Abstract
Background:
Brugada syndrome (BrS) is an inheritable cardiac disease associated with syncope, malignant ventricular arrhythmias and sudden cardiac death. The largest proportion of mutations in BrS is found in the
SCN5A
gene encoding the α-subunit of cardiac sodium channels (Nav1.5). Causal
SCN5A
mutations are present in 18–30% of BrS patients. The additional genetic diagnostic yield of variants in cardiac sodium channel β-subunits in BrS patients was explored and functional studies on 3 novel candidate variants were performed.
Methods and Results:
The
SCN1B-
SCN4B
genes were screened, which encode the 5 sodium channel β-subunits, in a
SCN5A
negative BrS population (n=74). Five novel variants were detected; in silico pathogenicity prediction classified 4 variants as possibly disease causing. Three variants were selected for functional study. These variants caused only limited alterations of Nav1.5 function. Next generation sequencing of a panel of 88 arrhythmia genes could not identify other major causal mutations.
Conclusions:
It was hypothesized that the studied variants are not the primary cause of BrS in these patients. However, because small functional effects of these β-subunit variants can be discriminated, they might contribute to the BrS phenotype and be considered a risk factor. The existence of these risk factors can give an explanation to the reduced penetrance and variable expressivity seen in this syndrome. We therefore recommend including the
SCN1-4B
genes in a next generation sequencing-based gene panel.
Brugada syndrome (BrS) is associated with syncope, malignant ventricular arrhythmias and sudden cardiac death (SCD).1
BrS is diagnosed based on a ST-segment elevation with a type 1 (or coved type) morphology on the electrocardiogram (ECG). This ST-segment elevation can occur spontaneously or can be evoked during a class I antiarrhythmic drug (AAD) test.1–4
BrS has a low prevalence of 1-5 per 10,000 in Europe and the USA, and an increased prevalence of 1-2.5 per 1,000 in South-East Asia.5,6
BrS is a genetic disease characterized by a dominant inheritance, reduced penetrance and variable expressivity.7
The predominant gene is
SCN5A
encoding Nav1.5, the pore-forming α-subunit of the cardiac sodium channels. Loss-of-function mutations in
SCN5A
can be found in 18–30% of the BrS patients.1
To date, 19 additional genes have been associated with BrS.8–10
These genes are considered to be minor BrS genes because mutations in them are rare.
The cardiac sodium channel consists of the Nav1.5 and associated regulatory proteins. Among these are the β-subunits, a family of 5 proteins: β1, β1b, β2, β3 and β4. These β-subunits interact directly with the Nav1.5 protein and are necessary for its proper function and expression.11
The β-subunits are encoded by 4 genes:
SCN1B
(encoding 2 isoforms: β1 and β1b),
SCN2B, SCN3B
and
SCN4B.
All β-subunits have been associated with BrS except for β4, which has been associated with long QT syndrome (LQTS).12–16
In this perspective, we consider these β-subunits suitable candidate genes for genetic testing in BrS patients.
In this study, we want to evaluate the genetic diagnostic yield of
SCN1-4B
genes by screening a
SCN5A
mutation-negative BrS population of 74 patients. We found 5 novel β-subunit variants, of which 4 were potentially disease causing. Functional studies were performed on 3 selected variants.
Methods
Population
This study was performed in accordance with the Declaration of Helsinki and approved by the Ethical committee of the UZ Brussel. Written informed consent was obtained from all patients.
We studied 74 unrelated BrS patients without
SCN5A
mutation(s) at the outpatient clinic of the UZ Brussels. Clinical diagnosis of BrS was made based on the appearance of a type 1 or coved type ECG recording characterized by a ST-segment elevation of ≥2 mm followed by a negative T-wave in at least 1 precordial lead (V1, V2) positioned in the 2nd, 3rd
or 4th
intercostal space.1–3
Blood samples for genetic analysis were collected between December 2007 and December 2011. Detailed clinical data were obtained, including symptoms, cardioverter defibrillator (ICD) implantation, standard 12-lead ECG and other cardiac rhythm registrations, results of a class I AAD provocation test, inducibility of sustained ventricular tachycardia/fibrillation (VT/VF) during electrophysiology study (EPS), and family history. ICD implantations at our center were performed conform to the international recommendations at the time of the implant.1,17–20
Genetic Screening
Total genomic DNA was isolated from whole blood samples by using standard techniques (Chemagen; PerkinElmer, Zaventem, Belgium).
After excluding mutations in
SCN5A
all exons, including intron-exon boundaries, of
SCN1B,
SCN2B,
SCN3B
and
SCN4B
were sequenced in both directions. In the case of
SCN1B,
we included the extended third exon to cover variants in β1b. Primers, PCR conditions and Sanger sequencing are described in
Supplementary File 1-1.
Results were compared to the reference sequence (NM_001037 for β1, NM_199037 for β1b, NM_004588 for β2, NM_018400 for β3 and NM_174934 for β4) using SeqPilot (version 4.0.1 build 502; JSI Medical systems GmbH, Kippenheim, Germany) and Variant Reporter v1.1 (Life Technologies). Human Genome Variant Society (HGVS) nomenclature was used (Alamut version 2.3 rev. 1; Interactive Biosoftware, Rouen, France).
Novel variants were submitted to the Leiden Open Variation Database (LOVD, http://www.LOVD.nl)
In Silico Prediction of the Variants
Variants were compared to online databases: 1,000 genomes, dbSNP, HGMD, LOVD, Cardiac arrhythmia database and literature (consulted October 2014). Minor allele frequency (MAF) was obtained from 1,000 genomes (1,000 genomes browser released October 2013; Ensembl v73, consulted June 2014). Variants were analyzed with different prediction tools: (1) Functional effects: GVGD classes (version October 2013), SIFT (version May 2009, consulted in June 2014) and Mutation Taster (built NCBI37/Ensembl69); (2) Conservation of amino acids: orthologue alignments (hg19, through Ensembl Compara database); and (3) Splice predictions: SpliceSiteFinder-like, MaxEntScan, GeneSplicer, Known constitutive signals, ESEFinder, RESCUE-ESE.
All prediction tools were consulted via Alamut (performed August 2014).
Variants were classified according to the system suggested by Hofman et al.21
This classification is based on the results of various prediction tools and categorizes the variants according to these results into 5 classes: (1) not pathogenic; (2) unlikely pathogenic (VUS1; variant of unknown significance 1); (3) unknown pathogenicity (VUS2); (4) likely pathogenic (VUS 3); and (5) pathogenic.
For novel variants in untranslated regions (UTRs), sequence comparison between different species was performed using Clustal Omega v2.122
(consulted in March 2014).
To predict putative microRNA binding, microRNA.org (August 2010 release, via Alamut, consulted in August 2014) and TargetScanHuman 6.2 prediction of microRNA targets23
(consulted in March 2014), were used.
Vectors and Site-Directed Mutagenesis
The β1b-WT vector is a pcDNA3.1 vector containing green fluorescent protein (GFP)13
and the wild-type
SCN1Bb
sequence.24
The β1b-del-ext vector was created based on the β1b-WT vector; genomic DNA of a control sample was used as a template to amplify the extended 3’ terminal region, and the deletion was introduced using site-directed mutagenesis (QuikChange II XL Site-Directed Mutagenesis Kit; Agilent Technologies, Santa Clara, CA, USA).
