Regenerated Endothelium and Its Senescent Response to Aggregating Platelets
Article ID: CJ-16-0179

Details
Article ID: CJ-16-0179
This essay summarizes 30 years of work attempting to understand why regenerated endothelium becomes dysfunctional. It focuses on the activation of endothelial nitric oxide synthase (eNOS) and the production of NO in response to platelet products and thrombin, which represents a first-line protection against vasospasm and atherosclerosis. Serotonin and adenosine diphosphate released by aggregating platelets are coupled to the activation of eNOS by different G-proteins. The endothelium-dependent relaxation that they cause is modulated non-selectively by the lipid content in the diet. When the endothelium regenerates after mechanical disruption, the newly formed endothelial cells selectively lose their Gi-mediated coupling and become less responsive to serotonin and thrombin. Accelerated senescence and the emergence of adipocyte-fatty acid binding protein leading to increased oxidative stress play a key role in the genesis of the dysfunction of regenerated endothelium. The consequent local NO deficiency not only favors the occurrence of vasospasm but sets the stage for the occurrence of atherosclerosis.
When Professor Hiroaki Shimokawa, the Editor-in-Chief of the Circulation Journal, approached me on behalf of the Japanese Circulation Society to deliver the Mikamo Lecture, honoring indeed a giant Japanese cardiologist and scientist, and also invited me to write the present article, I obviously was deeply honored and accepted with great pleasure and pride. I naturally assumed to be expected to cover the role of the endothelial cells in local vasomotor control in health and disease, as this has been the major interest of my group(s) for nearly 40 years. Indeed, endothelial cells generate nitric oxide (NO) and other mediators/signals that help to determine the degree of relaxation/contraction of the vascular smooth muscle cells that surround them. In addition, NO not only prevents abnormal constrictions (vasospasm) of the arteries favoring intraluminal clot formation, but also inhibits platelet aggregation, reduces the expression of adhesion molecules at the surface of the endothelial cells (and hence the adhesion and penetration of white blood cells [macrophages]), inhibits the formation of oxidized low-density lipoproteins (LDL) and blunts both the release and action of endothelium-derived vasoconstrictor prostanoids and of endothelin-1 (Figure 1). When endothelial cells become dysfunctional (with aging, obesity and disease [diabetes and hypertension, in particular]), the protective role of NO is reduced and the inflammatory response that leads to atherosclerosis is initiated.1–11 When reflecting on what aspect of abnormal endothelial function to cover best in my lecture, and thus in this essay, I