Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy – Three Decades of Progress –
Article ID: CJ-15-0288

Details
Article ID: CJ-15-0288
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) is a rare, inherited cardiomyopathy characterized by ventricular arrhythmias, sudden cardiac death, and right ventricular dysfunction. Since the first major description of this disease, much has been learned about ARVD/C. One of the main breakthroughs was the discovery that mutations in desmosomal proteins are the most frequent genetic basis of ARVD/C. Today, genetic testing plays an important role in both the diagnosis of ARVD/C and cascade family screening. Much has also been learned concerning the optimal approaches to diagnosis. The 2010 Task Force Diagnostic criteria for ARVD/C represent the standard for diagnosis today. We have also learned much about the importance of proband status and the 24-h PVC count to assess sudden death risk, and the importance of exercise both in the development of ARVD/C in susceptible individuals and in defining the course of the disease. From a treatment perspective, placement of ICDs in specific subsets of patients with ARVD/C who are at increased risk of sudden death is important. The techniques of VT ablation have also evolved over time and are valuable components of our management strategies for the ARVD/C patient today. This review will provide an update on ARVD/C, with specific attention to some of the contributions to this field reported by the Johns Hopkins ARVD/C Program.
The Johns Hopkins ARVD/C Program was founded in 1999, following the tragic death of a 32-year-old patient with arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). The 12-lead ECG of this patient is shown in Figure 1, demonstrating the typical ARVD/C findings including an epsilon wave. She presented with syncope, an electrophysiologic study (EPS) revealed inducible ventricular tachycardia (VT), ARVD/C was diagnosed, and an implantable cardioverter defibrillator (ICD) was recommended. However, the patient died suddenly. Of particular note is that her father died suddenly playing squash at the age of 32 years. Subsequent family screening revealed that the patient’s younger brother also had mild evidence of ARVD/C with an ECG revealing T wave inversion (TWI) in the right precordial leads. An MRI revealed mild evidence of ARVD/C, a Holter recording revealed less than 100 premature ventricular contractions (PVCs), and no VT was inducible at EPS. A decision was made to place an ICD in this patient and he was advised to avoid exercise. He has done well since then with no ICD therapies, and no evidence of progression of his ARVD/C. Genetic testing has revealed that he harbors a mutation in PKP2. In response to this tragic case, and the very limited understanding of ARVD/C in 1999, the ARVD/C program at Johns Hopkins was founded. The goals of this program were threefold. First, it sought to further the scientific understanding of ARVD/C, a second goal was to help educate the medical and patient communities about ARVD/C, and a final goal was to serve as a referral center for ARVD/C patients. Since the initiation of the program much has been learned about ARVD/C.

Twelve-lead ECG from a patient with advanced ARVDCC. T wave inversion is present in leads V1 through V5. Also present in this ECG is an epsilon wave clearly visible in lead V1.
ARVD/C is an inherited cardiomyopathy that is characterized by ventricular arrhythmias (VAs), an increased risk of sudden cardiac death (SCD), and abnormalities of right (and less commonly left) ventricular structure and function. Structural involvement of the right ventricle (RV) predominates and a left-dominant form is rarely seen. In most patients in the United States, structural abnormalities of the RV predominate. The pathologic hallmark of ARVD is myocyte loss with fibrofatty replacement (Figure 2). Since the first detailed clinical description of the disorder in 1982,1 significant advances have been made in our understanding of all aspects of this disease. Over the past decade, mutations predominantly in desmosomal proteins have been determined to be the genetic basis of ARVD/C. Today, a pathogenic mutation can be identified in more than 60% of affected individuals.2,3 Consistent with this, genetic testing has emerged as an important diagnostic tool and plays a critical role in cascade family screening. The purpose of this review is to summarize the current state of knowledge regarding the natural history, clinical presentation, diagnosis, and treatment of patients with ARVD/C.

(A) Gross specimen of an explanted heart showing a massive right ventricle (RV) with extensive fat that has replaced the entire RV epicardium leaving a thin rim of fibrotic endocardium. Also note the relative sparing of the left ventricle and the interventricular septum. (B) Near total fatty replacement of the RV wall with loss of myocytes renders the RV translucent. (C) Transmural low-resolution histopathologic image shows the fat replacement extending from epicardium to the endocardium. (D) Trichrome staining reveals fibrous tissue interspersed with fat tissue characteristic of this disease. (Reproduced with permission from Tandri H, et al.35)
ARVD/C is an inherited condition with an estimated prevalence of 1 per 5,000. Patients usually present during the second to fifth decade of life with palpitations, lightheadedness, syncope, or sudden death.3,4 It is exceedingly rare to manifest clinical signs or symptoms of ARVD/C prior to 12 years of age. It is also uncommon to develop clinical signs or symptoms of ARVD/C after the age of 60 years. Symptoms correlate with the presence of VA. Heart failure (HF) is an uncommon and late manifestation of the disease. Figure 3 summarizes the major presenting clinical features, clinical course, and arrhythmia and survival outcomes of 439 index patients (mean age at presentation 36±14 years).4 Of them, 419 (95%) presented with symptoms and the remaining 20 (5%) were asymptomatic but came to medical attention because of abnormal tests in diverse settings. A total of 48 index patients (11%) presented with a cardiac arrest, among whom 25 were resuscitated and 23 died with the diagnosis established at autopsy (median age at cardiac arrest, 25 years, interquartile range 21, range 13–70). An additional 220 index patients (50%) presented with a sustained VA. During the clinical disease course, an ICD was implanted in 212 (87%) of the 245 index patients with a sustained VA or resuscitated SCD and in 139 (81%) of the 171 index patients presenting without sustained VA; 65 index patients did not have an ICD implanted during follow-up (33 with and 32 without sustained VA at presentation). Sustained VA during follow-up was seen in more than two-thirds of index patients (301, 72%). Of the 65 index patients without an ICD, 31 (48%) experienced a sustained VA during follow-up.