A β3-WT-containing pReceiver-M61 vector with GFP (GeneCopoeia Inc, Rockville, MD, USA) was used as a template to create the mutated β3-Gln101Lys (site-directed mutagenesis).
The
SCN4B
prom-MT vector was constructed using the Gluc-ON™Promoter Reporter Clone containing the
SCN4B
promoter region (c.−1333 to c.−1; GeneCopoeia Inc) using site-directed mutagenesis.
A detailed description of plasmid construction can be found in
Supplementary File 1-1.
The pXOOM-Kv4.3 vector was a kind gift from Prof. Kristine Callø (University of Copenhagen, Denmark).
The pcDNA3.1-Nav1.5 vector was described previously.13,24
Cells and Transfection
Human embryonic kidney cells (HEK cells, tsa201) and H9c2 cells (rat cardiomyocyte-like cells) were maintained in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine Serum, 1% antibiotics (penicillin/streptavidin) and 1% Glutamax (all from Invitrogen, Carlsbad, CA, USA) at 37℃ and 5% CO2.
HEK cells were used for patch clamp experiments. Cells were transfected using GeneCellin transfection reagent (BioCellChallenge, Toulon, France) in experiments with Nav1.5 or Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) in experiments with Kv4.3. The following molar ratios were used: Nav1.5:β1b-WT or β1b-del-ext 1:2.3; Kv4.3:β1b-WT or β1b-del-ext 1:2.3, and Nav1.5:β3-WT or β3-Gln101Lys 1:4; to ensure saturation of the α-subunit with β-subunits.24
A maximum of 2 µg DNA per transfection was used.
For promoter studies, HEK cells transfected with polyethyleneimine (Polysciences, Eppelheim, Germany; 1 µg or 3 µg of DNA) or H9c2 cells transfected with Lipofectamine 2000 (0.5 µg of DNA) were used.
Electrophysiological Studies
Experiments were performed at room temperature, 48 h after transfection using standard whole-cell patch clamp techniques. An Axopatch 200B amplifier and Digidata 1440A acquisition system (Molecular Devices, Sunnyvale, CA, USA) were used to obtain the data.
Data were filtered at 5 kHz and sampled at 5–20 kHz; 80–90% series resistance compensation was used. Recording voltages were not corrected for junction potential.
Solutions and patch clamp protocols are described in
Supplementary File 1-1.
Luciferase Assays
The medium of transfected HEK or H9c2 cells was harvested 48 or 72 h after transfection and frozen at −20℃ until the luciferase assay was performed.
The luciferase assay was performed according to the manufacturer’s instructions (Secrete-Pair™ Dual Luminescence and Gaussia Luciferase Assay Kits; GeneCopoeia Inc) using a GloMax 96 Microplate Luminometer (Promega, Madison, WI, USA) or a Spectramax M3 (Molecular Devices).
Next Generation Sequencing (NGS)
Library preparation was performed by using the KAPA Low-Throughput Library preparations kit (KAPA Biosystems Inc, Wilmington, MA, USA) or NebNext Ultra DNA Library prep Kit for Illumina (New England Biolabs Inc). Target enrichment was performed by using the SeqCap EZ Human Exome v3.0 kit (Roche NimbleGen Inc, Madison, WI, USA). Sequencing (paired-end 2×200 base pairs) was performed on an Illumina HiSeq 1500 (Illumina Inc) sequencing platform at 75× average coverage.
Reads were mapped to the human genome (Hg19, BWA-mem 0.7.5). Variants were called using GATK version 2.8 and annotated with Annovar (version August 2013; ljb23 version 2.3 database).
Eighty-eight arrhythmia genes were analyzed (gene panel available on request). Variant filtering was performed by using Microsoft Excel (Microsoft Office 2007) and confirmed by Sanger sequencing. To exclude NGS sequencing errors, 3 in-house control samples of healthy individuals were used. A more detailed description can be found in
Supplementary File 1-1.
Statistical Analysis
P-values were calculated using the N−1 χ2
test for binary data or the Student’s t-test for continuous data of the population screening. OriginPro 8 software (OriginLab Corporation, Northhampton, MA, USA) was used to analyze the patch clamp data. An ANOVA with the Bonferroni post-hoc test was used to determine significance.
Values are given as mean±standard error of mean (SEM) unless indicated otherwise. Results are considered significant when P<0.05.
Results
Population Screening
Our patient population consisted of 74 unrelated
SCN5A
mutation-negative BrS patients (65% male, average age±SD 47.69±14.56 years), 20% of the patients displayed a spontaneous type 1 ECG. Patient characteristics were stratified based on the variants present in the β-subunits (Table 1). No significant differences were observed between the populations (Table 1).
Table 1.
Patient Characteristics
| Characteristic |
Variables of the
SCN5A-negative
BrS study
population |
All SCN1–4B
variants (n=74) |
SCN1B
variants (n=47) |
SCN2B
variants (n=51) |
SCN3B
variants (n=67) |
SCN4B
variants (n=7) |
| |
Mean age (years) |
47.69±14.56 |
46.87±12.49 |
46.65±14.37 |
47.72±14.74 |
47.43±18.47 |
| |
Age range (years) |
[10–81] |
[21–75] |
[10–81] |
[10–81] |
[21–81] |
| |
Male, n (%) |
48 (65) |
30 (64) |
31 (61) |
42 (63) |
5 (71) |
| Ethnic group, n (%) |
Caucasian |
65 (88) |
39 (83) |
43 (84) |
65 (97) |
6 (86) |
| African |
3 (4) |
3 (6) |
3 (6) |
3 (4) |
1 (14) |
| Asian |
2 (3) |
2 (4) |
2 (4) |
2 (3) |
0 (0) |
| Unknown |
4 (5) |
3 (6) |
3 (6) |
3 (4) |
0 (0) |
| ECG features, n (%) |
Spont. type 1 ECG |
13 (18) |
11 (23) |
10 (20) |
12 (18) |
0 (0) |
| Dynamic ECG |
24 (32) |
17 (36) |
20 (39) |
21 (31) |
2 (29) |
| Pos. Prov. Test |
72 (97) |
46 (98) |
49 (96) |
65 (97) |
7 (100) |
Clinical presentation,
n (%) |
Syncope |
37 (50) |
22 (47) |
25 (49) |
32 (48) |
2 (29) |
| Spont. AF |
11 (15) |
7 (15) |
9 (18) |
11 (16) |
0 (0) |
| Spont. VF |
7 (9) |
4 (8) |
7 (14) |
7 (10) |
0 (0) |
| Spont. VT |
6 (8) |
4 (8) |
4 (8) |
5 (7) |
0 (0) |
| ACA |
4 (5) |
3 (6) |
4 (8) |
4 (6) |
0 (0) |
| EPS performed |
62 (84) |
41 (87) |
42 (82) |
56 (84) |
6 (86) |
| EPS positive |
12 (16) |
8 (17) |
6 (12) |
10 (15) |
1 (14) |
| ICD |
44 (59) |
28 (60) |
28 (55) |
38 (57) |
2 (29) |
| Family history, n (%) |
BrS |
23 (31) |
15 (32) |
16 (31) |
20 (30) |
3 (43) |
| SCD |
39 (53) |
23 (49) |
29 (57) |
36 (54) |
5 (71) |
Syncope, VT/VF,
ACA |
18 (24) |
7 (15) |
12 (24) |
16 (24) |
1 (14) |
Age is indicated as mean±SD. Values in parentheses (except for age range) are percentages. ACA, aborted cardiac arrest; AF, atrial fibrillation; BrS, Brugada syndrome; ECG, electrocardiogram; EPS, electrophysiological study; ICD, implantable cardioverter defibrillator; pos. prov. test, a positive class I AAD provocative test; SCD, sudden cardiac death; spont, spontaneous; VF, sustained ventricular fibrillation; VT, sustained ventricular tachycardia.
