Abstract
Background:
Hypoxic pulmonary hypertension (HPH) is characterized by pulmonary vascular remodeling. Intracellular Ca2+
concentration ([Ca2+]i) is an essential signal for myocyte proliferation. Whether chronic hypoxia (CH) affects the basal [Ca2+]i
and Ca2+
entry through store- and/or receptor-operated calcium channels (SOCC, ROCC), and whether canonical transient receptor potential (TRPC) proteins are involved in CH-induced Ca2+
influx and proliferation in pulmonary venous smooth muscle cells (PVSMCs) is examined.
Methods and Results:
Rats were exposed to CH. PVSMCs were isolated from distal pulmonary veins. In freshly isolated PVSMCs, CH increased the basal [Ca2+]i; removal of Ca2+
or application of SKF-96365 reversed the elevated [Ca2+]i, whereas nifedipine had no effect. Receptor-operated Ca2+
entry (ROCE) was expressed in PVSMCs. In freshly isolated PVSMCs from CH rats, ROCE was enhanced, whereas store-operated Ca2+
entry had no alteration. Furthermore, real-time polymerase chain reaction and western blotting showed that mRNA and protein expression level of TRPC6, but neither TRPC1 nor TRPC3, in pulmonary venous smooth muscle (PV) from CH rats and PVSMCs exposed to CH was greater than in normal PV and PVSMCs. The knockdown of TRPC6 in hypoxic PVSMCs with siRNA inhibited the enhanced ROCE and attenuated CH-induced PVSMCs proliferation.
Conclusions:
The enhanced Ca2+
entry through ROCC, due to upregulated TRPC6, is a novel pathogenic mechanism contributing to the increased basal [Ca2+]i
in PVSMCs and excessive PVSMC proliferation during the development of HPH.
Pulmonary veins (PV) not only serve as conduits through which oxygenated capillary blood drains into the left atrium of the heart in pulmonary circulation, but they also play important roles in regulating both distention and recruitment of blood flow from alveolar wall capillaries and thus ventilation-perfusion matching in the lung. Studies reveal that PV exhibit vasoconstriction in response to various vasoconstrictor stimuli, such as endothelin, thromboxane, platelet-activating factor, leukotrienes and hypoxia. Exposure to chronic hypoxia (CH) causes vascular remodeling not only in pulmonary arteries (PA) but also in PV in a number of species including rat, sheep and humans,1–4
and leads to hypoxic pulmonary hypertension (HPH). Despite the numerous progresses in elucidating the underlying mechanisms of CH-induced pulmonary arterial remodeling, less attention has been paid to the PV side in searching for the cellular and molecular mechanism of chronically hypoxic pulmonary venous remodeling.
In pulmonary vascular smooth muscle cells, the increase in intracellular Ca2+
concentration ([Ca2+]i) is an essential signal for vasoconstriction and myocyte proliferation. The elevation of [Ca2+]i
could result from Ca2+
influx from an extracellular source through Ca2+
channels, such as L-type voltage-dependent Ca2+
channels (VDCC), store-operated Ca2+
channels (SOCC) and receptor-operated Ca2+
channels (ROCC), or Ca2+
release from internal storage sites, such as the sarcoplasmic reticulum (SR). VDCC can be activated by membrane depolarization and blocked by VDCC blockers such as nifedipine and verapamil.5
Recent evidence suggests that SOCC mediates store-operated Ca2+
entry (SOCE) and can be activated by depletion of Ca2+
stores using cyclopiazonic acid (CPA) or thapsigargin,6
whereas ROCC mediates receptor-operated Ca2+
entry (ROCE) and can be activated directly by diacylglycerol (DAG) via a protein kinase C-independent mechanism.7
As a membrane-permeable analogue of DAG, 1-oleolyl-2-acetyl-sn-glycerol (OAG) is commonly used as a ROCC activator.8
Canonical transient receptor potential (TRPC) proteins constitute a family of seven (TRPC1-7) non-selective cation channels within the wider transient receptor potential (TRP) superfamily. TRPC proteins have recently emerged as the molecular components of SOCC and ROCC and play important roles in the control of smooth muscle function. Evidence indicates that multiple TRPC subtypes have been identified in rat and human pulmonary arterial smooth muscle cells (PASMCs). An increased expression of TRPC induced by CH is largely responsible for raising [Ca2+]i
in PASMCs, which in turn may cause increased tone, proliferation and migration of PASMCs, thus contributing to pulmonary arterial remodeling.9
Although we recently demonstrated that SOCE and TRPC are expressed in rat pulmonary venous smooth muscle,10
and acute hypoxia activates SOCE and increases [Ca2+]i
in rat pulmonary venous smooth muscle cells (PVSMCs),11
it is still unknown what the effects of CH on basal [Ca2+]i, TRPC expression and TRPC-dependent Ca2+
entry in PVSMCs are. Therefore, the present study was designed to examine the alteration of TRPC expression, SOCE, ROCE, and their involvement in the increased basal [Ca2+]i
caused by CH in rat PVSMCs.
Methods
Chronic Hypoxic Exposure
All procedures were approved by the Animal Care and Use Committee of Guangzhou Medical University. Adult male Wistar rats (weighing 250–350 g) were exposed to normoxia or hypoxia (10% O2) for 21 days, as described previously.12
Preparation of Distal PV and Cell Culture
Intrapulmonary veins were dissected from rat lungs. PVSMCs were isolated from intrapulmonary veins and smooth muscle α-actin immunofluorescence of PVSMCs was performed, as described previously.10,11,13,14
PVSMCs from normoxic and chronically hypoxic rats were transiently (~24 h) cultured at 37℃ under 21% O2/5% CO2
and 4% O2/5%CO2, respectively.
Measurement of [Ca2+]i
The [Ca2+]i
was measured in PVSMCs using fura-2 fluorescence microscopy, as previously described.10–12
Measurement of SOCE and ROCE
The SOCE and ROCE were both assessed in 2 ways; Ca2+
restoration and Mn2+
quenching, as described previously.10,15
Real-Time Quantitative Polymerase Chain Reaction (PCR)
Total RNA was extracted from de-endothelialized distal PV smooth muscle and PVSMCs. Real-time quantitative PCR was performed, as described previously.10
Western Blotting
Proteins were extracted from PV smooth muscle and PVSMCs. TRPC1, TRPC3 and TRPC6 were detected using Western blotting, as previously described.10
Cell Proliferation Assay
The proliferation assay of PVSMCs was performed by using BrdU incorporation, as previously described.16
RNA Interference
The PVSMCs were transfected with siRNA specific for TRPC1 (siTRPC1), TRPC6 (siTRPC6) or non-targeting control siRNA (siNT), as previously described.17
(See the Supplementary File for more details about the Methods used.)