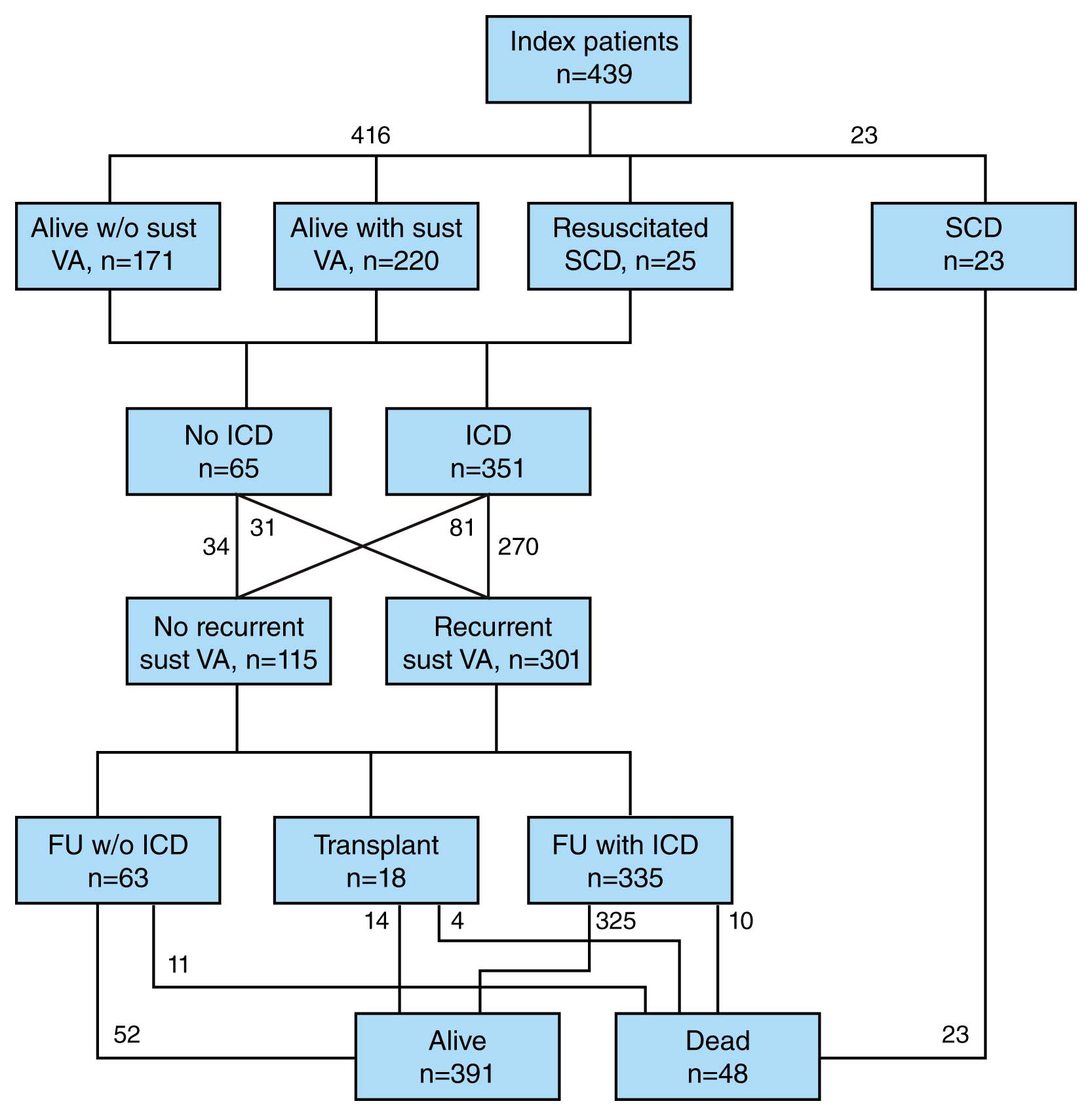
Schematic representation of the presentation, clinical course and outcome in arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) index patients. The majority present with sustained ventricular arrhythmias (VA) and receive an implantable cardioverter defibrillator (ICD) during follow-up (FU). (Reprinted with permission from Groeneweg J, et al.3)
Among the index patients with an ICD, 10 died (3%; median follow-up 7 years): 2 of SCD, 3 of HF, 2 of a combination of HF and arrhythmias, and 3 of non-cardiac causes. Among the index patients without an ICD, 11 died (17%; median follow-up 5 years): 10 of these 11 patients died of SCD and 1 of HF. The SCD incidence was higher in index patients without an ICD than in those with an ICD (16% vs. 0.6%, P<0.001). During long-term follow-up, 54 index patients (13%) developed symptomatic HF and 18 (4%) had a cardiac transplantation; 4 patients who underwent transplantation died during follow-up. Overall, 391 of the 416 index patients (94%) who presented alive were alive at last follow-up.
Figure 4 shows the results of Kaplan-Meier survival analysis by age for these 416 index patients for the following outcomes: (1) any ARVD/C-related symptoms, (2) sustained VA, (3) cardiac mortality, and (4) cardiac transplantation. Of particular note is that all patients presented in their teenage years or later (range 10–78 years). The development of symptoms correlated closely with the development of a sustained VA, with cardiac mortality and the need for cardiac transplantation being low.
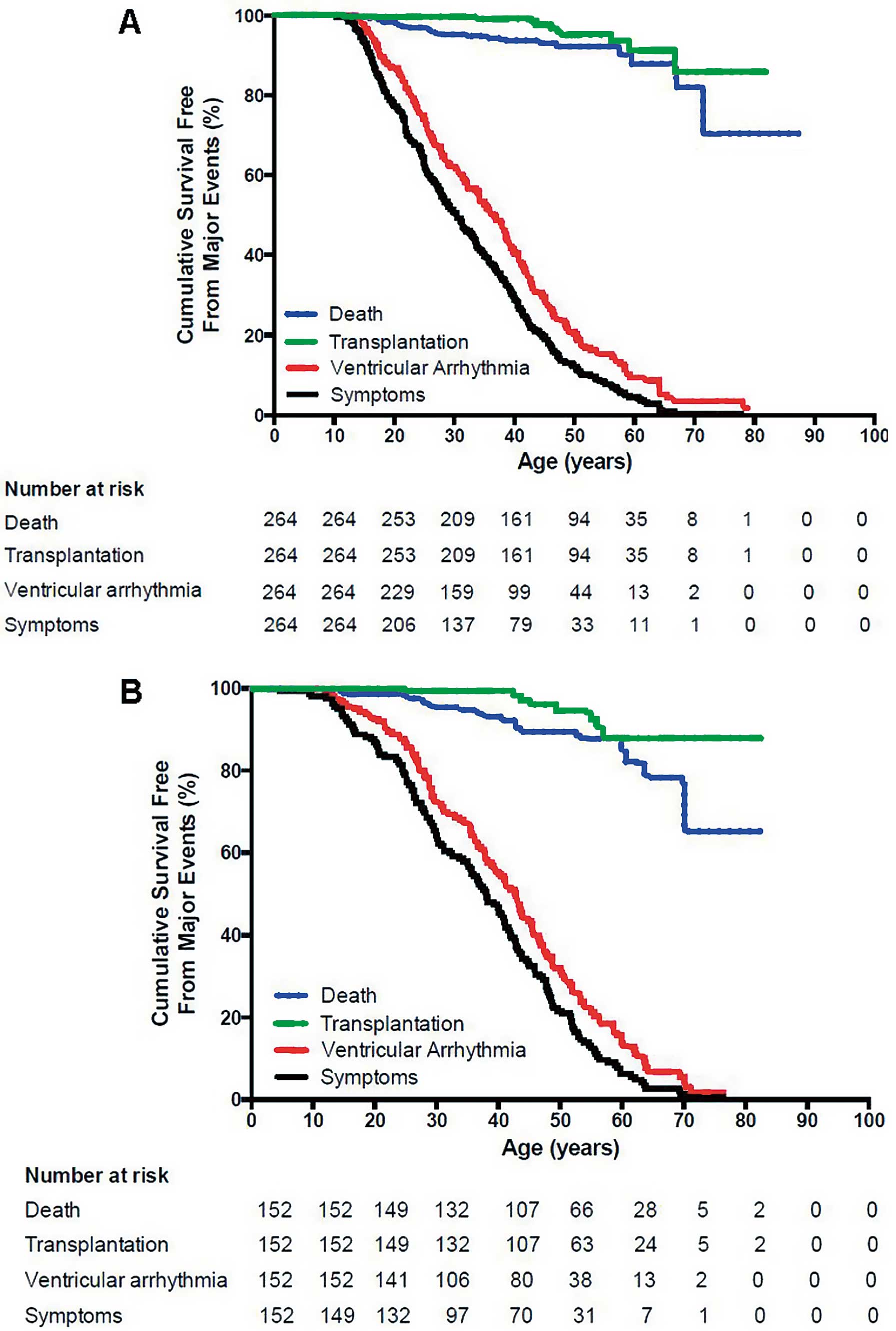
Survival free from any arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C)-related symptoms, sustained ventricular arrhythmias (VA), cardiac death and cardiac transplantation in ARVD/C index patients with (A) pathogenic mutations and (B) without identified mutations. Symptoms (P=0.005) and sustained VA (P=0.020) occurred more often at younger age in index patients with mutations. Survival free from cardiac death (P=0.644) and transplantation (P=0.704) was similar in both groups. (Reprinted with permission from Groeneweg J, et al.3)
Although ARVD/C is predominantly a disease of the RV,5–8 it is now well established that involvement of the left ventricle (LV) may occur, particularly when MRI is used to detect subtle abnormalities in LV function and also in patients with advanced disease. Left-dominant arrhythmogenic cardiomyopathy also is defined by early disease of the LV, often affecting the posterolateral LV wall, in the absence of significant RV systolic dysfunction. Left-dominant disease is more commonly seen in patients with desmoplakin mutations.8
In most cases, ARVD/C is inherited in an autosomal-dominant pattern with significantly variable penetrance and expressivity. Among probands diagnosed with this disease, screening of first-degree relatives identifies other affected individuals in approximately 50% of cases. In a minority of cases, ARVD/C is inherited in an autosomal recessive pattern as part of a cardiocutaneous syndrome such as Naxos disease or Carvajal syndrome, which is characterized by woolly hair and palmoplantar keratodermia.