A total of 30 sequence variations were found (Table 2). A study of the literature and various databases did not reveal any known mutations among the variants. Five of these 30 variants were novel variants.
Table 2.
Variants Found in the Study Population
| Gene |
cDNA |
Protein |
rs-number |
MAF study
population |
MAF general
population |
Hofman
classification |
| SCN1B |
| exon 1 |
40+15G>T |
|
rs72556351 |
0.122 |
0.168 |
Not pathogenic |
| exon 2 |
207+55G>A |
|
rs147990128 |
0.007 |
0.003 |
Not pathogenic |
| exon 3B |
622_623delCT$ |
Leu208Valfs*99 |
|
0.007 |
|
VUS2 |
| 629T>C |
Leu210Pro |
rs55742440 |
0.351 |
0.38 |
Not pathogenic |
| 744C>A |
Ser248Arg |
rs67701503 |
0.203* |
0.133 |
Not pathogenic |
| 749G>C |
Arg250Thr |
rs67486287 |
0.203* |
0.129 |
Not pathogenic |
| 769G>A |
Gly257Arg |
rs72558028 |
0.007 |
NA |
VUS1 |
| *85G>A |
|
rs375266320 |
0.007 |
NA |
VUS2 |
| exon 4 |
501T>C |
Ile167Ile |
rs16969930 |
0.027 |
0.014 |
Not pathogenic |
| exon 5 |
591–25T>G |
|
rs28365107 |
0.014 |
0.006 |
Not pathogenic |
| 591–14C>A |
|
rs28365109 |
0.014 |
0.006 |
Not pathogenic |
| *5+31G>A |
|
rs28365108 |
0.014 |
0.001 |
Not pathogenic |
| SCN2B |
| exon 1 |
–212C>T$ |
|
|
0.007 |
|
VUS2 |
| 70+11_70+12insTC |
|
rs72544143 |
0.014 |
0.008 |
Not pathogenic |
| exon 2 |
237+27A>G |
|
rs645675 |
0.027** |
0.201 |
Not pathogenic |
| exon 4 |
449–12C>A |
|
rs8192613 |
0.588** |
0.463 |
Not pathogenic |
| *38C>T |
|
rs8192614 |
0.068 |
0.072 |
Not pathogenic |
| SCN3B |
| exon1 |
–763G>A |
|
rs12420563 |
0.169 |
0.148 |
Not pathogenic |
| –692G>C |
|
rs7483687 |
0.169 |
0.16 |
Not pathogenic |
| –234G>A |
|
rs3851104 |
0.169 |
0.153 |
Not pathogenic |
| –181A>G |
|
rs3851103 |
0.169 |
0.154 |
Not pathogenic |
| 55+44C>T |
|
rs3851102 |
0.162 |
0.158 |
Not pathogenic |
| exon 3 |
301C>A$ |
Gln101Lys |
|
0.007 |
|
VUS2 |
| 390G>A |
Ala130Ala |
rs148484744 |
0.007 |
0.002 |
Not pathogenic |
| 438C>T |
Thr146Thr |
rs1275085 |
0.108 |
0.068 |
Not pathogenic |
| exon 5 |
585–45G>C |
|
rs1148110 |
0.372* |
0.289 |
Not pathogenic |
| SCN4B |
| exon 1 |
–137T>C$ |
|
|
0.007 |
|
VUS2 |
| exon 2 |
174C>T |
Cys58Cys |
rs45539032 |
0.014 |
0.031 |
Not pathogenic |
| exon 3 |
295G>A$ |
Asp99Asn |
|
0.007 |
|
Not pathogenic |
| exon 5 |
639C>T |
Asn213Asn |
rs72544155 |
0.020 |
0.001 |
Not pathogenic |
The minor allele frequency (MAF) of the study, which was significantly different to the general population, is indicated: *P<0.05, **P<0.01. Novel variants are indicated with a $-sign. The MAF of variant c.449–12C>A in SCN2B and c.558–45G>C in SCN3B correlates with the C and G frequencies, respectively. Variants in SCN1B exon 3 are only given as variants in exon 3B (NM_199037) because they all occur in the intronic region of β1 (NM_001037). MAF, minor allele frequency; VUS1, unlikely pathogenic; VUS2, unclear pathogenicity (according to Hofman et al21).
When compared to the normal population, the MAF of 5 single nucleotide polymorphisms (SNPs) were found to be statistically different (Table 2). All variants were fed into multiple in silico prediction tools. These results were used to classify the variants according to the system suggested by Hofman et al (Table 2).21
Additional In Silico Prediction Analysis of Candidate Variants
Further in silico analysis was performed on novel variants and variants classified as VUS 2 (unknown pathogenicity).
The 2 base pair deletion (Figure 1A) in β1b (SCN1Bb, c.622_623delCT, p.Leu208Valfs*99) causes a frameshift, which abolishes the normal stop codon and introduces a novel stop codon further downstream. This completely changes the C-terminal part of the protein starting at amino acid 208. The second variant in β1 could potentially affect both isoforms of
SCN1B.
In β1, this variant (c.448+444G>A,
Figure 1A) was not predicted to have an effect on splicing. In β1b, the same variant is located in the 3’UTR (c.*85G>A,
Figure 1A). Prediction tools for microRNA binding did not reveal any potential effect on the electrophysiology of the heart (Supplementary File 1-2). In addition, this nucleotide was not conserved (Figure 1B). The
SCN2B
c.−212C>T variant (Figure 1A) is a conserved nucleotide (Figure 1C). The β3 missense variant (c.301C>A, p.Gln101Lys,
Figure 1A) involves a conserved amino acid. This change is predicted to be disease causing by all in silico prediction tools. The
SCN4B
promoter variant (c.−137T>C,
Figure 1A) is a conserved nucleotide, located in a stretch of 11 conserved nucleotides (Figure 1D).

The novel variant in exon 3 of β4, c.295G>A (p.Asp99Asn,
Figure 1A), is predicted to be not pathogenic (Table 2).
