cancer. Cardiovasc Res 2019; 115: 844–853.
- 35.
Lloyd-Jones DM, Hong Y, Labarthe D, Mozaffarian D, Appel LJ, Van Horn L, et al. Defining and setting national goals for cardiovascular health promotion and disease reduction: The American Heart Association’s strategic Impact Goal through 2020 and beyond. Circulation 2010; 121: 586–613.
- 36.
Rasmussen-Torvik LJ, Shay CM, Abramson JG, Friedrich CA, Nettleton JA, Prizment AE, et al. Ideal cardiovascular health is inversely associated with incident cancer: The Atherosclerosis Risk In Communities study. Circulation 2013; 127: 1270–1275.
- 37.
Ogunmoroti O, Allen NB, Cushman M, Michos ED, Rundek T, Rana JS, et al. Association between Life’s Simple 7 and noncardiovascular disease: The Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc 2016; 5: e003954.
- 38.
Foraker RE, Abdel-Rasoul M, Kuller LH, Jackson RD, Van Horn L, Seguin RA, et al. Cardiovascular health and incident cardiovascular disease and cancer: The Women’s Health Initiative. Am J Prev Med 2016; 50: 236–240.
- 39.
Lau ES, Paniagua SM, Liu E, Jovani M, Li SX, Takvorian K, et al. Cardiovascular risk factors are associated with future cancer. JACC CardioOncol 2021; 3: 48–58.
- 40.
Van Sloten T, Valentin E, Climie RE, Deraz O, Weiderpass E, Jouven X, et al. Association of midlife cardiovascular health and subsequent change in cardiovascular health with incident cancer. JACC CardioOncol 2023; 5: 39–52.
- 41.
Mann DL, Felker GM. Mechanisms and models in heart failure: A translational approach. Circ Res 2021; 128: 1435–1450.
- 42.
Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer 2015; 15: 563–572.
- 43.
Kamo T, Akazawa H, Komuro I. Pleiotropic effects of angiotensin II receptor signaling in cardiovascular homeostasis and aging. Int Heart J 2015; 56: 249–254.
- 44.
George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: Old dog, new tricks. Nat Rev Cancer 2010; 10: 745–759.
- 45.
Pinter M, Jain RK. Targeting the renin-angiotensin system to improve cancer treatment: Implications for immunotherapy. Sci Transl Med 2017; 9: eaan5616.
- 46.
Rhodes DR, Ateeq B, Cao Q, Tomlins SA, Mehra R, Laxman B, et al. AGTR1 overexpression defines a subset of breast cancer and confers sensitivity to losartan, an AGTR1 antagonist. Proc Natl Acad Sci U S A 2009; 106: 10284–10289.
- 47.
Cortez-Retamozo V, Etzrodt M, Newton A, Ryan R, Pucci F, Sio SW, et al. Angiotensin II drives the production of tumor-promoting macrophages. Immunity 2013; 38: 296–308.
- 48.
Libby P, Kobold S. Inflammation: A common contributor to cancer, aging, and cardiovascular diseases: Expanding the concept of cardio-oncology. Cardiovasc Res 2019; 115: 824–829.
- 49.
Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol 2020; 17: 269–285.
- 50.
Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019; 139: 1289–1299.
- 51.
Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity 2019; 51: 27–41.
- 52.
Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017; 390: 1833–1842.
- 53.
Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med 2014; 20: 1472–1478.
- 54.
Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med 2014; 371: 2477–2487.
- 55.
Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 2014; 371: 2488–2498.
- 56.
Khetarpal SA, Qamar A, Bick AG, Fuster JJ, Kathiresan S, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential reshapes age-related CVD: JACC review topic of the week. J Am Coll Cardiol 2019; 74: 578–586.
- 57.
Sano S, Wang Y, Walsh K. Clonal hematopoiesis and its impact on cardiovascular disease. Circ J 2018; 83: 2–11.
- 58.
Tall AR, Fuster JJ. Clonal hematopoiesis in cardiovascular disease and therapeutic implications. Nat Cardiovasc Res 2022; 1: 116–124.
- 59.
Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med 2017; 377: 111–121.
- 60.
Zuriaga MA, Fuster JJ. Emerging role of acquired mutations and clonal hematopoiesis in atherosclerosis: Beyond conventional cardiovascular risk factors. Circ J 2023; 87: 394–400.