Linkage mapping and candidate gene evaluation studies performed in patients with the autosomal-dominant form of ARVD/C initially was not productive because of the significant variability in penetrance and expressivity of the disease. It was the evaluation of patients with Naxos syndrome, a disease with 100% penetrance by adolescence, that identified a disease-causing mutation: a homozygous deletion of 2 base pairs found in the plakoglobin gene located in the 17q21 locus.9 The gene encodes a key component of desmosomes, which are complex intercellular adhesion structures found in stratified epithelial cells of the skin as well as in myocytes. Desmosomes are composed of 3 major groups of proteins: Cadherins are transmembrane proteins that provide the actual mechanical coupling between individual cells and include desmoglein and desmocollin. Desmoplakin is a plakin-family protein that serves to anchor the desmosomal structure to the intermediate filaments of the cell. Lastly, Armadillo proteins, including plakoglobin and plakophilin, link desmoplakin and the cadherin tails.
The identification of defective desmosomal proteins in Naxos syndrome led to studies investigating their role in other arrhythmogenic cardiomyopathies. The Carvajal syndrome, another cardiocutaneous syndrome that has left-dominant arrhythmogenic cardiomyopathy, was shown to be associated with a recessive mutation in desmoplakin.10 Other genetic mutations were subsequently identified in the autosomal-dominant form of ARVC/D and include the desmoplakin (DSP), desmoglein (DSG)-2, desmocollin (DSC)-2 and plakophillin (PLN)-2 genes.2,8 Mutations in several extra-desmosomal genes, such as those encoding transforming growth factor β3, cardiac ryanodine receptor, Titin, and transmembrane protein 43 (TMEM43), have been also implicated in specific types of atypical forms of ARVC/D. In the United States, a desmosomal protein mutation can be identified in approximately 60% of ARVD/C patients.3,8
The genetic findings of a large transatlantic cohort of patients with ARVD/C, selected according to the presence of an ARVD/C-related mutation, were recently reported.8 The study population consisted of 577 patients from 241 unrelated families and was derived from the Johns Hopkins ARVD/C registry (259 patients: 102 probands, 157 family members) and the Dutch ARVD/C cohort (318 patients: 128 probands, 190 family members). The registries were statistically similar in terms of sex, race and proband/family member ratios. Men constituted 55% of the overall population, with the average age at presentation being 35±17 years. The majority of participants (463, 80%) carried a single copy of a PKP2 mutation (164 probands, 299 family members), with significantly fewer heterozygous carriers of mutations in other ARVD/C-associated genes (31 DSG2, 31 PLN, 19 DSP, 8 DSC2, 2 JUP, and 1 TMEM43). A total of 22 individuals (4%) carried 2 or more pathogenic mutations (compound heterozygote, homozygote, or digenic mutation carriers). Among carriers of single mutations, premature truncating, splice site, and missense mutations were identified in 342 (60%), 130 (23%), and 83 (14%) patients, respectively. Patients with SCD/ventricular fibrillation (VF) at presentation (n=36) were younger (median 23 vs. 36 years; P<0.001) than those presenting with sustained monomorphic VT. Among 541 subjects presenting alive, over a mean follow-up of 6±7 years, 12 (2%) died, 162 (30%) had sustained VT/VF, 78 (14%) manifested LV dysfunction (ejection fraction <55%), 28 (5%) experienced HF and 10 (2%) required cardiac transplantation. Patients (n=22; 4%) with >1 mutation had significantly earlier occurrence of sustained VT/VF (mean age 28±12 years), lower VT/VF-free survival (P=0.037), more frequent LV dysfunction (29%), HF (19%) and cardiac transplantation (9%) as compared with those with only 1 mutation. DSP mutation carriers experienced >4-fold occurrence of LV dysfunction (40%) and HF (13%) than PKP2 carriers. Missense mutation carriers had similar death/transplant-free survival and VT/VF penetrance (P=0.137) as compared with those with truncating or splice site mutations. Men are more likely to be probands (P<0.001), symptomatic (P<0.001) and have earlier and more severe arrhythmic expression.
