Triiodothyronine as a Therapeutic Candidate for Cardiac Metabolism in the Failing Heart
Article ID: CJ-14-1140

Details
Article ID: CJ-14-1140
Cardiac energy metabolism plays a major role in heart failure (HF).1 Although many strategies have been attempted as metabolic interventions for the treatment of HF, a clinically suitable method has not yet been developed.2,3 Therefore, all available devices and drugs need to be used within a limited therapeutic window in order to treat the increasing number of patients with severe HF. In this issue of the Journal, Kajimoto et al4 firstly report a strategy for restoring triiodothyronine (T3) to improve metabolic flux and enhance cardiac function during the weaning of extracorporeal membrane oxygenation (ECMO), which is commonly used to treat acute HF and postoperative cardiopulmonary failure,5 because it reduces the oxygen consumption requirements of the heart and readjusts the balance between energy production and utilization. Despite positive effects on failing hearts, ECMO has been shown to promote an inflammatory response that reduces cardiac contractions. Furthermore, ECMO-associated increases in inflammatory cytokines disrupt systemic thyroid hormone homeostasis, which leads to a reduction in already low circulating levels of T3. Because cardiac myocytes cannot metabolize thyroxine to T3, the level of T3 in the blood affect myocyte function. Serum-free T3 is taken up by transport proteins and genomically or nongenomically regulates cardiac function. Many cardiac key proteins, such as α-myosin heavy chains or sarcoplasmic reticulum Ca2+-ATPase, are controlled transcriptionally through T3-binding thyroid hormone nuclear receptors. T3 also mediates various membrane ion channels and has been shown to affect various intracellular signal pathways (Figure). However, its role in cardiac metabolism has not yet been elucidated in detail.
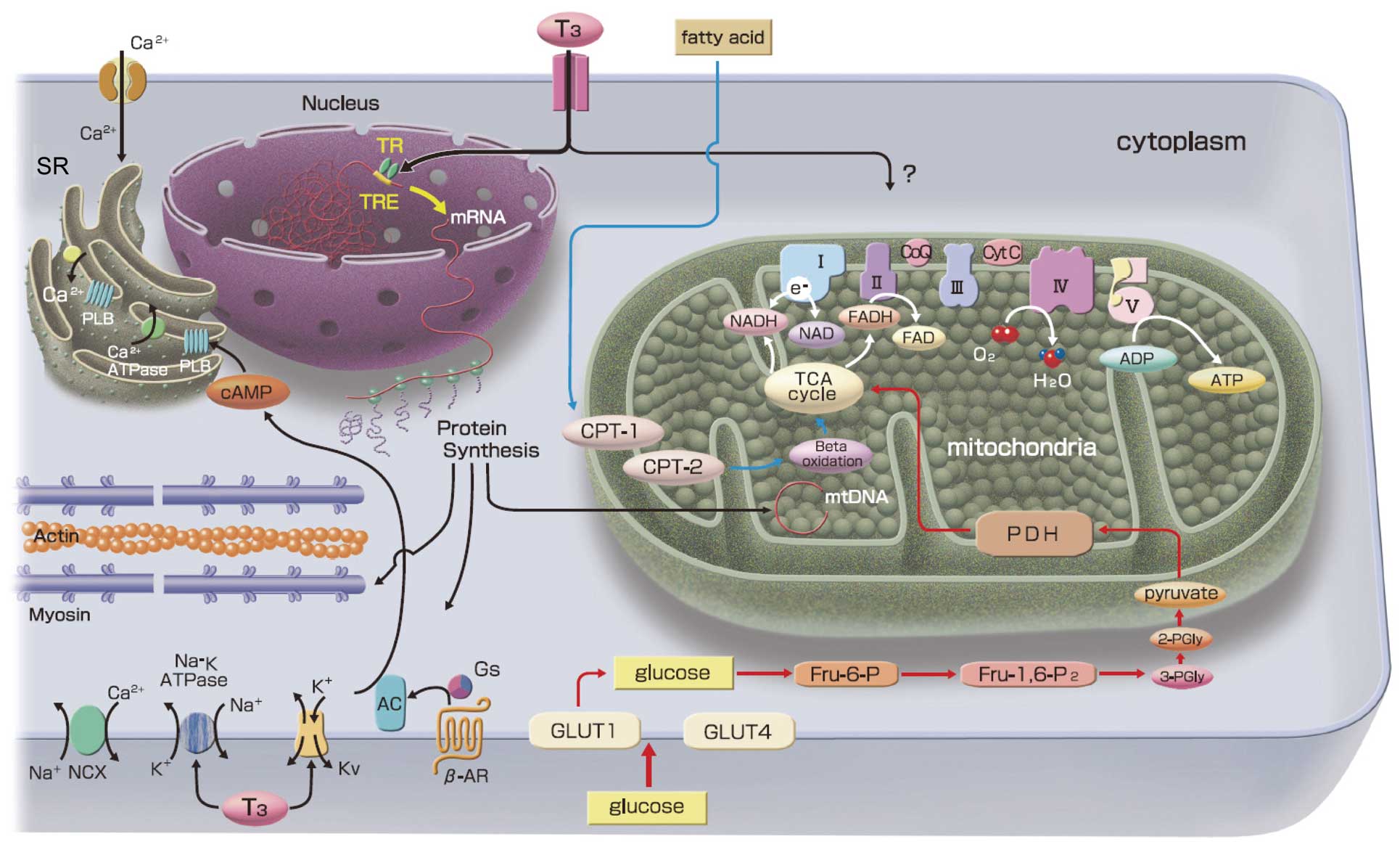
Cardiac metabolism and the effects of triiodothyronine (T3) on cardiac myocytes. Fatty acid and glucose account for the vast majority of ATP production in the heart. In addition to the genomic effects of T3, several cardiac responses are mediated via nongenomic mechanisms. Mitochondrial biogenesis is upregulated by T3 thorough genomic effects. Direct nongenomic action on mitochondria or their metabolic pathway remains to be established. β-AR, β-adrenergic receptor; Kv, voltage gated potassium channels; Na-K ATPase, sodium potassium ATPase; mtDNA, mitochondrial DNA; NCX, sodium-calcium exchanger; SR, sarcoplasmic reticulum; TR, thyroid hormone receptor; TRE, thyroid hormone response elements.
Article p ????