- 61.
Yura Y, Sano S, Walsh K. Clonal hematopoiesis: A new step linking inflammation to heart failure. JACC Basic Transl Sci 2020; 5: 196–207.
- 62.
Sikking MA, Stroeks S, Waring OJ, Henkens M, Riksen NP, Hoischen A, et al. Clonal hematopoiesis of indeterminate potential from a heart failure specialist’s point of view. J Am Heart Assoc 2023; 12: e030603.
- 63.
Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J Am Coll Cardiol 2018; 71: 875–886.
- 64.
Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res 2018; 123: 335–341.
- 65.
Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J 2020; 41: 933–939.
- 66.
Sano S, Wang Y, Yura Y, Sano M, Oshima K, Yang Y, et al. JAK2 (V617F)-mediated clonal hematopoiesis accelerates pathological remodeling in murine heart failure. JACC Basic Transl Sci 2019; 4: 684–697.
- 67.
Meijers WC, Maglione M, Bakker SJL, Oberhuber R, Kieneker LM, de Jong S, et al. Heart failure stimulates tumor growth by circulating factors. Circulation 2018; 138: 678–691.
- 68.
Avraham S, Abu-Sharki S, Shofti R, Haas T, Korin B, Kalfon R, et al. Early cardiac remodeling promotes tumor growth and metastasis. Circulation 2020; 142: 670–683.
- 69.
Awwad L, Aronheim A. Cardiac dysfunction promotes cancer progression via multiple secreted factors. Cancer Res 2022; 82: 1753–1761.
- 70.
Koelwyn GJ, Newman AAC, Afonso MS, van Solingen C, Corr EM, Brown EJ, et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat Med 2020; 26: 1452–1458.
- 71.
Argiles JM, Lopez-Soriano FJ, Stemmler B, Busquets S. Cancer-associated cachexia: Understanding the tumour macroenvironment and microenvironment to improve management. Nat Rev Clin Oncol 2023; 20: 250–264.
- 72.
Barkhudaryan A, Scherbakov N, Springer J, Doehner W. Cardiac muscle wasting in individuals with cancer cachexia. ESC Heart Fail 2017; 4: 458–467.
- 73.
Zaorsky NG, Churilla TM, Egleston BL, Fisher SG, Ridge JA, Horwitz EM, et al. Causes of death among cancer patients. Ann Oncol 2017; 28: 400–407.
- 74.
Anker MS, Sanz AP, Zamorano JL, Mehra MR, Butler J, Riess H, et al. Advanced cancer is also a heart failure syndrome: A hypothesis. Eur J Heart Fail 2021; 23: 140–144.
- 75.
Springer J, Tschirner A, Haghikia A, von Haehling S, Lal H, Grzesiak A, et al. Prevention of liver cancer cachexia-induced cardiac wasting and heart failure. Eur Heart J 2014; 35: 932–941.
- 76.
Stevens SC, Velten M, Youtz DJ, Clark Y, Jing R, Reiser PJ, et al. Losartan treatment attenuates tumor-induced myocardial dysfunction. J Mol Cell Cardiol 2015; 85: 37–47.
- 77.
Karlstaedt A, Barrett M, Hu R, Gammons ST, Ky B. Cardio-oncology: Understanding the intersections between cardiac metabolism and cancer biology. JACC Basic Transl Sci 2021; 6: 705–718.
- 78.
Pirozzi CJ, Yan H. The implications of IDH mutations for cancer development and therapy. Nat Rev Clin Oncol 2021; 18: 645–661.
- 79.
Akbay EA, Moslehi J, Christensen CL, Saha S, Tchaicha JH, Ramkissoon SH, et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev 2014; 28: 479–490.
- 80.
Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, et al. Oncometabolite D-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci USA 2016; 113: 10436–10441.
- 81.
Xu R, Rai A, Chen M, Suwakulsiri W, Greening DW, Simpson RJ. Extracellular vesicles in cancer: Implications for future improvements in cancer care. Nat Rev Clin Oncol 2018; 15: 617–638.
- 82.
Gargiulo P, Marzano F, Salvatore M, Basile C, Buonocore D, Parlati ALM, et al. MicroRNAs: Diagnostic, prognostic and therapeutic role in heart failure: A review. ESC Heart Fail 2023; 10: 753–761.