The genetics of ARVD/C has provided support for the hypothesis that ARVD/C is a disease of desmosomal dysfunction. The pathogenic mechanisms are not fully clear, but several theories have been advanced. Defective desmosomal proteins may lead to impaired mechanical coupling between individual cells, leading to myocyte uncoupling especially under conditions that increase myocardial strain. The resulting inflammation, fibrosis and adipocytosis may be a nonspecific response to injury similar to that seen in other forms of myocardial damage. This pathogenic model can explain the observation that prolonged strenuous exertion, which increases myocardial strain, significantly increases the risk of an earlier clinical onset of the disease and augments the risk of SCD. It also explains why the RV, which is more distensible than the LV because of its thinner wall and asymmetric shape, is more often involved in ARVC, especially in the early stages. Furthermore, defects in mechanical coupling of myocytes may also lead to impairment in electrical coupling. Other hypotheses concerning the mechanisms of ARVD/C involve complex intracellular signaling pathways, which is an active area of investigation.
Diagnosis of ARVD/C relies on a scoring system with major and minor criteria based on the demonstration of a combination of defects in RV morphology and function, characteristic depolarization/repolarization ECG abnormalities, characteristic tissue pathology, VAs, family history, and the results of genetic testing. The original 1994 Task Force Criteria (TFC) were recently revised to improve upon the subjectivity of some of the original criteria and to increase sensitivity by integrating new knowledge and technology (Table).11 These criteria should now be relied on to make a diagnosis of ARVD/C. Patients are diagnosed as having ARVD/C if they have “4 points” with a major criteria equal to 2 points and a minor criteria equal to 1 point. Patients whose score totals “3 points” are classified as having probable ARVD/C, whereas those with “1 or 2 points” are classified as not meeting the criteria for ARVD/C.
| 1 Global or regional dysfunction and structural alterations |
| Major |
| 2nd echo criteria |
| Regional RV akinesia, dyskinesia, or aneurysm AND 1 of the following measured at end diastole |
| PLAX RVOT ≥32 mm or |
| PSAX RVOT ≥36, |
| Fractional area change ≤33% |
| MRI criteria |
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction AND 1 of the following |
| Ratio of RV end-diastolic volume to BSA ≥100, 110 ml/m2 (male) or ≥100 ml/m2 |
| RV ejection fraction >40% ≤45% |
| RV angiography criteria |
| Regional RV akinesia, dyskinesia, or aneurysm |
| Minor |
| 2nd echo criteria |
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction AND 1 of the following measured at end diastole |
| PLAX RVOT ≥29 <32 mm or |
| PSAX RVOT ≥32 <36 |
| Fractional area change >33% ≤40% |
| MRI criteria |
| Regional RV akinesia or dyskinesia or dyssynchronous RV contraction AND 1 of the following |
| Ratio of RV end-diastolic volume to BSA ≥110 ml/m2 (male) or ≥100 ml/m2 |
| RV ejection fraction ≤40% |
| 2 Tissue characterization of wall |
| Major |
| Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, |
| with or without fatty replacement of tissue on endomyocardial biopsy |
| Minor |
| Residual myocytes 60–75% by morphometric analysis (or 50–65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample |
| with or without fatty replacement of tissue on endomyocardial biopsy |
| 3 Repolarization abnormalities |
| Major |
| Inverted T waves in right precordial leads (V1, V2, and V3) or beyond in individuals >14 years of age (in the absence of complete RBBB QRS ≥120 ms) |
| Minor |
| Inverted T waves in V1 and V2 in individuals >14 years of age (in the absence of complete RBBB) or in V4, V5, and V6 |
| Inverted T waves in leads V1, V2, V3, and V4 in individuals >14 years of age in the presence of a complete RBBB |
| 4 Depolarization/conduction abnormalities |
| Major |
| Epsilon wave (reproducible low-amplitude signals between end of QRS complex to onset of T wave) in the right precordial leads (V1–3) |
| Minor |
| Late potentials by SAECG in ≥1 of 3 parameters in the absence of a QRSd ≥110 ms on standard ECG |
| Filtered QRS duration (fQRS) ≥114 ms |
| Duration of terminal QRS <40 microV ≥38 ms |
| Root-mean-square voltage of terminal 40 ms ≤ micro V |
| Terminal activation duration ≥55 ms measured from the nadir of the end of the QRS, including R’, in V1, V2, or V3 in absence of complete RBBB |
| 5 Arrhythmias |
| Major |
| Nonsustained or sustained VT of LBBB morphology with superior axis |
| Minor |
| Nonsustained or sustained VT of RVOT configuration, LBBB morphology with inferior axis or of unknown axis |
| >500 PVCs per 24 h (Holter) |
| 6 Family history |
| Major |
| ARVD/C in first-degree relative who meets Task Force Criteria |
| ARVD/C confirmed pathologically at autopsy or surgery in first-degree relative |
| Identification of pathogenic mutation categorized as associated or probably associated with ARVD/C in the patient under evaluation |
| Minor |
| History of ARVD/C in first-degree relative in whom it is not possible to determine whether the family member meets Task Force Criteria |
| Premature sudden death (<35 years of age) due to suspected ARVD/C in a first-degree relative |
| ARVD/C confirmed pathologically or by current Task Force Criteria in second-degree relative |
*Adapted from Marcus FI, et al.11 ARVD/C, Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy; BSA, body surface area; LBBB, left bundle branch block; PVC, premature ventricular contraction; RBBB, right bundle branch block; RVOT, right ventricular outflow tract; SAECG, signal-averaged ECG; VT, ventricular tachycardia.
When considering these guideline recommendations it is important to note that there is no single gold-standard diagnostic test. The initial evaluation of all patients should include a physical examination and clinical history including family history of arrhythmias or sudden death, ECG, signal-averaged ECG (SAECG) 24-h Holter monitoring, and comprehensive noninvasive imaging of both ventricles. MRI can be of great value but is also a common cause of misdiagnosis.12 If this noninvasive workup is suggestive but not diagnostic of ARVD/C, further testing should be considered to establish the diagnosis, including EPS. Although endomyocardial biopsy was used in the past, it is rarely done today, which reflects current, extensive experience with MRI. Similarly, right ventriculography is rarely needed because of the availability of high-quality echocardiography and MRI.