Symptoms and Family Information
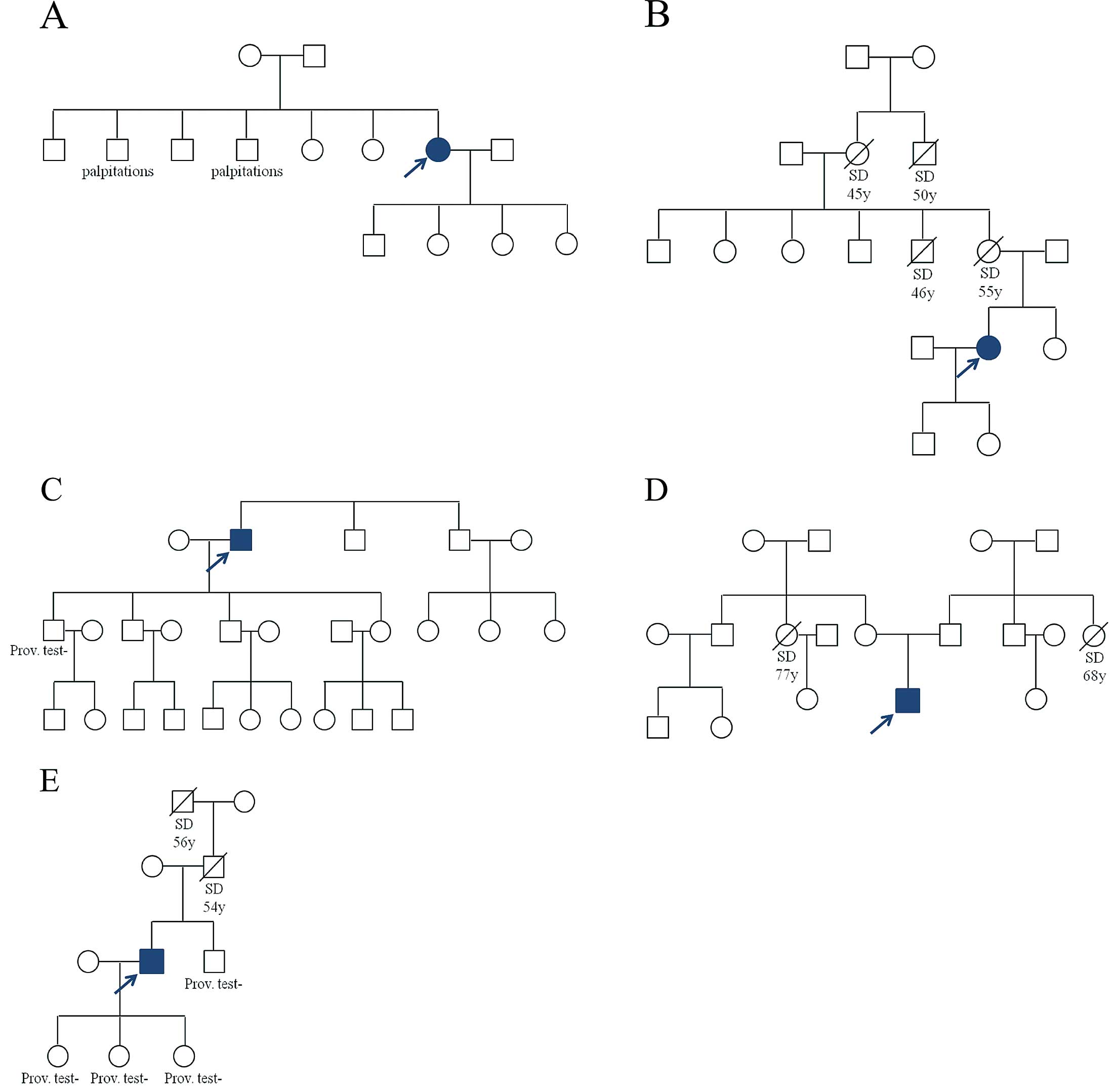
The patient carrying the deletion in β1b complained of dizziness and presyncope during a febril episode. Two of her brothers suffer from palpitations (Figure 2A). They could not be examined in our hospital as they live abroad. The
SCN1Bb
c.*85G>A variant was found in a patient with a history of syncope during exercise. The patient reported multiple cases of SCD in the family (Figure 2B).
The patient with the
SCN2B
promoter variant had a syncope while sitting at the breakfast table. Neither his brothers nor children display cardiac symptoms. One son tested negative for BrS (Figure 2C).
The β3-p.Gln101Lys variant was found in a patient who had syncope while driving his car and during a sauna visit. The patient displayed a spontaneous type 2 BrS ECG and was diagnosed after a positive class I AAD provocation test. He has neither siblings nor children (Figure 2D). The patient reported SCD of a maternal and paternal aunt at the age of 77 and 68 years, respectively.
A second promoter variant (SCN4B,
c.−137T>C) was found in a patient who had an episode of syncope during the day, sitting at his desk. Both his father and grandfather died suddenly in the fifth decade of their life. His brother and 3 daughters all tested negative for BrS (Figure 2E).
Based on the results of the prediction tools and family information, we selected 3 novel variants for further functional study: β1b pLeu208Valfs*99, β3 Gln101Lys and
SCN4B
c.−137T>C.
Functional Effects of β1b-del-ext
Cellular electrophysiology was performed to study the effect of β1b p.Leu208Valfs*99 on the sodium current (INa). HEK cells were transfected with Nav1.5 in combination with wild-type (Nav1.5+β1b-WT) or p.Leu208Valfs*99 (Nav1.5+β1b-del-ext) β1b. No significant difference was seen in the peak INa
density (Table 3, Figures 3A,B). The inactivation voltage-dependence showed a significant negative shift (P<0.01) and the activation curve displayed a small positive shift (P<0.01,
Table 3, Figure 3C) due to the variant. There was a significant difference in the slope factor of the activation voltage-dependence (P<0.01,
Table 3, Figure 3C). These alterations shifted the window current (Figure 3D).
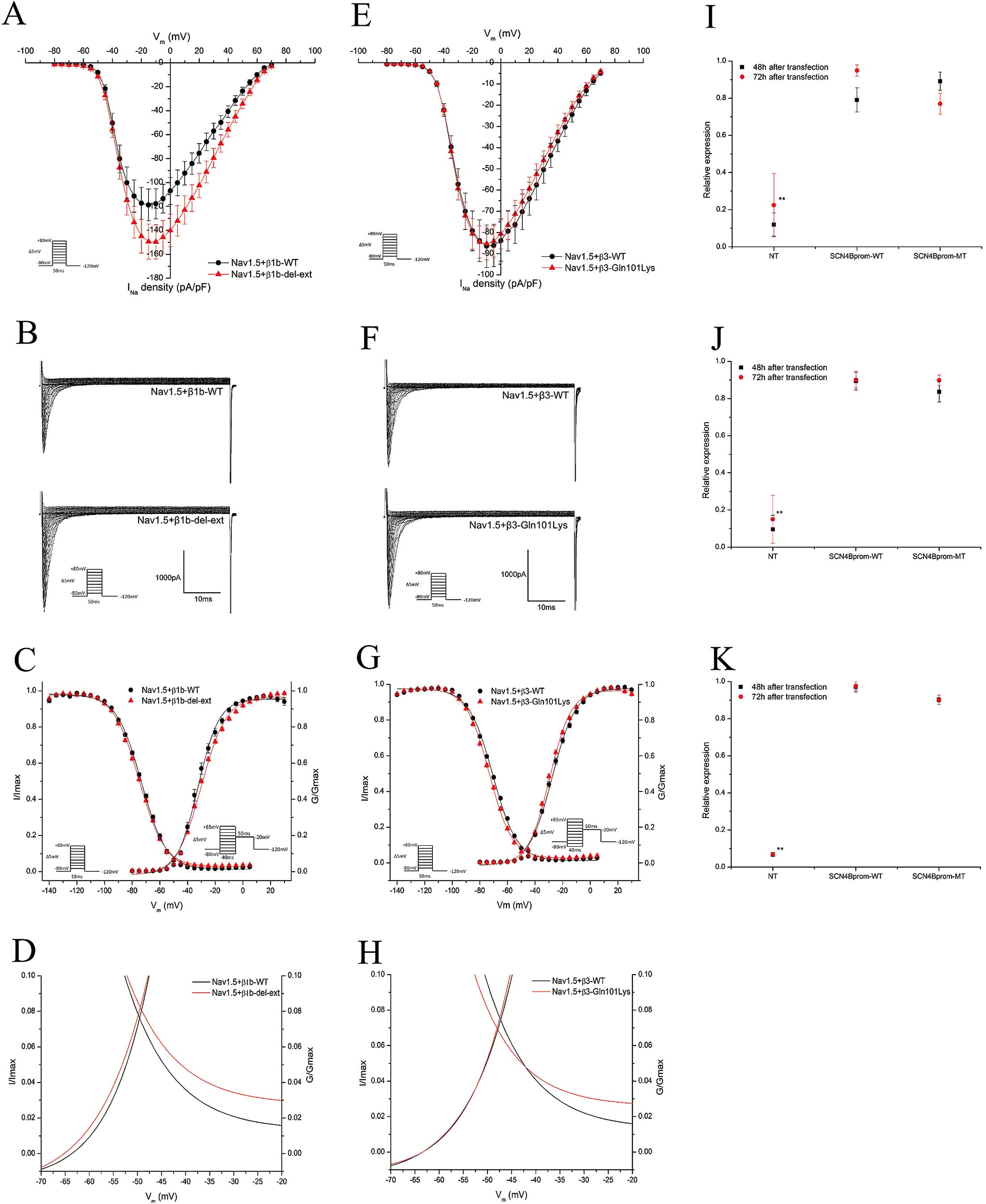
Table 3.