decided to focus on a peculiar type of dysfunction, the one exhibited by the regenerated endothelium following traumatic injury, as this has the highest clinical relevance for patients undergoing angioplasty or receiving a heart transplant. To be honest, the selection of this particular topic was prompted by the fact that my continued interest in the matter has been triggered by the initial outstanding experiments performed by Hiroaki Shimokawa when I had the privilege to work with him at the Mayo Clinic in Rochester, MN, USA. Over the years, we have revisited the problem to obtain a better understanding of why regenerated endothelial cells become so typically dysfunctional and believe we have reached a better understanding of the molecular events involved. In reviewing these initial and subsequent experiments, I will restrict most of the discussion to endothelium-derived NO, because of its predominant vasoprotective role. Endothelial cells do much more than only releasing this mediator, but those aspects have been covered elsewhere.6,9,11–14
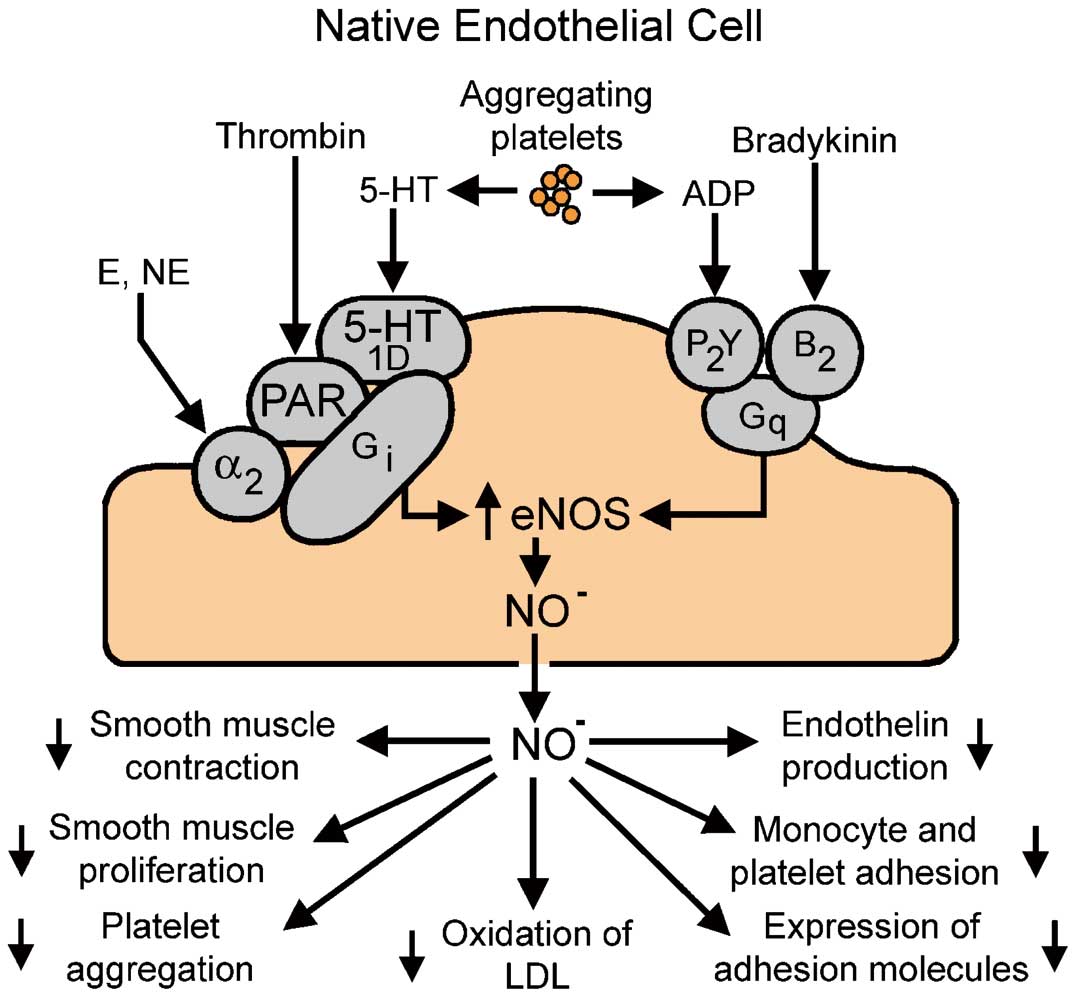
G-protein-mediated signal transduction processes in a normal, native endothelial cell. Activation of the cell causes the release of nitric oxide (NO), which has important protective effects in the vascular wall. ADP, adenosine diphosphate; α, α-adrenergic receptor; B, bradykinin receptor; E, epinephrine; eNOS, endothelial NO synthase; G, coupling proteins; LDL, low-density lipoproteins; NE, norepinephrine; PAR, proteinase-activated receptor; 5-HT, serotonin receptor; P, purinergic receptor. Downward arrows indicate reduced effect/activity. (Modified with permission from Acta Physiologica.9)
The historical experiments by Furchgott and Zawadzki demonstrated the endothelium-dependency of relaxation to acetylcholine in isolated arteries.15 Subsequently, the pivotal role of endothelial NO synthase (eNOS) as a source of NO, the main mediator of such endothelium-dependency was established, opening up a new era in vascular biology.2,8,9,11,15 However, although under peculiar circumstances (eg, lower temperatures) endothelial cells can produce acetylcholine in amounts sufficient to stimulate eNOS in an autocrine manner,11 little evidence is available to suggest that the cholinergic neurotransmitter truly participates in the endothelial control of vascular tone. Hence, early on, the quest was initiated to determine whether or not natural substances more likely to come in contact with endothelial cells shared with acetylcholine the potential to evoke endothelium-dependent relaxation. It soon became apparent that the endothelial cell membrane is endowed with a number of receptor proteins and ion channels that when activated/opened by circulating hormones (eg, catecholamines, vasopressin, insulin, adiponectin) or local autacoids (eg, bradykinin, histamine, angiotensin 