The standard 12-lead ECG is abnormal in the majority of patients with ARVD (Figure 1).13–15 Because of this observation, the finding of a normal ECG in a patient with ARVD/C renders the diagnosis of ARVD/C extremely unlikely.14 TWI in the right precordial leads (V1–3) is the most common ECG manifestation of ARVD/C and is a major diagnostic criteria (Table). TWI in leads V1 and V2 is a minor criterion. A complete or incomplete right bundle branch block (RBBB) pattern is a common finding in ARVD/C patients, especially in those with severe structural disease, and its presence obscures the interpretation of the known depolarization abnormalities.15 In patients with incomplete RBBB, TWI through V3 still appears to be the feature with optimal sensitivity and specificity. However, in patients with complete RBBB, an R/S ratio <1 in V1 seems to provide the optimal sensitivity and specificity in diagnosing ARVD/C.15 TWI beyond V3 in patients with complete RBBB is also a feature of ARVD/C. Epsilon waves, which are distinct low-frequency deflections in the ECG that occur following the QRS and before the T wave (Figure 1), are uncommon and a marker of advanced ARVD/C. The new diagnostic criteria for ARVD/C include terminal activation delay, which is defined as a prolonged duration of time (≥55 ms) from the S wave to the termination of the QRS complex as a minor criterion for ARVD.11
The SAECG provides increased sensitivity in the detection of activation delay. It is defined as abnormal if 1 or more of the following criteria are present: (1) filtered QRS duration (fQRS) >114 ms, (2) terminal low-amplitude (<40µV) signal duration ≥38 ms, and (3) root-mean-square voltage of the terminal 40 ms of the filtered QRS complex (RMS40) <20 µV.11 The likelihood of an abnormal result seems to correlate with the degree of structural abnormalities. A positive SAECG is now considered a minor criterion.11
Ambulatory Holter monitoring is a valuable test for the evaluation of patients with suspected ARVD/C. The presence of frequent ventricular ectopic beats or sustained/nonsustained VT (NSVT), particularly of left bundle branch block (LBBB) morphology, is consistent with the diagnosis of ARVD/C. It is generally accepted that less than 200 PVCs per 24 h is within normal limits. The presence of ≥500 PVCs per 24 h is commonly observed in patients with ARVD/C and is considered a minor criterion.11 Recently, it was reported that the number of PVCs on 24-hour Holter monitoring is directly proportional to a patient’s chance of developing appropriate ICD therapy among those with an ICD implanted for primary prevention.16,17 The day-to-day variability in PVC frequency observed in patients with ARVD/C has also explored.18 Although some degree of PVC variability was observed, the degree of variability was acceptable for risk stratification based on the number of PVCs per 24 h. Holter monitoring can also be done on a yearly basis to assist in evaluating a patient’s arrhythmic risk. If 24-h Holter monitoring does not capture any arrhythmias, further options include extending the duration of Holter monitoring, implanting a loop recorder or exercise testing to induce VT.
Assessment of ventricular structure and function is critical for the diagnosis. Echocardiography is the most widely available method of imaging. Findings on a transthoracic echocardiogram that are suggestive of ARVC/D include global or segmental wall motion abnormalities with cavity dilation, hypertrophic RV trabeculation and systolic dysfunction. RV outlet tract (RVOT) dilation (diameter >30 mm) has been reported to have the highest sensitivity and specificity of the echocardiographic parameters in diagnosing ARVD.11 Table lists the echocardiographic parameters that have been incorporated into the TFC.11 Representative MRI in a patient with ARVD/C are shown in Figure 5. Cardiac MRI (CMR) is an imaging modality that has the benefit of providing tissue characterization and identification of intramyocardial fat and fibrosis in addition to assessment of ventricular structure and function. MRI is the imaging modality of choice in evaluating the RV in ARVD/C.5–7 The ability to provide multiplanar images of the RV allows for accurate quantitative assessment of RV global function and assessment of RV regional function. The noninvasive nature of MRI makes it ideal for the evaluation and follow-up of asymptomatic first-degree relatives. The major and minor criteria for ARVD/C on MRI are listed in Table. However, It is important to recognize that MRI is the most common reason for misdiagnosis of ARVD/C.11 Some common reasons for misdiagnosis of ARVD/C were recently identified and include the presence of a myocardial tether, a moderator band, and pectus excavatum.6 It is also important to recognize that ARVD/C does not affect the RV apex early in the course of the disease, which is a common misconception resulting from an erroneous initial report of a triangle of dysplasia.1 It was recently reported that the triangle of dysplasia consists of the inflow tract, the outflow tract, and the posterolateral LV.19

Cardiac MRI of a patient with ARVC. Bright blood images in the right ventricular outflow tract (RVOT) plane obtained in end diastole (A) and end systole (B) show microaneurysms (arrows) in the RV free wall with persistent bulge in both phases. Short-axis bright blood images obtained in end diastole (C) and end systole (D) demonstrate dyskinesis (bulge in systole, arrows) at the acute angle of the RV and hypokinesis in the inferior wall (arrowheads).
An endomyocardial biopsy may be obtained to demonstrate the characteristic histopathology of ARVD/C if the diagnosis is unclear or to rule out a competing diagnosis. However, the test appears to have low diagnostic sensitivity for several reasons, including patchy distribution of the disease. One study reported that immunohistochemical analysis of desmosomal protein distribution in endomyocardial biopsy specimens has diagnostic value.20 Although there was initial hope that this test may prove to be invaluable in the evaluation of patients, enthusiasm fell rapidly when it was recognized that sarcoidosis and giant cell myocarditis can cause patterns similar to that observed in ARVD/C.21 The diagnostic role of immunohistochemical analysis of biopsy samples as a diagnostic tool in ARVD/C is no longer supported. Table shows the pathological parameters that have been incorporated into the revised TFC.11 Despite the potential value of endomyocardial biopsy, biopsy is rarely performed today.
The role of genetic testing in the diagnosis of ARVD/C is now well established and is considered a major diagnostic criterion.11 More than 60% of ARVC/D probands are found to have a mutation.2,3 Thus, lack of an identifiable mutation does not exclude the disease. However, at present the most important utility of this test is in the cascade screening of family members. Asymptomatic gene carriers are likely to require life-long monitoring because of age-dependent penetrance, whereas non-carriers are unlikely to have the disease and serial screening is generally not required. Exercise restriction also needs to be considered, especially in those who harbor a disease-causing mutation.