Patch Clamp Results
| |
I–V curve |
Inactivation voltage-dependence |
Activation voltage-dependence |
Recovery from inactivation |
τ of inactivation |
Voltage-dependent slow inactivation |
| Peak INa (pA/pF) |
n |
V1/2 (mV) |
Slope (k) |
n |
V1/2 (mV) |
Slope (k) |
n |
τ (ms) |
n |
τ (ms) |
n |
V1/2 (mV) |
Slope (k) |
n |
| Nav1.5+β1b-WT |
−118.68±13.24 |
17–20 |
−73.40±0.16* |
8.95±0.14 |
19–20 |
−32.38±0.31* |
7.59±0.28* |
20 |
4.37±0.07 |
16–18 |
11.37±1.09 |
20 |
−84.03±1.62 |
6.76±1.40 |
14–15 |
| Nav1.5+β1b-del-ext |
−149.36±14.72 |
18–19 |
−75.03±0.14* |
9.13±0.12 |
30–31 |
−29.93±0.18* |
8.97±0.16* |
19 |
4.38±0.05 |
19–21 |
13.44±0.78 |
19 |
−88.80±1.76 |
4.27±1.70 |
13 |
| Nav1.5+β3-WT |
−86.46±10.02 |
22–23 |
−70.69±0.13* |
8.71±0.12 |
23–29 |
−27.54±0.18* |
8.50±0.17* |
21–23 |
4.05±0.06 |
20–21 |
12.41±0.57 |
21–23 |
−84.02±2.05 |
5.01±1.70 |
17 |
| Nav1.5+β3-Gln101Lys |
−85.32±7.60 |
24–25 |
−73.21±0.15* |
8.32±0.13 |
25–27 |
−29.43±0.16* |
7.77±0.14* |
25 |
3.93±0.07 |
23–25 |
12.66±1.00 |
25 |
−84.37±1.95 |
4.77±1.58 |
17–18 |
Values represent mean±SEM. *P<0.01. Voltage-dependence of activation, inactivation and voltage-dependent slow inactivation parameters were calculated using a Boltzmann function, where V1/2
is the half-maximal value (Supplementary File 1-1). For recovery from inactivation, the ratio of the current values of the second pulse and the first pulse were fitted with an exponential function to determine the time constant, τ. INa, sodium current.
No difference could be found in the recovery from inactivation, voltage-dependent and time-dependent slow inactivation, or the τ of inactivation between the wild-type and variant β1b (Table 3,
Supplementary File 1-3).
Given the known interaction between β1b and Kv4.3, responsible for the transient outward current (Ito) during the notch phase of the cardiac action potential,12
we analyzed the effects of β1b-del-ext on the Kv4.3 channel function. The plasmid expressing Kv4.3 was transfected into HEK cells in combination with the wild-type (Kv4.3+β1b-WT) or variant (Kv4.3+β1b-del-ext) β1b; however, no significant differences were found (Supplementary File 1-3).
Functional Effects of β3-Gln101Lys
To assess the functional effects of β3-Gln101Lys on the Nav1.5 function, HEK cells were transfected with Nav1.5 and β3 wild-type (Nav1.5+β3-WT) or β3-Gln101Lys (Nav1.5+β3-Gln101Lys). No difference in the peak INa
density was observed (Table 3, Figures 3E,F). Analyzing the inactivation voltage-dependence, a shift to more negative potentials was observed with β3-Gln101Lys (P<0.01,
Table 3, Figure 3G). β3-Gln101Lys also shifted the activation curve to more negative potentials (P<0.01,
Table 3, Figure 3G). These differences in activation and inactivation caused small changes in the window current (Figure 3H). No differences were observed when comparing recovery from inactivation, τ of inactivation, and voltage-dependent or time-dependent slow inactivation (Table 3,
Supplementary File 1-3).
Promoter Study of β4, c. −137T>C
HEK cells transfected with either the wild-type (SCN4B
prom-WT) or variant promoter (SCN4B
prom-MT) were studied 48 h and 72 h after transfection (Figure 3I,J). These experiments did not reveal any significant difference in promoter activity. Because HEK cells are cells of a non-cardiac lineage, the experiment was repeated in rat cardiomyocyte-like H9c2 cells. These cells are more likely to express the correct transcription factors and therefore would better mimic the physiological conditions. However, similar to the HEK cells, no difference could be detected between the wild-type and the variant promoter (Figure 3K).