1–7, endothelin-1) initiate the stimulation of eNOS, the production of NO and thus endothelium-dependent relaxation.11 In my mind, from a cardiovascular pathophysiological point-of-view, the most relevant endothelium-dependent stimuli appear to be products associated with platelet aggregation and the subsequent coagulation of blood. As a matter of fact, adenosine triphosphate (ATP) and diphosphate (ADP), products released by aggregating platelets activating P2Y receptors, and thrombin, the terminal enzyme in the coagulating cascade activating proteinase-activated receptors (PAR), were among the first substances, potentially present in the vicinity of endothelial cells and demonstrated to evoke endothelium-dependent relaxation; the latter were later identified to result from eNOS activation and mediated by NO released by the enzyme.16–18 This was followed by the demonstration that 5-hydroxytryptamine (serotonin), a potent vasoconstrictor released by aggregating platelets, also evoked endothelium-dependent relaxation (mediated by 5HT1D serotonergic endothelial cell membrane receptors) in coronary arteries, but only so if it was administered intraluminally, or if the vasoconstrictor response to the monoamine was prevented by appropriate blockade (eg, with ketanserin) of the 5HT2 constrictor receptors of the vascular smooth muscle cells.17–20 The next logical step was to examine whether or not aggregating platelets per se could initiate endothelium-dependent relaxation and this soon turned out to be the case for both animal and human platelets because they release serotonin and ADP in amounts sufficient to activate the endothelial production of NO (Figure 2).17,18,20,21 Hence the scenario emerged that if platelet aggregation occurred in the vicinity of a coronary (or cerebral) artery lined with healthy endothelium, then the release of serotonin and ADP, especially if combined with the production of thrombin because of local activation of the coagulation cascade, will powerfully stimulate the endothelial cells to release NO (Figure 1). The endothelial mediator will diffuse to the underlying vascular smooth muscle cells and stimulate their soluble guanylyl cyclase, yielding increased levels of cyclic GMP, initiating relaxation and thus increasing blood flow and mechanically impeding the progression of the coagulation process. In addition, NO also diffuses towards the lumen where it exerts, in a powerful synergy with prostacyclin, an immediate feed-back inhibition on the aggregation process. When the endothelial barrier is interrupted by injury, the vasoconstrictor products (serotonin and thromboxane A2) released by the aggregating platelets can reach the vascular smooth muscle cells and cause their contraction (vasospasm), initiating the vascular phase of hemostasis.6,9,11,22 Further pharmacological analysis, using pertussis toxin (the established inhibitor of Gi-coupling proteins), demonstrated the endothelial cell membrane P2y and PAR receptors responding to serotonin and thrombin, respectively, are coupled to the activation of eNOS by Gi-proteins. However, the P2y-purinoceptors responding to ADP (and ATP) and those responding to bradykinin are coupled to eNOS activation by Gq-proteins (Figures 1,3).11,23–27