The diagnosis of ARVD/C should be considered in any patient who does not have known heart disease and who presents with frequent PVCs or symptomatic VT, especially if there is LBBB morphology with superior axis, the LBBB VT ECG pattern is not typical of idiopathic RVOT VT, or if the VT occurs in a patient with TWI in the right precordial leads. The most common conditions that must be considered in the differential diagnosis include idiopathic RVOT VT, VT arising from the aortic root, and cardiac sarcoidosis. Several studies have recently compared the morphology of VT or PVCs with a LBBB inferior (LBI) axis morphology in patients with idiopathic RVOT VT with that of patients with ARVC/D. A recent study examining patients with PVCs or VT with a LBI axis morphology confirmed that a QRSd >120 ms in lead V1 during VT or with PVCs favors the diagnosis of ARVD/C as compared with idiopathic RVOT VT.22 Although it is important to consider the diagnosis of ARVD/C in a patient with LBI axis PVCs or VT, it is not recommended that these patients undergo a complete evaluation for ARVD/C. If the ECG and echocardiogram are normal and there is no family history of sudden death, an exhaustive evaluation is not advised. If the patient prefers a curative ablative strategy, an EPS and ablation would be the next step. If at the time of EPS multiple forms of VT are induced, the potential diagnosis of ARVD/C would be reconsidered.
Another novel marker of ARVD/C that has been recently reported is a characteristic response to high-dose isoproterenol infusion.23,24 In a recent study, a total of 412 consecutive patients referred for evaluation of PVCs or ARVD/C underwent this test and 35 of them had been diagnosed with ARVD/C based on the revised 2010 TFC. The isoproterenol infusion consisted of intravenous administration of very high dose (45 mcg/min) isoproterenol for 3 min. A continuous ECG was recorded during and for up to 10 min following the infusion. The test was interpreted as being “positive” if polymorphic PVCs (>3 morphologies) and at least 1 couplet were observed or if sustained or nonsustained monomorphic or polymorphic VT with a predominantly LBBB morphology not typical of RVOT VT was observed. The isoproterenol test was positive in 74/412 (18%) patients, in 32/35 (91.4%) patients with an established ARVD/C diagnosis and in 42/377 (11.1%) patients without ARVD/C. The resultant sensitivity, specificity, positive and negative predictive values of isoproterenol testing to identify patients diagnosed with ARVD/C based on the 2010 TFC were 91.4%, 88.9%, 43.2%, and 99.1%, respectively. Monomorphic sustained or nonsustained RVOT VT were observed in 0/35 ARVD/C patients vs. 27/377 (7.2%) of patients without ARVD/C. During a median follow-up of 5 years, 6 of the initial non-ARVD/C group met the diagnostic criteria for ARVD/C. Each of these individuals had had a positive isoproterenol response. None of the patients who had typical RVOT VT developed ARVD/C during follow-up. Based on these data, it was determined that survival free from a diagnosis of ARVD/C was lower in the negative isoproterenol group vs. the positive isoproterenol response group. Genetic screening was performed in 30 of the 41 patients eventually diagnosed with ARVD/C (with a pathogenic mutation identified in 10) and in 24/371 non-ARVD/C patients. The concept of using a high-dose isoproterenol test as a screening/diagnostic test for ARVD/C is novel and potentially of great importance. However, at this point I think it should not be considered to be a diagnostic test. Whether in the long run it will prove to be useful for diagnosis and/or risk stratification will require further study.
Cardiac sarcoidosis should be suspected in patients who have evidence of conduction system abnormalities, especially in the presence of other extracardiac symptoms. In rare situations, an endomyocardial biopsy may become necessary to differentiate between the 2 disorders. The infrequency with which endomyocardial biopsy is required reflects the fact that cardiac sarcoidosis can usually be suspected based on the presence of conduction system abnormalities, mediastinal lymphadenopathy, tissue diagnosis of extracardiac sarcoidosis, or the presence of septal scar.25
A minority of ARVD/C patients first present with symptoms of RV systolic HF. The differential diagnosis in such patients includes RV infarction or pulmonary hypertension. Dilated cardiomyopathy must be considered if there is evidence of biventricular failure. Patients with dilated cardiomyopathy who have early significant ventricular ectopy should be evaluated for possible biventricular arrhythmogenic cardiomyopathy. Cardiac transplantation is rarely required in patients with ARVD/C.26
Management of patients with ARVD/C has 4 components. The first is getting the diagnosis correct. The second is accurate risk stratification for SCD and deciding whether to place an ICD. The third component of management is minimizing ICD therapies. And the final component of management is preventing progression of the disease. Each of these will be examined in detail.
Establishing an Accurate DiagnosisAs noted, the first step in management is making the diagnosis. Misdiagnosis of ARVD/C is common because of lack of awareness of the 2010 TFC and misinterpretation of MRI.
Risk Stratification and Deciding When to Implant an ICDPrevention of SCD is the primary goal of management. Several studies of ARVD/C patients who received an ICD have reported appropriate interventions during follow-up in more than 50% of patients.17,27 Predictors of appropriate therapy included a prior sustained VA, syncope, and the extent of structural heart disease.
The incidence of appropriate ICD therapy among 84 patients who had an ICD implanted for primary prevention was recently investigated.17 Over a mean follow-up of 4.7±3.4 years, appropriate ICD therapy was experienced by 40 (48%) patients, of whom 16 (19%) received interventions for rapid VT/VF. Proband status, inducibility at EPS, presence of NSVT, and PVC count >1,000/24 h were identified as significant predictors of appropriate ICD therapy (Figure 6). The 5-year survival free from appropriate ICD therapy for patients with 1, 2, 3 and 4 risk factors was 100%, 83%, 21% and 15%, respectively. Inducibility at EPS and NSVT remained as significant predictors on multivariable analysis. These findings are important because they demonstrate that nearly half of the ARVD/C patients treated with an ICD for primary prevention experienced appropriate ICD interventions. Inducibility at EPS and NSVT are independent strong predictors of appropriate ICD therapy. More frequent ventricular ectopy is associated with progressively more common ICD therapy. Incremental risk of VAs and ICD therapy was observed in the presence of multiple risk factors.