Results
Basal [Ca2+]i
in PVSMCs
Because the main PV approaching the left atrium was thought to contain cardiac myocytes,18
and the intrapulmonary PV constitutes the major site of vasomotor responses to hypoxia,1
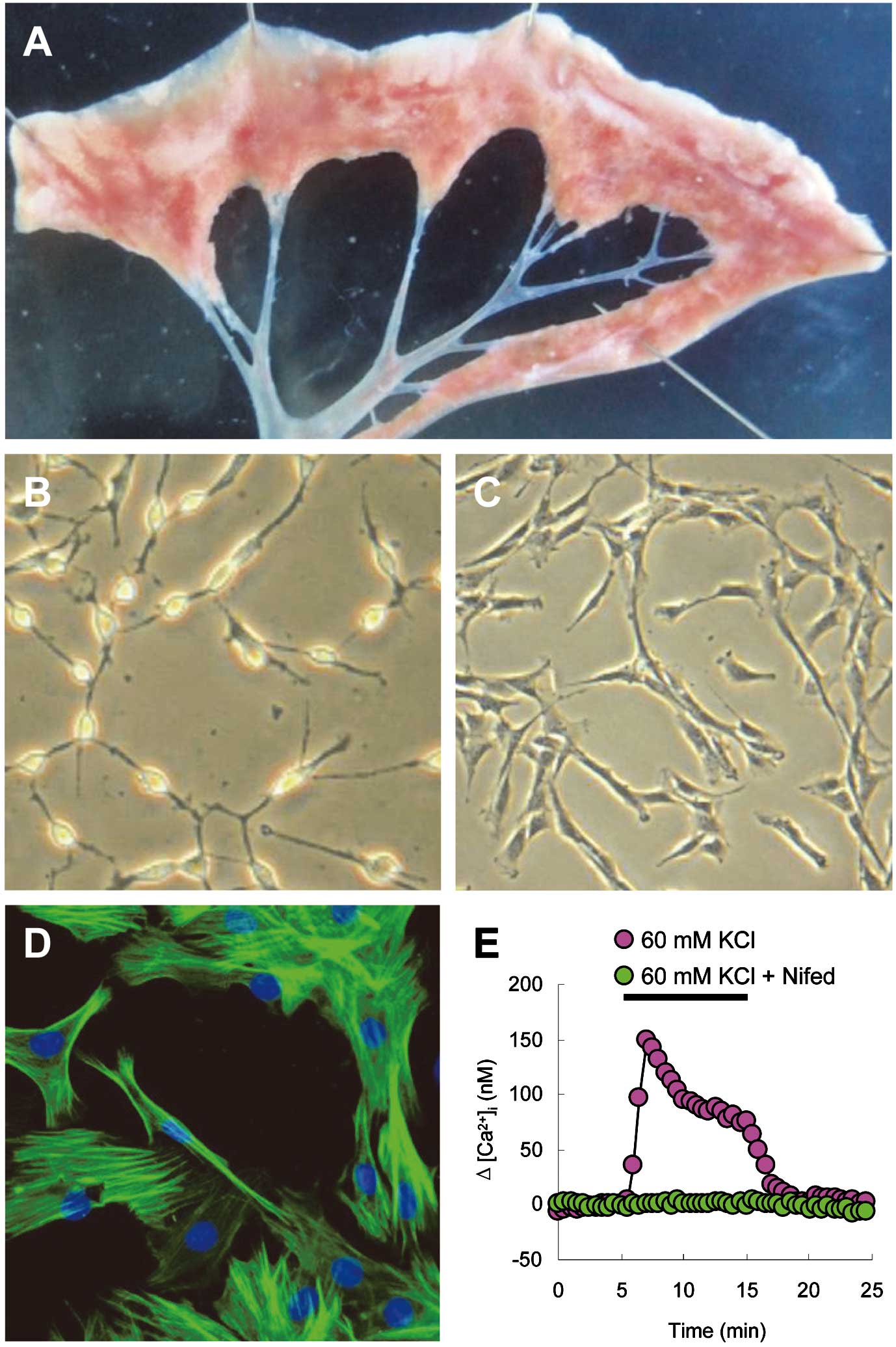
the veins used in this study were isolated from >4th generation intrapulmonary venous branches (Figure 1A). Observed with a phase-contrast microscope, fresh PVSMCs (cultured within 24 h) were phase bright and started to lose their round shape with thin fibers (Figure 1B). When cultured for 3–4 days, PVSMCs appeared spindle-shaped with elongated fibers (Figure 1C). Immunofluorescence of smooth muscle α-actin showed specific green staining of actin fiber bundles in >98% of cultured cells with blue staining for nuclei (Figure 1D). PVSMCs showed a clear increase in [Ca2+]i
in response to 60 mmol/L KCl, whereas the L-type VDCC antagonist, nifedipine (5 μmol/L), completely blocked the response to KCl, indicating the presence of functional VDCC (Figure 1E).
As shown in
Figures 2A
and
2B, CH caused a marked increase in basal [Ca2+]i
from 104.7±6.2 nmol/L (n=7 experiments in 178 cells) in PVSMCs from normoxic rats to 196.6±10.4 nmol/L (n=7 experiments in 181 cells; P<0.0001) in cells from chronically hypoxic animals. In PVSMCs from chronically hypoxic rats, the removal of extracelluar Ca2+
induced an apparent decrease in basal [Ca2+]i
from baseline to 117.8±4.2 nmol/L (n=7 experiments in 181 cells; P<0.0001), whereas the removal of extracelluar Ca2+
did not alter the basal [Ca2+]i
(104.7±6.2 to 103.7±5.5 nmol/L, n=7 experiments in 178 cells ) in cells from normoxic rats (Figures 2A,2B).