Traces of isometric tension recordings in rings (with and without endothelium) of the same canine coronary artery studied in vitro. The author’s platelets (70,000/µl) were added either to quiescent preparations (upper 2 traces) or during sustained contractions (lower 2 traces) to prostaglandin F2α (PGF2α, 2×10–6 mol/L). The presence of the endothelium prevents the vasoconstrictor response to the platelets (Upper) because the platelet products cause the release of NO (Lower). (Data from Houston DS, et al.18)
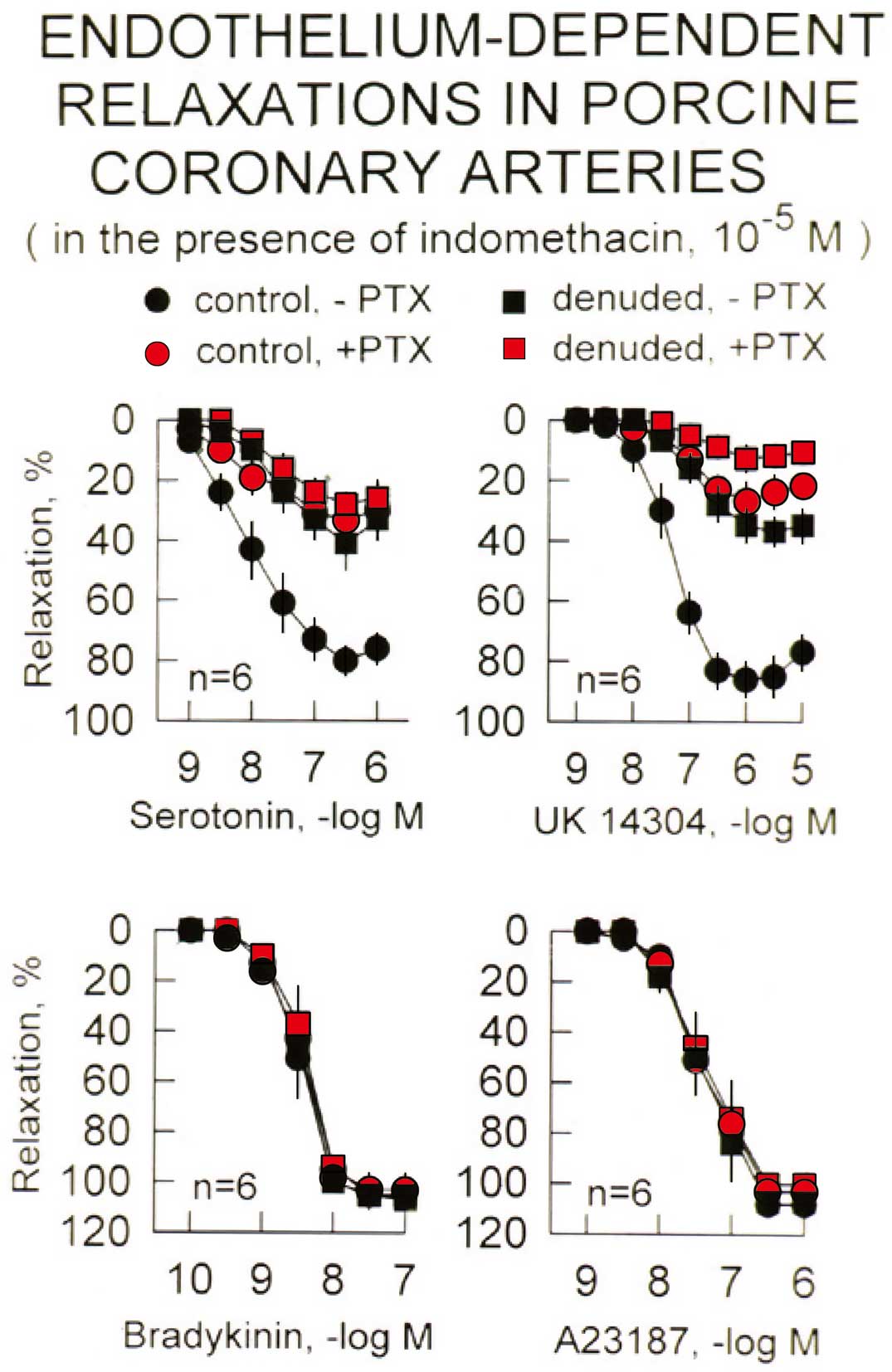
Cumulative concentration-relaxation curves to serotonin (Upper, Left), the α2-adrenergic agonist UK14304 (Upper, Right), bradykinin (Lower, Left) and the Ca2+-ionophore A23187 (Lower, Right) in rings of porcine coronary arteries with endothelium during sustained contractions to prostaglandin F2α. Rings of arteries lined with native (control) or regenerated (denuded: 4 weeks after endothelium removal and subsequent regeneration) endothelium of the same hearts were studied in the absence and presence of the Gi-protein inhibitor, pertussis (PTX). Relaxation is expressed as percent decrease in tension from the contraction evoked by prostaglandin F2α. Data shown as mean±SEM. The data demonstrate the selective reduction in the response to Gi-protein dependent responses (serotonin and UK14304) by PTX, while that to Gq-dependent activation of eNOS (bradykinin) or the Ca2+-ionophore are unaffected. Note that the responses to serotonin and UK14304 in rings with regenerated endothelium are not affected by PTX, and are not different from those in control preparations with native endothelium treated with the inhibitor of Gi-proteins. (Data from Shimokawa H, et al.24)
The ability of aggregating platelets to induce NO-mediated responses is not limited to the coronary arteries, although it is particularly pronounced in those blood vessels. Thus, the endothelium-dependent relaxation response to aggregating platelets, mediated by both serotonin and ADP, has been reported in the basilar, carotid, femoral, iliac, pulmonary and renal arteries, as well as in the femoral and jugular veins.28–32 They are potentiated by the chronic intake of ω-3 polyunsaturated fatty acids (eg, contained in cod-liver oil) but blunted by high fat/high cholesterol diets; these chronic modulations are not selective for Gi-mediated responses but affect the endothelium-dependent relaxation response to ADP and/or bradykinin in a similar manner.28,30,33–37