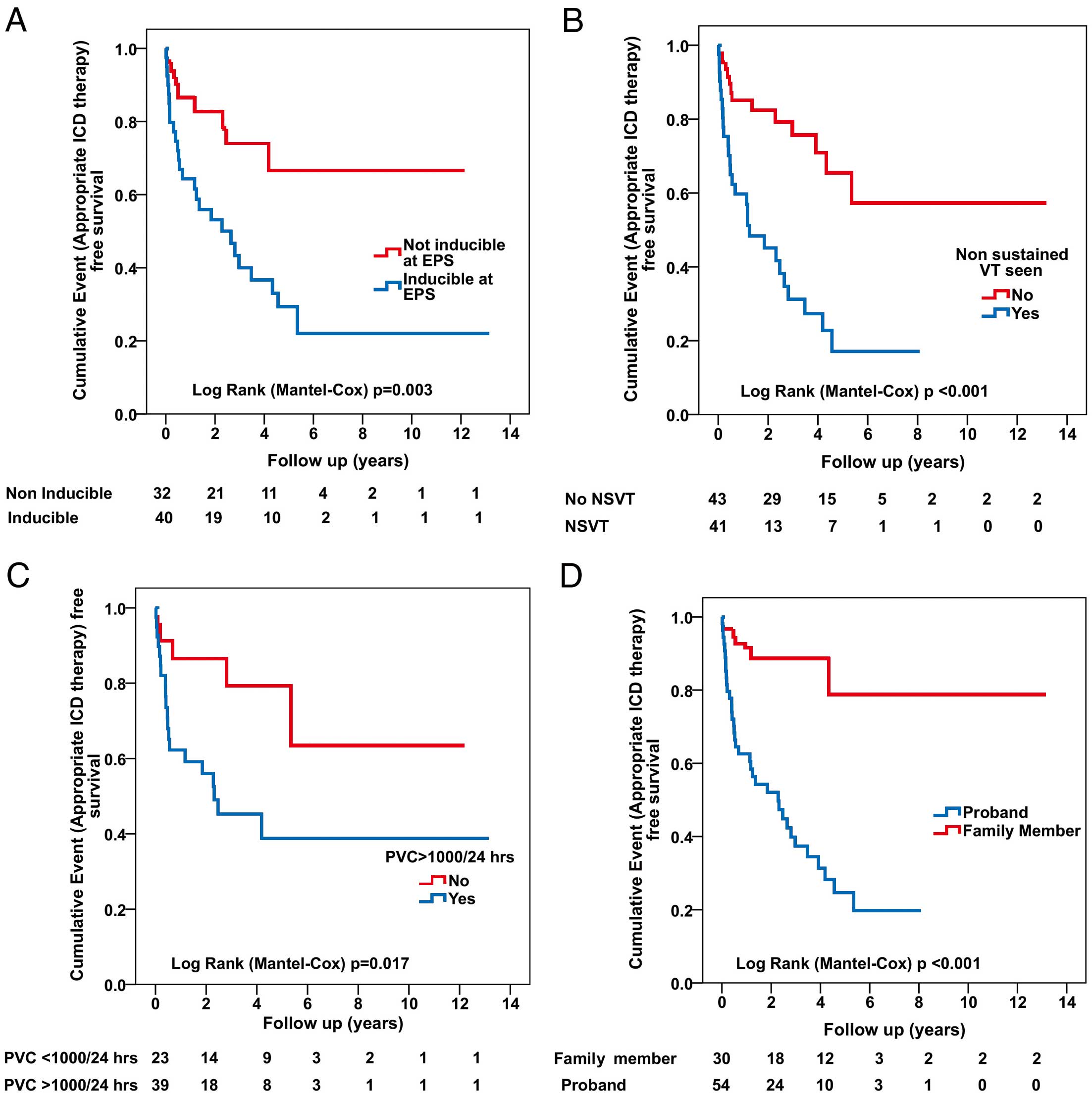
(A–D) Incidence of sustained ventricular arrhythmias in patients with ARVD/C receiving an implantable cardioverter defibrillator (ICD) for primary prevention based on 4 variables: inducibility of ventricular tachycardia (VT) at electrophysiologic study (EPS), frequency of premature ventricular contractions (PVC), nonsustained VT (NSVT), and proband status. (Adapted with permission from Bhonsale A, et al.17)
At present ICD placement is recommended for all probands who meet the TFC, especially if they have a history of sudden death, sustained VT or arrhythmogenic syncope, or have a high degree of ventricular ectopy and/or NSVT. Clinicians should be more circumspect about recommending implantation of an ICD in a family member who has been diagnosed with ARVD/C as a result of cascade screening. These individuals are being identified at a much earlier stage in their disease than was possible previously. With exercise restriction and the use of a β-blocker, their risk of SCD appears to be reduced to a level below which placement of an ICD should be routinely advised. If a decision is made not to implant an ICD close monitoring and follow-up are emphasized.
Minimizing Symptoms and Preventing ICD TherapiesPharmacologic Therapy Pharmacologic therapy plays an important role in the management of patients with ARVD/C. This is the case despite the few studies done to define the safety and efficacy of pharmacologic therapy. β-blockers are recommended for almost all patients with ARVD/C, which reflects several lines of evidence. First, β-blockers are the only pharmacologic therapy that has been shown to reduce risk of SCD in populations of patients without ARVD/C. Second, the great majority of VAs and sudden death episodes are triggered by exercise. And third, high-dose isoproterenol results in the triggering of VT in patients with ARVD/C.23,24 Angiotensin-converting enzyme (ACE) inhibitors are also advised for most patients with ARVD/C and especially those with evidence of significant disease. There are no published trials in patients with ARVD/C, but there is a wealth of data supporting the role of ACE in a broader population of patients with cardiomyopathy. When it comes to membrane-active antiarrhythmic agents, sotolol is used most commonly and if ineffective, amiodarone is chosen. In rare cases, flecainide, propafenone, or dofetilide is used. For most patients with ARVD/C and symptomatic VAs and/or ICD therapies therapy with at least one antiarrhythmic medication will be attempted before considering VT ablation. Some patients have a strong bias against antiarrhythmic drug therapy and move quickly to VT ablation. Whereas others would much prefer more extensive attempts at antiarrhythmic drug therapy before considering VT ablation.
Catheter Ablation Catheter ablation is another important option for treatment of patients with ARVD/C who have VT. It is important to recognize that unlike patients with idiopathic VT in which catheter ablation is curative, the role of catheter ablation in patients with ARVD/C is to improve quality of life by decreasing the frequency of episodes of VT. The standard approach at Johns Hopkins is to consider catheter ablation following a trial of antiarrhythmic therapy.