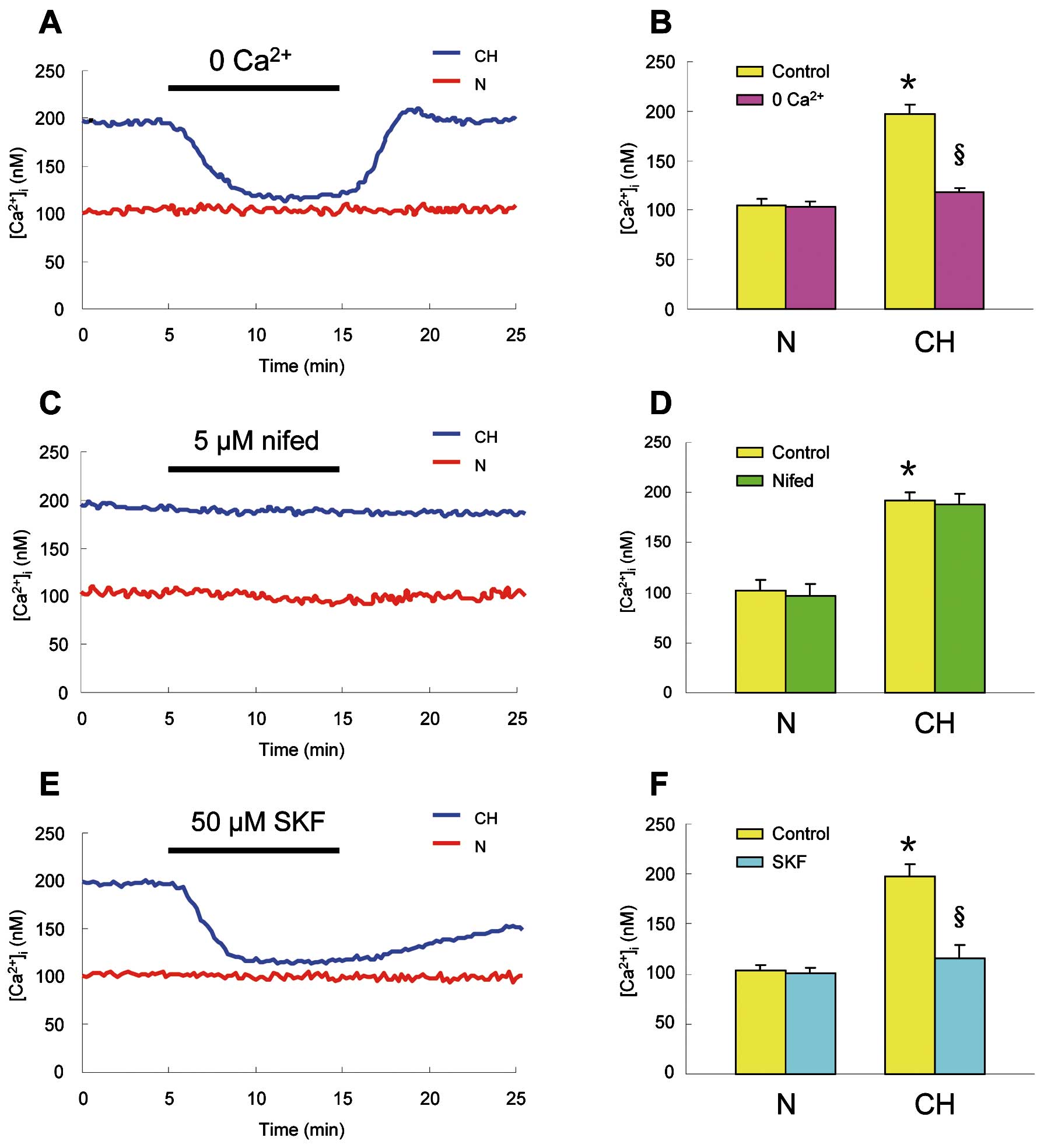
In PVSMCs isolated from chronically hypoxic rats and perfused for 10 min with KRB solution containing 5 μmol/L nifedipine to block the L-type VDCC, nifedipine did not alter the resting [Ca2+]i
(191.6±9.2 to 188.8±9.6 nmol/L, n=8 experiments in 206 cells), which was similar to that which occurred in PVSMCs from normoxic rats (102.8±10.4 to 97.1±11.2 nmol/L, n=7 experiments in 169 cells) (Figures 2C,2D). However, the application of 50 μmol/L SKF-96365 to inhibit non-selectively SOCC and ROCC caused a large decrease in the resting [Ca2+]i
from baseline to 116.0±13.2 nmol/L (n=7 experiments in 171 cells; P<0.0001;
Figures 2E,2F) in PVSMCs from chronically hypoxic rats. In contrast, SKF-96365 did not cause an alteration in the resting [Ca2+]i
(102.5±6.5 to 100.2±6.6 nmol/L, n=6 experiments in 152 cells ) in PVSMCs from normoxic animals (Figures 2E,2F).
SOCE in PVSMCs
SOCE was first assessed by measuring the [Ca2+]i
response to extracellular Ca2+
restoration. Perfusion of PVSMCs with Ca2+-free KRB solution containing 5 μmol/L nifedipine and 10 μmol/L CPA caused an initial small transient increase in [Ca2+]i, which reached a peak and returned to baseline after 5–10 min (Figure 3A), indicating calcium release from the SR. The peak change in [Ca2+]i
induced by CPA was similar in PVSMCs from normoxic and chronically hypoxic rats (△[Ca2+]i=136.1±6.5 nmol/L, n=8 experiments in 193 cells for normoxic; and 142.0±10.6 nmol/L, n=8 experiments in 188 cells for chronically hypoxic cells) (Figure 3B). Subsequent restoration of extracellular Ca2+
to 2.5 mmol/L induced a second large increase in [Ca2+]i, which went up quickly to a peak of △[Ca2+]i=273.1±37.0 nmol/L (n=8 experiments in 193 cells) in PVSMCs from chronically hypoxic rats. This peak change in [Ca2+]i
was also similar to that measured in PVSMCs from normoxic rats (△[Ca2+]i=263.2±24.3 nmol/L; n=8 experiments in 188 cells;
Figures 3A,3B).

SOCE was further evaluated by the rate of Mn2+-quenched fura-2 fluorescence. In PVSMCs from normoxic rats perfused with Ca2+-free KRB solution containing both nifedipine and CPA, Mn2+
administration (200 μmol/L) resulted in a 40.0±4.3% decrease in fura-2 fluorescence (n=7 experiments in 169 cells;
Figures 3C,3D). In PVSMCs from chronically hypoxic rats, fura-2 fluorescence decreased by 40.8±4.2% (n=7 experiments in 172 cells;
Figures 3C,3D), which was similar to that observed in normoxic cells, suggesting Ca2+
entry through SOCC in PVSMCs from chronically hypoxic rats was similar to that in PVSMCs from normoxic rats.