Arrhythmia Gene Panel Analysis
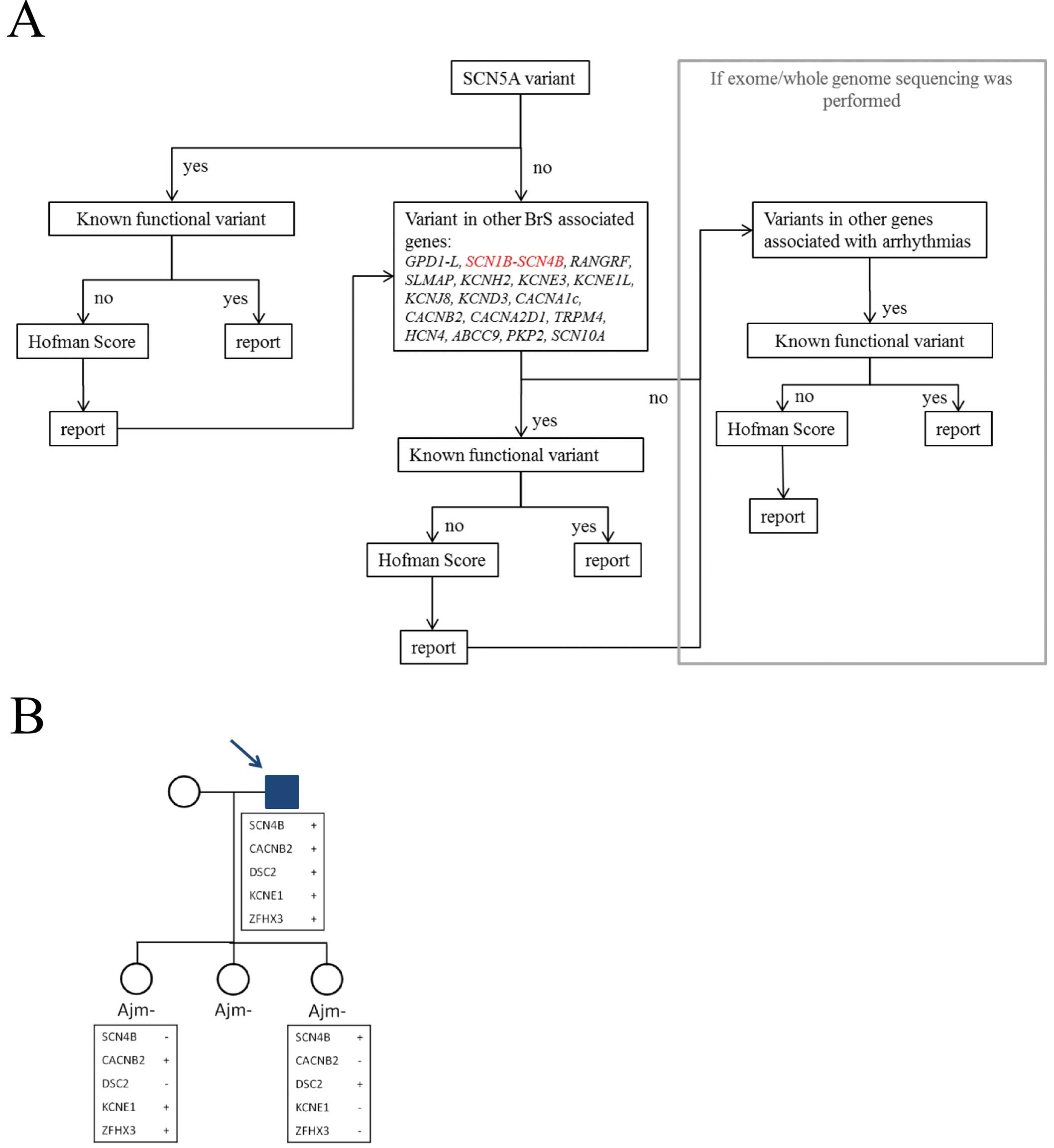
Because functional analysis did not indicate any of the variants as the sole cause of BrS, the patients were screened for a set of 88 cardiac arrhythmia genes using massive parallel screening (MPS). Only coding variants or variants affecting splice sites with a MAF of less than 5% were considered. A relative high MAF cut-off value was used to avoid the exclusion of variants associated with BrS, because they can be found in 4.35% of the overall population.25
In a first step, we analyzed the 20 genes already described in BrS; subsequently the other arrhythmia associated genes were studied (Figure 4A).
MPS detected 15 additional variants (Table 4): 5 novel and 10 previously described. All variants were further classified according to Hofman et al (Table 4).21
Six variants were classified as VUS1 (unlikely pathogenic), 7 as VUS2 (unclear pathogenicity) and 5 as not pathogenic.
Table 4.
Results of MPS
| Chromosome (chr) |
Reference
allele |
Alternative
allele |
Gene |
Effect |
cDNA |
Protein |
rs number |
MAF |
Associated
disease |
Described
variant |
Score |
| Patient 1 |
| chr1 |
T |
C |
RYR2 |
Missense |
13969T>C |
Tyr4657His |
– |
– |
CPVT, ARVD, SD |
|
VUS1 |
| chr2 |
A |
C |
SNTG2 |
Missense |
149A>C |
Glu50Ala |
rs142024310 |
0.008 |
regulation of SCN5A |
|
VUS1 |
| chr2 |
T |
C |
SCN7A |
Missense |
3805A>G |
Met1269Val |
– |
– |
arrhythmia |
|
VUS2 |
| chr10 |
C |
T |
ANK3 |
Missense |
6955G>A |
Asp2319Asn |
rs140463162 |
0.003 |
arrhythmia |
|
VUS1 |
| chr14 |
– |
G |
MYH7 |
Splicing |
3337–3dupG |
– |
rs45504498 |
NA |
HCM, DCM, LVNC, SD |
|
VUS1 |
| chr16 |
A |
C |
ZFHX3 |
Missense |
5516T>G |
Val1839Gly |
– |
– |
arrhythmia |
|
VUS2 |
| chr18 |
A |
C |
DSG2 |
Missense |
2098A>C |
Ser700Arg |
– |
– |
ARVD, DCM, SD |
|
VUS2 |
| chr19$ |
CT |
– |
SCN1B |
Frameshift |
622_623delCT |
Leu208Valfs*99 |
– |
– |
BrS, AF, CCD, SD |
|
VUS2 |
| Patient 2 |
| chr1 |
G |
A |
GJA5 |
Missense |
793C>T |
Pro265Ser |
rs148311482 |
0 |
AF, SD |
ToF |
VUS1 |
| chr7 |
C |
T |
AKAP9 |
Missense |
139C>T |
His47Tyr |
rs35669569 |
0.003 |
LQTS, SD |
BV |
Not pathogenic |
| chr11$ |
G |
T |
SCN3B |
Missense |
301C>A |
Gln101Lys |
– |
– |
BrS, IVF, ERS, SD |
|
VUS2 |
| chr15 |
C |
T |
HCN4 |
Missense |
2275G>A |
Val759Ile |
rs62641689 |
0.003 |
BrS, IVF, ERS, SSS |
SIDS, SUDEP |
Not pathogenic |
| chr16 |
C |
G |
ZFHX3 |
Missense |
6042G>C |
Gln2014His |
rs62051555 |
0.022 |
arrhythmia |
|
Not pathogenic |
| Patient 3 |
| chr10 |
C |
G |
CACNB2 |
Missense |
1651C>G |
Arg551Gly |
rs61733968 |
0.007 |
BrS, SQTS, SD |
BV? |
VUS1 |
| chr11$ |
A |
G |
SCN4B |
Promoter function |
–137T>C |
– |
– |
– |
LQTS |
|
VUS2 |
| chr16 |
C |
A |
ZFHX3 |
Missense |
1563G>T |
Lys521Asn |
rs62640007 |
0.001 |
arrhythmia |
|
Not pathogenic |
| chr18 |
C |
T |
DSC2 |
Missense |
2393G>A |
Arg798Gln |
rs61731921 |
0.029 |
ARVD, SD |
BV |
Not pathogenic |
| chr21 |
C |
A |
KCNE1 |
Missense |
200G>T |
Arg67Leu |
– |
– |
LQTS, JLNS, SD |
|
VUS2 |
Variants were filtered according to the arrhythmia gene panel. The variants that were studied with patch clamp or promoter studies are indicated with a $-sign. ARVD, arrhythmogenic right ventricular dysplasia; BV, benign variant; BV?, probably benign variant; found in a technical control; CCD, cardiac conduction disease; CPVT, catecholaminergic polymorphic ventricular tachycardia; DCM, dilated cardiomyopathy; ERS, exercise-related syncope; HCM, hypertrophic cardiomyopathy; IVF, idiopathic ventricular fibrillation; JLNS, Jervell and Lange-Nielsen syndrome; LQTS, long QT syndrome; LVNC, left ventricular non-compaction; MPS, massive parallel screening; SD, sudden death; SIDS, sudden infant death syndrome; SQTS, short QT syndrome; SSS, sick sinus syndrome; SUDEP, sudden unexplained death in epilepsy; ToF, tetralogy of Fallot. Variants are classified according to Hofman et al.21 Other abbreviations as in Tables 1,2.