Kajimoto et al4 focused on fatty acid (FA) metabolism and lactate oxidation, both of which are very important in the failing myocardium. By using 13C-labeled lactate and 13C-labeled FA, they showed that the administration of T3 supplements released the inhibition of lactate oxidation following ischemia-reperfusion injury without impairing the oxidation of FA, thereby allowing for increases in [ATP]/[ADP] in the myocardium. The principle of metabolic analyses by gas chromatography-mass spectrometry (GC-MS) using [13C6, 15N]-L-leucine and [2-13C]-pyruvate was described in their recent study.6 They have also reported several metabolic analyses in a swine model,6–8 to which they administered physiological levels of FA and pyruvate without inotropic agents.
The findings of the present study will promote investigations focusing on T3 and mitochondria. T3 is a primary regulator of mammalian mitochondrial biogenesis9 and may influence mitochondrial function both indirectly and directly. As an indirect role, T3 binds to TRs, thereby upregulating the transcriptional regulators of mitochondrial biogenesis, such as nuclear respiratory factor 1 (NRF-1) and peroxisome proliferator-activated receptor gamma coactivator 1-α (PGC1-α). NRF-1 and PGC1-α then upregulate the transcription of nuclear-encoded mitochondrial transcriptional machinery such as mitochondrial transcription factor A (Tfam), which stimulates mitochondrial DNA replication and mitochondrial biogenesis. T3 has been shown to regulate the mitochondrial DNA copy number in cardiac mitochondria from induced-hypothyroid rats (Figure).10 T3 also directly regulates mitochondrial function through the T3-binding adenine nucleotide transporter and p43; however, direct regulation of mitochondria is tissue-specific and cardiac mitochondria are mainly regulated by an indirect mechanism via the nucleus.11,12 In Kajimoto et al’s study, the administration of T3 recovered the metabolic flexibility of the cardiac mitochondria and upregulated the usage of lactate in the border zone of myocardial ischemia. This improvement may prove to be significant following further research involving a larger number of animals.
These findings prompted consideration of the development of new strategies to treat HF. Firstly, although Kajimoto et al used an immature swine model, their clinically relevant protocol can be developed for the treatment of humans. We might be able to observe other positive effects in human hearts, such as regeneration,13 or novel genomic and nongenomic actions on cardiac myocytes.
Secondly, the administration of T3 supplements to patients with failing hearts may open the use of metabolic therapy for chronic HF. A previous study showed that 22% of HF patients with preserved ejection fraction had abnormal thyroid function,14 and subclinical hypothyroidism may be a risk for cardiovascular events.15
The use of T3 may represent the initiation of metabolic therapy for the treatment of HF, which could be a breakthrough for curing such patients.