ROCE in PVSMCs
ROCE was also evaluated by recording the Ca2+
transients and the rate of Mn2+-quenched fura-2 fluorescence. In the presence of 5 μmol/L nifedipine, perfusion of normoxic PVSMCs with Ca2+-free KRB solution containing 100 μmol/L OAG for 10 min did not cause an alteration of [Ca2+]i
(Figures 4A,4B), indicating no calcium release from the SR. But subsequent restoration of extracellular Ca2+
to 2.5 mmol/L caused an apparent increase of [Ca2+]i
that reached a peak of △[Ca2+]i=176.0±20.3 nmol/L (n=7 experiments in 165 cells, P<0.001 vs. the value in the absence of OAG), followed by a lower plateau before it returned to baseline by perfusion of cells with KRB solution. When OAG was omitted in the perfusate, the change in [Ca2+]i
did not happen (Figures 4A,4B). In the Mn2+
quenching study, perfusion of normoxic PVSMCs for 10 min with Ca2+-free KRB solution containing Mn2+
resulted in a 30.3±1.4% (n=8 experiments in 193 cells) decrease in fura-2 fluorescence in the presence of nifedipine and OAG, whereas there was only a 14.5±1.3% decrease in the absence of OAG (n=8 experiments in 197 cells; P<0.0001 vs. the value in the presence of OAG;
Figures 4C,4D). These data indicate the occurrence of ROCE that was induced by OAG in PVSMCs.

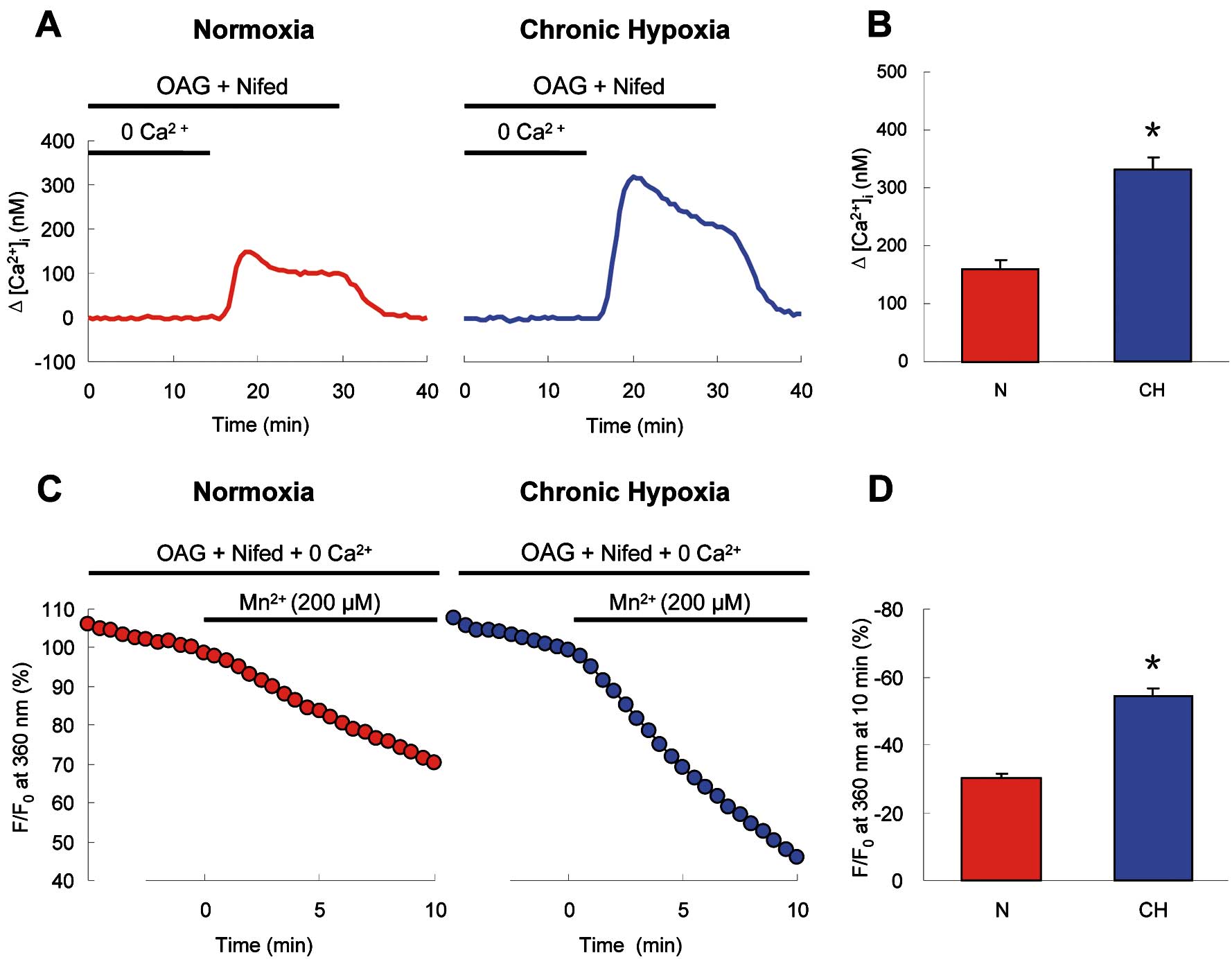
In chronically hypoxic PVSMCs, after perfusion with 100 μmol/L OAG in nifedipine containing Ca2+-free KRB solution, restoration of extracellular Ca2+
to 2.5 mmol/L caused a large increase of [Ca2+]i
(△[Ca2+]i=332.0±21.1 nmol/L, n=7 experiments in 173 cells) that was twice the magnitude of the [Ca2+]i
increase elicited in normoxic PVSMCs (△[Ca2+]i=160.6±15.7 nmol/L, n=7 experiments in 165 cells; P<0.001;
Figures 5A,5B). Moreover, Mn2+
quenching also showed that OAG-induced Ca2+
entry was significantly enhanced in chronically hypoxic PVSMCs. The rate of OAG-induced Mn2+
quenching was increased from 29.7±1.5% (n=8 experiments in 185 cells) in normoxic to 54.4±2.5% (n=8 experiments in 191 cells, P<0.00001) in chronically hypoxic PVSMCs (Figures 5C,5D). These results clearly indicate that the functional activities of ROCC were enhanced in PVSMCs from chronically hypoxic rats.