In patient 1, we found 8 variants, including the β1b p.Leu208Valfs*99 (Table 4). Four variants were classified as possibly pathogenic: 3 missense mutations (SCN7A,
ZFHX3
and
DSG2) and the β1b deletion. Except for
SCN1Bb, none of the genes have been described in BrS; no functional studies concerning these variants were described.
The β3-p.Gln101Lys carrying patient (patient 2) had 4 additional missense variants (GJA5,
AKAP9,
HCN4
and
ZFHX3); none of them are classified as possibly pathogenic (Table 4). The
HCN4
gene is associated with BrS8
and other arrhythmias. The variant found here has not been described in BrS, but in sudden infant death syndrome (SIDS) and sudden unexplained death in epilepsy.26,27
Its BrS disease-causing effect is questionable due to a lack of further functional evidence and the non-pathogenic Hofman score.
Besides the studied promoter variant, 4 additional missense variants were found in patient 3 (CACNB2,
ZFHX3,
DSC2
and
KCNE1). The
CACNB2
gene is associated with BrS;8
however, this particular mutation seems to be benign, as it was present in 1 of our technical control samples.
KCNE1
is associated with LQTS.26
Changes of this particular amino acid (p.Arg67) to other amino acids have been found in patients with LQTS and sudden unexplained death;28–31
however, the variant described here has not been reported yet. Two variants were predicted to be possibly pathogenic: the
SCN4B
promoter variant and the
KCNE1
missense variant. Additional familial segregation analysis was performed in the proband and 2 of his daughters (Figure 4B). Each variant observed in the proband was also present in one of the 2 BrS negative daughters, but never in both daughters; one daughter carried 2 of the 5 variants and the other daughter carried the 3 other variants.
Discussion
Screening of the 4 β-subunits leads to a potential supplemental genetic diagnostic yield of 5.40% (4 out of 74 patients) based on exclusion of common variants, the results of in vitro prediction tools and Hofman classification. The prevalence of potential disease-associated variants in the β-subunit genes is thus comparable to other minor BrS genes. This correlates with what has been described in the literature.32
A functional study of the variant β1b showed a significant shift of the activation and inactivation voltage-dependence compared to the wild-type. However, no significant decrease in INa
was found. Based on these results, we assume that this mutation is not the major cause of BrS in this patient.
It is surprising that the deletion, creating a completely different and extended C-terminus, only causes small effects on the Nav1.5 function. The frameshift changes 2 amino acids already associated with an impaired function of β1b:12,24
a known BrS and SIDS-associated mutation12
(p.Arg214Gln) and a LQTS-associated mutation24
(p.Pro213Tyr). Both missense mutations have been shown to nearly obliterate the effect of β1b on Nav1.5,12,24
indicating the importance of the C-terminus.
β1b can also associate with the cardiac potassium channel Kv4.3,12
which is responsible for Ito.
However, no differences in Ito
caused by the variant β1b compared to the wild type β1b were detected.
The β3-Gln101Lys electrophysiology studies displayed negative shifts in activation and inactivation voltage-dependence. However, this was not further supported by a decrease of INa, indicating that the global effect of this variant is too small to be the single cause of BrS in this patient. This was unexpected since all in silico prediction tools classified this variant as disease causing. Two mutations associated with BrS have been described in the β3 protein: p.Lys10Pro14
and p.Val110Ile15; the latter is located close to the region of our variant, which suggested that this might be an important region.
The β4-subunit has not been associated with BrS yet, but it is linked with LQTS.16
Little is known about the structure and function of the promoter regions of the β-subunits. The
SCN4B
c.−137T>C promoter variant was selected based on its location in a stretch of 11 conserved nucleotides. Neither HEK cells nor H9c2 cells showed any difference in expression caused by this variant, which suggests that this nucleotide and/or region is not involved in the promoter function of
SCN4B.
Although Juang et al33
demonstrated a high concordance rate (83.3%) between in silico prediction tools and the results of in vitro functional experiments for
SCN5A, this rate does not seem applicable to the β-subunit genes as shown by our study; this is possibly due to the fact that β-subunits are not as well characterized and studied as the α-subunit.33
MPS analysis revealed that, at least in patients 1 and 2, the other identified variants were not better potential disease-causing candidates. Further familial segregation analysis could add support, but unfortunately no family samples were accessible.
In patient 3, the familial segregation analysis revealed discordant results, indicating that none of the variants alone are the major cause of BrS in the patient. These findings can, however, not rule out a cumulative effect of the variants in the father.
Limitations of the Study
Only limited familial segregation studies could be performed due to the small pedigrees and restricted accessibility of family samples. Patch clamp recordings were performed at room temperature for technical reasons.
Conclusions
Our study demonstrates that the classification of variants, based on prediction tools alone, should be interpreted with caution. It stresses the importance of familial segregation analysis and functional studies. Unfortunately, it is often not possible to assess multiple family members. Certainly in such situations, functional studies are of great added value.
Our electrophysiological experiments demonstrate that the variants studied here cannot solely explain the existence of BrS in these patients. This supports the concept of BrS as a complex genetic disorder rather than a clear monogenic disease.34
The discordant familial segregation in patient 3 adds support to the hypothesis of an additive and/or synergistic effect of genetic variants. The genetic background, possibly together with environmental factors, could explain the reduced penetrance and variable expressivity that is seen in BrS.
We suggest including these
SCN1-4B
genes together with the other minor and major BrS genes in MPS-based gene panels and whole exome genetic analysis (Figure 4A).
Acknowledgments
We would like to thank Kristof Endels for his technical support, Ben Caljon and Didier Croes of the BRIGHTcore (BRussels Interuniversity Genomics High Throughput core, www.brightcore.be) for their help with the MPS, and Pedro Beltran-Alvarez for his help with the construction of the expression vectors.
This work was supported by UMCOR financing: Wetenschappelijk fonds Willy Gepts 2012 and 2010; UZ Brussels financing: WOK Heart Rhythm Management Centre, and WOK Centre for Medical Genetics; Vrije Universiteit Brussel (VUB) financing: REGE basisfinanciering and OZR2434; and Erasmus mobility 2013/2014.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Files
Supplementary File 1
1. Methods
Table S1.
Primers and PCR conditions for the SCN1B to SCN4B genes
2. Potential changes in microRNA binding due to the β1b *85G>A variant
Table S2.