Three studies have evaluated the outcomes of catheter ablation of VT in patients with ARVD/C.28–30 The first study28 reported a cohort of 24 patients with ARVD/C who underwent VT ablation. These 24 patients underwent 48 ablation procedures at 29 different electrophysiology centers. The cumulative VT-free survival was 75% at 1.5 months, 50% at 5 months, and 25% at 14 months. The immediate success of the procedure had no bearing on recurrence nor did the use of assisted mapping techniques, or repetition of the procedure. The second study29 reported an expanded series of 87 patients who underwent a total of 175 VT ablation procedures. Over a follow-up of 88+66 months, the freedom from VT was 47%, 21%, and 15% at 1, 5, and 10 years, respectively. The cumulative freedom from VT following epicardial VT ablation was 64% and 45% at 1 and 5 years, respectively. Importantly, the burden of VT decreased following ablation from a median of 0.16 VT episodes per month pre-ablation to 0.08 episodes per month post-ablation. Most recently, a single center experience with epicardial VT ablation in patients with ARVD/C was published.30 It included 30 ARVD/C patients who underwent endo-/epicardial mapping and epicardial catheter ablation of VT. ICD interrogations were evaluated for VT recurrence: 8 (27%) patients experienced VT recurrence after epicardial RFA and the VT-free survival was 83%, 76%, and 70% at 6, 12 and 24 months, respectively. A significant reduction in the VT burden was observed. No complications occurred. Most VT recurrences were in the first year after RFA, during exercise, had fast cycle lengths, and required ICD shock for termination. We consider this to be an important treatment modality. For most patients it represents a second-line therapy but rarely a patient prefers to proceed with VT ablation prior to antiarrhythmic drug therapy.
Exercise Restriction An important recommendation for patients with ARVD/C is exercise restriction. The association of exercise stems from the observation that SCD in ARVD/C patients often occurs during exertion. Kirchhof et al showed that endurance training of transgenic plakoglobin-deficient mice was associated with RV enlargement, RV conduction slowing and VAs as compared to wild-type hearts.31 The results of this and other studies support the hypothesis that repeated strenuous exercise is an important factor increasing the chance of development of ARVD/C, especially in patients with a desmosomal mutation.
The first study evaluating the role of exercise in ARVD/C patients inheriting a pathogenic desmosomal mutation was performed by James et al (Figure 7).32 They studied 87 probands and family members from the Johns Hopkins ARVD/C Registry who were identified as carrying a single copy of a pathogenic mutation in a desmosomal gene. All patients participated in an exercise interview detailing their exercise history for leisure/recreation, work, and transportation. Participants were classified as an endurance athlete if they performed ≥50 h/year of vigorous intensity sports with a high dynamic demand as defined by the 36th Bethesda Conference Classification of Sports (Task Force 8).32 The study had several key findings. First, ARVD/C patients who were endurance athletes became symptomatic at an earlier age. Second, endurance exercise and a higher duration of exercise participation were both associated with increasing likelihood of developing manifest ARVD/C. Third, endurance exercise was associated with worse survival from VA and HF. Lastly, those individuals who continued to participate in the top quartile for hours of annual exercise after presentation had worse survival from first VA compared with individuals who reduced their exercise after presentation. The study thus confirmed the role of exercise as a disease modifier in ARVD/C patients with a desmosomal mutation.

Association of exercise history with the likelihood of a diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C). The likelihood of meeting the ARVD/C diagnostic criteria at last follow-up is associated with increasing hours per year of exercise (P<0.001) and participation in endurance athletics (P<0.001). Hrs/yr, hours per year; TFC, 2010 Task Force diagnostic Criteria. (Adapted with permission from James CA, et al.32)
The role of exercise in ARVD/C patients without a mutation was subsequently studied.33 The role of duration and intensity of exercise participation was examined among 43 probands without a desmosomal mutation (non-desmosomal) and compared with 39 probands with desmosomal mutations all of whom met the 2010 TFC. It was found that among non-desmosomal patients, those participating in the highest quartile of MET-Hrs/year exercise presented at a significantly earlier age, had worse structural abnormalities on CMR and significantly poorer survival from a VA during follow-up. Next, exercise participation stratified by mutation status and family history was compared and it was found that although non-desmosomal ARVD/C patients had done a similar duration of annual exercise as desmosomal carriers; their exercise was significantly more intense, with significantly higher participation in endurance sports and expending greater MET-Hrs/year of exercise as compared with the desmosomal patients. Lastly, ARVD/C non-desmosomal patients with no family history performed the highest MET-Hrs/year exercise compared with non-desmosomal patients with a family history and being desmosomal mutation carriers. Because the non-desmosomal patients without family history did the highest intensity exercise, it was concluded that exercise may be the key environmental factor driving disease pathogenesis in that subgroup. Interestingly, when annual pre-presentation MET-Hrs and endurance exercise participation during youth (age ≤25) were compared, it was found that higher intensity exercise during youth was associated with earlier age of presentation in both desmosomal and non-desmosomal ARVD/C patients.
The widely used “36th Bethesda Conference Eligibility Recommendations for Competitive Athletes with Cardiovascular Abnormalities” guidelines recommend avoidance of all competitive activities for patients with probable or definite diagnosis of ARVD/C, except for class I-A sports with a low cardiovascular demand. The American Heart Association has a grading system for recommendations regarding participation in recreational sports, ranking them on a scale of 0 to 5.34 In general, ARVD/C patients are prohibited from participating in all competitive sports and recreational sports requiring ≥4 MET’s. Based on the growing body of literature described here and the clinical experience at Johns Hopkins, there is a strong belief that patients with ARVD/C or those at risk for developing ARVD/C should limit exercise.
Preventing ProgressionThe final consideration in patients with ARVD/C is preventing disease progression. It is important to note that no studies have examined the extent and rate of progression of ARVD/C. In my experience the rate of progression is slow, but it is important to note that ARVD/C is a progressive disease. The most important tool in preventing disease progression is restriction of exercise and the considerable data supporting this relationship have been described.
It is rare that patients with ARVD require cardiac transplantation.25 Details of 18 ARVD/C patients who underwent cardiac transplantation for ARVD/C were recently reported. The average age at first ARVD symptoms was 24±13 years and the average age at cardiac transplant was 40±14 years. These patients often had clinical onset of disease at a relatively young age. At 1 year after transplantation, survival was 94% and 88% were alive at an average post-transplant follow-up of 6.2±4.8 years.
ARVD/C is an inherited cardiomyopathy characterized clinically by VA, SCD, and structural abnormalities of the RV. Because of the significant heterogeneity in its manifestation, the diagnosis of ARVD/C is challenging and requires a multifaceted approach to patient evaluation. The management of ARVD/C is primarily aimed at reducing the burden of symptomatic arrhythmias and decreasing the incidence of SCD. ICDs significantly reduce mortality in patients with ARVD/C. Antiarrhythmic drug therapy and VT ablation are used primarily to decrease the frequency of episodes of VT. Exercise is a very important environmental stimulus that triggers development of the disease in susceptible individuals and affects the clinical course. Restriction of exercise in patients with ARVD/C is strongly recommended.
The Johns Hopkins ARVD/C Program is supported by the St. Jude Medical Foundation and Medtronic Inc, Dr Francis P. Chiaramonte Private Foundation Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, the Wilmerding Endowments, and the Dr Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins.
Research support was provided by St Jude Medical and Medtronic.