TRPC Expression in Distal PV Smooth Muscle and PVSMCs
Real-time quantitative PCR showed that TRPC1 and TRPC3 mRNA expression in distal PV smooth muscle from chronically hypoxic rats and hypoxic PVSMCs cultured in 4% O2
for 60 h was similar to that from normoxic PV smooth muscle and PVSMCs (Figures 6A,6B). In contrast, TRPC6 mRNA expression in PV smooth muscle from chronically hypoxic rats and hypoxic PVSMCs was significantly higher than that from normoxic PV smooth muscle and PVSMCs. Compared with normoxic PV smooth muscle and PVSMCs, CH (10% O2
21 days for rats, 4% O2
60 h for cells) enhanced TRPC6 mRNA expression by 168.5±9.1% (n=5; P<0.01) and 88.0±7.3% (n=5; P<0.01;
Figures 6A,6B) in distal PV smooth muscle and PVSMCs, respectively.

Western blotting showed TRPC1 and TRPC3 proteins in distal PV smooth muscle from chronically hypoxic rats, and hypoxic PVSMCs did not differ from normoxic PV smooth muscle (Figures 6C,6D) and PVSMCs (Figures 6E,6F). However, TRPC6 protein was significantly increased in both distal PV smooth muscle from chronically hypoxic rats (Figures 6C,6D) and hypoxic PVSMCs (Figures 6E,6F). Compared with normoxic controls, CH increased TRPC6 protein expression by 144.9±16.5% (n=5; P<0.01;
Figures 6C,6D) and 76.4±9.1% (n=5; P<0.01;
Figures 6E,6F) in distal PV smooth muscle and PVSMCs, respectively. These results clearly indicate that the expression of TRPC6, but neither TRPC1 nor TRPC3, was upregulated in distal PV smooth muscle after chronic exposure to hypoxia.
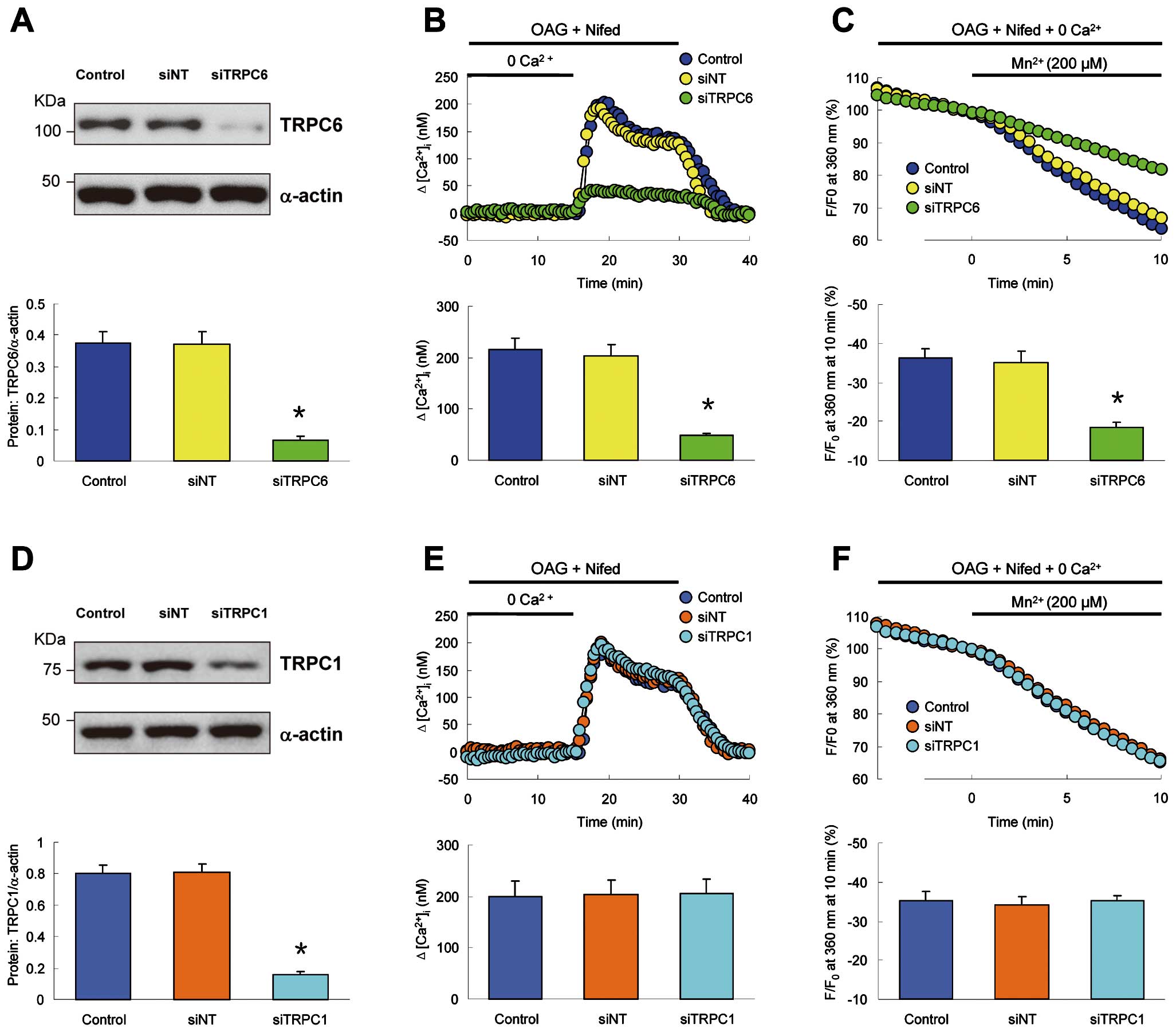
Downregulation of TRPC6 Inhibits ROCE in Hypoxic PVSMCs
To obtain direct evidence for the involvement of TRPC6 in CH-induced enhancement of ROCE, we used siRNA to knockdown TRPC6 and examined whether TRPC6 was necessary for ROCE in hypoxic PVSMCs. Treatment of hypoxic PVSMCs with siTRPC6 significantly decreased the protein level of TRPC6 (Figure 7A) and markedly inhibited the increase of [Ca2+]i
after extracellular Ca2+
restoration (Figure 7B) and the rate of Mn2+-quenched fura-2 fluorescence (Figure 7C) in the presence of nifedipine and OAG. However, downregulation of TRPC1 with siTRPC1 (Figure 7D) altered neither the increase of [Ca2+]i
after extracellular Ca2+
restoration (Figure 7E) nor the rate of Mn2+-quenched fura-2 fluorescence (Figure 7F) in the presence of nifedipine and OAG in hypoxic PVSMCs. These experiments provide clear evidence that it is TRPC6 that is necessary for the enhanced Ca2+
entry through ROCC in rat PVSMCs after CH exposure.