Potential changes in microRNA binding sites due to the β1b *85G>A variant
3. Additional patch clamp results
Figure S1.
Electrophysiological effects of β1b-del-ext on the α-subunit of the cardiac sodium channel (Nav1.5).
Table S3.
Electrophysiological effects of β1b-del-ext on the α-subunit of the cardiac sodium channel (Nav1.5)
Figure S2.
Electrophysiological effects of β1b-del-ext on the transient outward current-potassium channel (Kv4.3).
Table S4.
Electrophysiological effects of β1b-del-ext on the transient outward current-potassium channel (Kv4.3)
Figure S3.
Electrophysiological effects of β3-Gln101Lys on the α-subunit of the cardiac sodium channel (Nav1.5).
Table S5.
Electrophysiological effects of β3-Gln101Lys on the α-subunit of the cardiac sodium channel (Nav1.5)
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-15-0164
References
- 1.
Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: Report of the second consensus conference: Endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation 2005; 111: 659–670.
- 2.
Bayés de Luna A, Brugada J, Baranchuk A, Borggrefe M, Breithardt G, Goldwasser D, et al. Current electrocardiographic criteria for diagnosis of Brugada pattern: A consensus report. J Electrocardiol 2012; 45: 433–442.
- 3.
Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, et al. Executive summary: HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes. Europace 2013; 15: 1389–1406.
- 4.
Shimizu W. Update of diagnosis and management of inherited cardiac arrhythmias. Circ J 2013; 77: 2867–2872.
- 5.
Murakoshi N, Aonuma K. Epidemiology of arrhythmias and sudden cardiac death in Asia. Circ J 2013; 77: 2419–2431.
- 6.
Benito B, Brugada R, Brugada J, Brugada P. Brugada syndrome. Prog Cardiovasc Dis 2008; 51: 1–22.
- 7.
Di Diego JM, Cordeiro JM, Goodrow RJ, Fish JM, Zygmunt AC, Pérez GJ, et al. Ionic and cellular basis for the predominance of the Brugada syndrome phenotype in males. Circulation 2002; 106: 2004–2011.
- 8.
Nielsen MW, Holst AG, Olesen SP, Olesen MS. The genetic component of Brugada syndrome. Front Physiol 2013; 4: 179.
- 9.
Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol 2014; 64: 66–79.
- 10.
Hu D, Barajas-Martínez H, Terzic A, Park S, Pfeiffer R, Burashnikov E, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol 2014; 171: 431–442.
- 11.
Wilde AA, Brugada R. Phenotypical manifestations of mutations in the genes encoding subunits of the cardiac sodium channel. Circ Res 2011; 108: 884–897.
- 12.
Hu D, Barajas-Martinez H, Medeiros-Domingo A, Crotti L, Veltmann C, Schimpf R, et al. A novel rare variant in SCN1Bb linked to Brugada syndrome and SIDS by combined modulation of Nav1.5 and Kv4.3 channel currents. Heart Rhythm 2012; 9: 760–769.
- 13.
Riuró H, Beltran-Alvarez P, Tarradas A, Selga E, Campuzano O, Vergés M, et al. A missense mutation in the sodium channel β2 subunit reveals SCN2B as a new candidate gene for Brugada syndrome. Hum Mutat 2013; 34: 961–966.
- 14.
Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, et al. A mutation in the β3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype. Circ Cardiovasc Genet 2009; 2: 270–278.
- 15.
Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, et al. Novel SCN3B mutation associated with Brugada syndrome affects intracellular trafficking and function of Nav1.5. Circ J 2013; 77: 959–967.
- 16.
Medeiros-Domingo A, Kaku T, Tester DJ, Iturralde-Torres P, Itty A, Ye B, et al. SCN4B-encoded sodium channel beta4 subunit in congenital long-QT syndrome. Circulation 2007; 116: 134–142.
- 17.
Priori SG. Genetic testing to predict sudden cardiac death: Current perspectives and future goals. Indian Heart J 2013; 66(Suppl 1): S58–S60.
- 18.
Zipes DP, Camm AJ, Borggrefe M, Buxton AE, Chaitman B, Fromer M, et al. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: A report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines. Circulation 2006; 114: e385–e484, doi:10.1161/CIRCULATIONAHA.106.178233.
- 19.
JCS Joint Working Group. Guidelines for clinical cardiac electrophysiologic studies (JCS 2011): Digest version. Circ J 2013; 77: 497–518.
- 20.
JSC Joint Working Group. Guidelines for non-pharmacotherapy of cardiac arrhythmias (JCS 2011): Digest version. Circ J 2013; 77: 249–274.
- 21.
Hofman N, Tan HL, Alders M, Kolder I, de Haij S, Mannens MM, et al. Yield of molecular and clinical testing for arrhythmia syndromes: Report of 15 years’ experience. Circulation 2013; 128: 1513–1521.
- 22.
Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011; 7: 539.
- 23.
Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decreases the proficiency of Isy-6 and other miRNA’s. Nat Struct Mol Biol 2011; 18: 1139–1146.
- 24.
Riuró H, Campuzano O, Arbelo E, Iglesias A, Batlle M, Pérez-Villa F, et al. A missense mutation in the sodium channel β1b subunit reveals SCN1B as a susceptibility gene underlying LQT Syndrome. Heart Rhythm 2014; 11: 1202–1209.
- 25.
Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunsø S, Sadjadieh A, et al. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet 2013; 1992: 1–7.
- 26.
Evans A, Bagnall RD, Duflou J, Semsarian C. Postmortem review and genetic analysis in sudden infant death syndrome: An 11-year review. Hum Pathol 2013; 44: 1730–1736.
- 27.
Tu E, Waterhouse L, Duflou J, Bagnall RD, Semsarian C. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol 2011; 21: 692–698.
- 28.
Skinner JR, Crawford J, Smith W, Aitken A, Heaven D, Evans CA, et al. Prospective, population-based long QT molecular autopsy study of postmortem negative sudden death in 1 to 40 year olds. Heart Rhythm 2011; 8: 412–419.
- 29.
Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, et al. The genetic basis of long QT and short QT syndromes: A mutation update. Hum Mutat 2009; 30: 1486–1511.
- 30.
Kapplinger J, Tester D, Salisbury B, Carr J, Harris-Kerr C, Pollevick GD, et al. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION® long QT syndrome genetic test. Heart Rhythm 2009; 6: 1297–1303.
- 31.
Wang D, Shah KR, Um SY, Eng LS, Zhou B, Lin Y, et al. Cardiac channelopathy testing in 274 ethnically diverse sudden unexplained deaths. Forensic Sci Int 2014; 237: 90–99.
- 32.
Ricci MT, Menegon S, Vatrano S, Mandrile G, Cerrato N, Carvalho P, et al. SCN1B gene variants in Brugada Syndrome: A study of 145 SCN5A-negative patients. Sci Rep 2014; 4: 6470.
- 33.
Juang JMJ, Lu TP, Lai LC, Hsueh CH, Liu YB, Tsai CT, et al. Utilizing multiple in silico analyses to identify putative causal SCN5A variants in Brugada syndrome. Sci Rep 2014; 4: 3850.
- 34.
Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud J-B, Simonet F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet 2013; 45: 1044–1049.