TRPC6 Knockdown Attenuates CH-Induced Enhancement of Cell Proliferation in PVSMCs
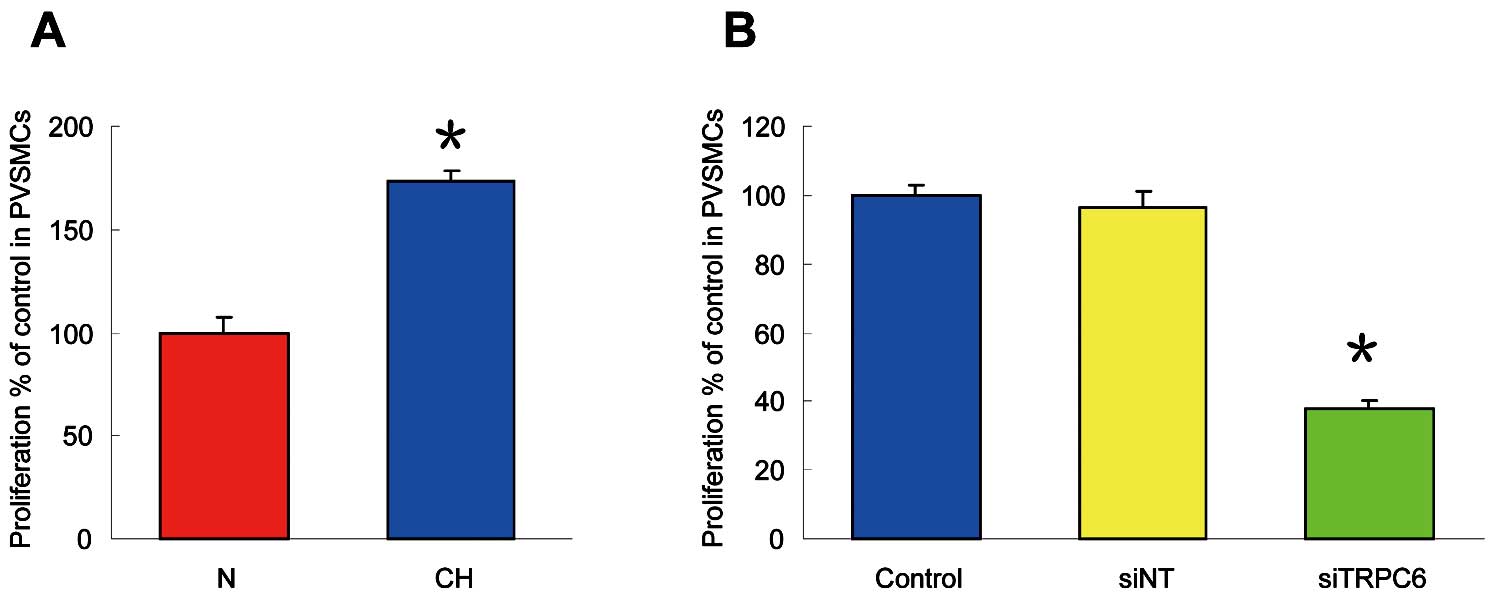
In comparison to normoxic cells, as seen in
Figure 8A, CH (4% O2
60 h) enhanced PVSMC proliferation by 72.9±5.3% (n=5; P<0.01). Treatment of hypoxic PVSMCs with siTRPC6 significantly attenuated cell proliferation by 62.2±2.6% (n=5; P<0.01;
Figure 8B). These data provide compelling evidence that TRPC6 is necessary for the enhanced cell proliferation in PVSMCs after CH exposure.
Discussion
In this study, we found that: (1) the basal [Ca2+]i
in PVSMCs from CH rats was increased, and removal of Ca2+
or application of SKF-96365 reversed the elevated [Ca2+]i, but nifedipine had no effect; (2) ROCE was expressed and enhanced in PVSMCs from CH rats, but SOCE had no alteration; (3) both mRNA and protein expression level of TRPC6, but neither TRPC1 nor TRPC3, in PV smooth muscle from CH rats and PVSMCs exposed to CH was greater than in normal PV smooth muscle and PVSMCs; (4) downregulation of TRPC6 with siRNA in hypoxic PVSMCs inhibited the enhanced ROCE, whereas downregulation of TRPC1 had no effect; (5) CH enhanced PVSMCs proliferation and TRPC6 knockdown attenuated cell proliferation. Collectively, these observations indicate that functionally upregulated TRPC6 and subsequently enhanced ROCE via ROCC in PVSMCs contribute to the augmented Ca2+
signaling and excessive PVSMCs proliferation in CH exposed animals.
The elevated resting [Ca2+]i
in smooth muscle cells is associated with contraction of pulmonary vessels and pulmonary vascular remodeling, thus contributing to HPH. In our study, we found that exposure to CH caused an elevated resting [Ca2+]i
in PVSMCs, which was similar to that in PASMCs.12,15
Consistent with PASMCs,15
in PVSMCs from chronically hypoxic rats, removal of extracellular Ca2+
could reverse the CH-induced increase in resting [Ca2+]i, whereas the L-type VDCC blocker nifedipine, given at a concentration that blocked the KCl-induced increase in [Ca2+]i, had no effect on resting [Ca2+]i, indicating that CH causes an influx of Ca2+
through not the L-type VDCC, but other independent pathways in PVSMCs. Moreover, in PVSMCs from normoxic rats, the non-selective cation channel blocker, SKF-96365, had no effect on resting [Ca2+]i, whereas in PVSMCs from chronically hypoxic rats, the blocker caused immediate reduction in [Ca2+]i
to a level similar to that measured in PVSMCs from normoxic animals. Because the non-selective cation channel blocker, SKF-96365, has been shown to block both SOCC and ROCC in various cell types, including smooth muscle cells,5,12,15
these results provide the hint that SOCE via SOCC and/or ROCE via ROCC may contribute to the CH-induced elevation of resting [Ca2+]i
in PVSMCs.
We have previously demonstrated the presence of SOCE in PVSMCs.10
In our subsequent experiments, we found that SOCE, measured via Ca2+
restoration following store depletion by CPA, and the rate of Mn2+
quenching of fura-2 fluorescence in cells exposed to CPA, were both similar in PVSMCs from normoxic and chronically hypoxic rats. Because nifedipine at 5 μmol/L and CPA at 10 μmol/L have been proven to be effective in blocking VDCC or depleting the Ca2+
store from the SR in various cell types,10,11,19
the similar rise in [Ca2+]i
following restoration of extracellular Ca2+
and the similar rate of Mn2+
quenching of fura-2 fluorescence in PVSMCs from normoxic and chronically hypoxic animals indicate similar Ca2+
influx through SOCC, clearly suggesting that SOCE did not contribute to the CH-induced increase in [Ca2+]i
in PVSMCs.
ROCC have been identified to be expressed in various cell types including smooth muscle cells.15,20
ROCC could be activated directly by DAG, resulting in Ca2+
entry through ROCC, also known as ROCE. OAG is commonly used as a DAG analogue to identify the presence and the role of ROCE in various cell types.8,15,20
In the presence of nifedipine, perfusion of PVSMCs with Ca2+-free KRB solution containing OAG did not cause an increase in [Ca2+]i, indicating that OAG could not induce Ca2+
release from the SR in PVSMCs. However, in the presence of nifedipine and OAG, restoration of extracellular Ca2+
caused a large increase of [Ca2+]i
in PVSMCs, reflecting Ca2+
influx through the ROCC. This response was dependent on OAG, as it did not occur in PVSMCs exposed to a Ca2+-free perfusate containing nifedipine but not OAG. Moreover, in cells not exposed to OAG, Mn2+
caused a slow decline in fura-2 fluorescence; whereas in cells exposed to OAG, Mn2+
caused a marked increase of this decline, also suggesting Ca2+
entry through the ROCC. Besides the functional presence of ROCC, we have previously demonstrated the expression of TRPC6 and TRPC3 in PVSMCs, which have been identified as major constituents for ROCC mediating ROCE in various cell types.10,21
Together, these results provide strong evidence indicating the presence of ROCE in PVSMCs.
Because neither Ca2+
influx through VDCC nor SOCE contributes to the elevated resting [Ca2+]i
in PVSMCs, another possibility is that the CH-induced increase of resting [Ca2+]i
results from increased Ca2+
entry through the ROCC. We found that ROCE, measured via Ca2+
restoration, was greater in PVSMCs from chronically hypoxic rats than in PVSMCs from normoxic rats. Moreover, in the presence of nifedipine and OAG, the addition of Mn2+
caused significantly faster quenching of fura-2 fluorescence in PVSMCs from chronically hypoxic rats. These results indicate clearly enhanced ROCE in PVSMCs from chronically hypoxic rats.
Both ROCC and SOCC are believed to be composed of TRPC proteins, with ROCC consisting of TRPC3, TRPC6 and TRPC7 and SOCC consisting of TRPC1, TRPC4 and TRPC5.20
A reasonable explanation for enhanced ROCE in PVSMCs is upregulation of TRPC proteins. In PV smooth muscle and PVSMCs, real-time quantitative PCR and western blotting demonstrated that TRPC6 expression was upregulated by CH, whereas the expression of TRPC1 and TRPC3 did not alter during CH. These results indicate that CH upregulated only TRPC6 expression in PVSMCs, suggesting TRPC6 contributes to the enhancement of ROCE in PVSMCs during CH. Although TRPC3 has also been known as a constituent for ROCC,22
the unchangeable expression of TRPC3 suggests TRPC3 did not contribute to the enhancement of ROCE in PVSMCs during CH. Moreover, the unchangeable expression of TRPC1 provides further evidence indicating that SOCE did not contribute to the CH-induced increase in [Ca2+]i
in PVSMCs. Our observation in PVSMCs is different from that of other reports involving PASMCs. Previous studies reported that CH upregulated both TRPC1 and TRPC6 in rat PASMCs.12,15
Real-time quantitative PCR detected only TRPC1 mRNA upregulation in murine hypoxic PASMCs.23
These data clearly revealed that TRPC upregulation by CH is subtype-specific and varies among species and cell preparations.
TRPC6 is usually believed to be a major component of ROCC, activated directly by DAG.21
OAG is commonly used as a DAG analogue.8,15,20
Using a specific siRNA knockdown strategy, we found that TRPC6 knockdown reduced OAG-induced ROCE but TRPC1 knockdown did not affect OAG-induced ROCE in PVSMCs cultured under hypoxic conditions, further confirming that TRPC6 mediates the enhanced ROCE in hypoxic PVSMCs. A recent study reports that TRPC6 upregulation was related to enhanced SOCE in rat hypoxic PVSMCs, which was not consistent with our observations.24
However, the direct evidence confirming that TRPC6 mediates SOCE using siRNA to knockdown TRPC6, and the relationship between TRPC6 and ROCE in PVSMCs, were both absent. Moreover, it is unclear if TRPC6 displays a different phenotype in in vitro cultured PVSMCs; for example, by heteromerization with TRPC1 or TRPC4, which has been implicated in SOCE,25
or if the enhanced SOCE in PVSMCs cultured in vitro for 60 h under hypoxic conditions results from altered expression of other TRP proteins including melastatin- and vanilloid-TRP under in vitro conditions, which have also been implicated in SOCE.26
These possibilities require further investigations.
In actual fact, in chronically hypoxic animals, the increased resting [Ca2+]i
via augmented Ca2+
entry through ROCC was directly associated with the functional upregulation of TRPC6 in PVSMCs. The knockdown of TRPC6 in hypoxic PVSMCs not only diminished ROCE but also inhibited CH-induced cell proliferation. These data provide compelling evidence that TRPC6 is necessary and sufficient for the increased resting [Ca2+]i
and ROCE and excessive PVSMC proliferation in chronically hypoxic animals, which suggests that the detailed mechanisms by which pulmonary vessels respond to CH were different between PV and PA. In rat PASMCs, CH-induced upregulation of both TRPC1 and TRPC6 was correlated with elevated [Ca2+]i
and enhanced SOCE and triggered proliferation.12,15
In summary, we initially identified the presence of ROCE in rat PVSMCs and demonstrated that upregulated expression of TRPC6 in PVSMCs and enhanced function of TRPC6 mediating Ca2+
entry through ROCC are new pathogenic mechanisms involved in the initiation and progression of proliferation of PVSMCs exposed to CH. Our study may provide the patho/physiological basis for targeting the TRPC6 protein or ROCC for future treatments of CH-related pulmonary hypertension, specifically pulmonary venous hypertension.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81000020), the Natural Science Foundation of Guangdong Province (2014A030313486), the Scientific Research Grant of Guangzhou City (201510010226), Guangzhou Department of Education Yangcheng Scholarship (10A025G) and the Science Foundation of State Key Laboratory (2011011).
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author(s).
Supplementary Files
Supplementary File 1
Methods
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-15-0067
References
- 1.
Takahashi H, Soma S, Muramatsu M, Oka M, Fukuchi Y. Upregulation of ET-1 and its receptors and remodeling in small pulmonary veins under hypoxic conditions. Am J Physiol Lung Cell Mol Physiol 2001; 280: L1104–L1114.
- 2.
Salinas CE, Blanco CE, Villena M, Giussani DA. High-altitude hypoxia and echocardiographic indices of pulmonary hypertension in male and female chickens at adulthood. Circ J 2014; 78: 1459–1464.
- 3.
Nergui S, Fukumoto Y, Do.e Z, Nakajima S, Shimizu T, Ikeda S, et al. Role of endothelial nitric oxide synthase and collagen metabolism in right ventricular remodeling due to pulmonary hypertension. Circ J 2014; 78: 1465–1474.
- 4.
Elias-Al-Mamun M, Satoh K, Tanaka S, Shimizu T, Nergui S, Miyata S, et al. Combination therapy with fasudil and sildenafil ameliorates monocrotaline-induced pulmonary hypertension and survival in rats. Circ J 2014; 78: 967–976.
- 5.
Ng LC, Gurney AM. Store-operated channels mediate Ca2+ influx and contraction in rat pulmonary artery. Circ Res 2001; 89: 923–929.
- 6.
Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca(2+) channels in native vascular smooth muscle cells. Circ Res 2001; 88: 84–87.
- 7.
Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 1999; 397: 259–263.
- 8.
Weissmann N, Sydykov A, Kalwa H, Storch U, Fuchs B, Mederos y Schnitzler M, et al. Activation of TRPC 6 channels is essential for lung ischaemia-reperfusion induced oedema in mice. Nat Commun 2012; 3: 649, doi:10.1038/ncomms1660.
- 9.
Shimoda LA, Wang J, Sylvester JT. Ca2+ channels and chronic hypoxia. Microcirculation 2006; 13: 657–670.
- 10.
Peng G, Lu W, Li X, Chen Y, Zhong N, Ran P, et al. Expression of store-operated Ca2+ entry and transient receptor potential canonical and vanilloid-related proteins in rat distal pulmonary venous smooth muscle. Am J Physiol Lung Cell Mol Physiol 2010; 299: L621–L630.
- 11.
Peng G, Ran P, Lu W, Zhong N, Wang J. Acute hypoxia activates store-operated Ca(2+) entry and increases intracellular Ca(2+) concentration in rat distal pulmonary venous smooth muscle cells. J Thorac Dis 2013; 5: 605–612.
- 12.
Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 2006; 98: 1528–1537.
- 13.
Peng G, Wang J, Lu W, Ran P. Isolation and primary culture of rat distal pulmonary venous smooth muscle cells. Hypertens Res 2010; 33: 308–313.
- 14.
Peng G, Wen X, Shi Y, Jiang Y, Hu G, Zhou Y, et al. Development of a new method for the isolation and culture of pulmonary arterial endothelial cells from rat pulmonary arteries. J Vasc Res 2013; 50: 468–477.
- 15.
Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, et al. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: A novel mechanism of hypoxic pulmonary hypertension. Circ Res 2004; 95: 496–505.
- 16.
Li L, Xu ZP, Liu GP, Xu C, Wang ZH, Li XG, et al. Expression of 1N3R-Tau isoform inhibits cell proliferation by inducing S phase arrest in N2a cells. PLoS One 2015; 10: e0119865, doi:10.1371/journal.pone.0119865.
- 17.
Lu W, Wang J, Peng G, Shimoda LA, Sylvester JT. Knockdown of stromal interaction molecule 1 attenuates store-operated Ca2+ entry and Ca2+ responses to acute hypoxia in pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 2009; 297: L17–L25.
- 18.
Michelakis ED, Weir EK, Wu X, Nsair A, Waite R, Hashimoto K, et al. Potassium channels regulate tone in rat pulmonary veins. Am J Physiol Lung Cell Mol Physiol 2001; 280: L1138–L1147.
- 19.
Wang J, Shimoda LA, Sylvester JT. Capacitative calcium entry and TRPC channel proteins are expressed in rat distal pulmonary arterial smooth muscle. Am J Physiol Lung Cell Mol Physiol 2004; 286: L848–L858.
- 20.
Tang C, To WK, Meng F, Wang Y, Gu Y. A role for receptor-operated Ca2+ entry in human pulmonary artery smooth muscle cells in response to hypoxia. Physiol Res 2010; 59: 909–918.
- 21.
Mohl MC, Iismaa SE, Xiao XH, Friedrich O, Wagner S, Nikolova-Krstevski V, et al. Regulation of murine cardiac contractility by activation of α(1A)-adrenergic receptor-operated Ca(2+) entry. Cardiovasc Res 2011; 91: 310–319.
- 22.
Roedding AS, Tong SY, Au-Yeung W, Li PP, Warsh JJ. Chronic oxidative stress modulates TRPC3 and TRPM2 channel expression and function in rat primary cortical neurons: Relevance to the pathophysiology of bipolar disorder. Brain Res 2013; 1517: 16–27.
- 23.
Malczyk M, Veith C, Fuchs B, Hofmann K, Storch U, Schermuly RT, et al. Classical transient receptor potential channel 1 in hypoxia-induced pulmonary hypertension. Am J Respir Crit Care Med 2013; 188: 1451–1459.
- 24.
Xu L, Chen Y, Yang K, Wang Y, Tian L, Zhang J, et al. Chronic hypoxia increases TRPC6 expression and basal intracellular Ca2+ concentration in rat distal pulmonary venous smooth muscle. PLoS One 2014; 9: e112007, doi:10.1371/journal.pone.0112007.
- 25.
Antigny F, Koenig S, Bernheim L, Frieden M. During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca2+ entry. J Cell Sci 2013; 126: 2525–2533.
- 26.
Yang XR, Lin MJ, McIntosh LS, Sham JS. Functional expression of transient receptor potential melastatin- and vanilloid-related channels in pulmonary arterial and aortic smooth muscle. Am J Physiol Lung Cell Mol Physiol 2006; 290: L1267–L1276.