Once endothelial cells have formed a monolayer (determined by “contact-inhibition”) in mature blood vessels, they apparently remain quiescent for years before apoptotic programming initiates their turnover. The latter is accelerated by aging, exposure to chronic increases in arterial pressure (hypertension) and prolonged oxidative stress (diabetes). The apoptotic cells detach and are removed by the flowing blood. Considerable disruption of the endothelial lining is also an unavoidable consequence of acute cardiological interventions such as angioplasty. Whatever the cause of the interruption of the endothelial monolayer, the denuded/injured parts of the intima are rapidly (within hours or days) recolonized by “regenerated” endothelial cells, originating from replicating neighboring cells (freed of contact-inhibition) and/or circulating endothelial progenitor cells.4,11,38–43 However, to judge from the experiments summarized next, such “regenerated” endothelial cells are dysfunctional, although they maintain the ability to release NO, having lost an important component of the physiological response to aggregating platelets.
Hiroaki Shimokawa introduced us to the technique whereby gentle (trying to minimize the impact on the media) angioplasty with a balloon catheter allows denuding of a portion (ie, 1–1.5 cm) of the porcine left ascending coronary of its endothelium. Within 7 days total relining of the endothelial surface has occurred. At 1 month after the in vivo angioplasty, the preparations containing regenerated endothelium exhibited a considerable reduction in relaxation response to aggregating platelets, serotonin or thrombin, while the remaining, reduced response remained unaffected by pertussis toxin. By contrast, decreases in tension evoked by ADP, bradykinin, and calcium ionophore A23187 (which causes an increase in endothelial Ca2+-concentration, which directly activates eNOS) were normal and, as expected, also unaffected by pertussis toxin, illustrating the maintained ability of the regenerated endothelium to produce NO. We concluded that selective dysfunction of Gi-dependent responses characterizes regenerated endothelial cells. In vitro, the selective endothelial dysfunction results in exaggerated vasoconstrictor responses to serotonergic activation by aggregating platelets, serotonin itself or ergonovine. Likewise, in vivo, this dysfunction unmasked severe angiographic hyperconstriction (vasospasm) in response to aggregating platelets or exogenous serotonin. The selective dysfunction of Gi-dependent responses persisted for at least 6 months (Figure 4), implying that it reflected a permanent weakness of the endothelium. It was delayed by dietary cod-liver oil and amplified by the chronic intake of a hypercholesterolemic diet, which resulted, within a period of 2 months, in the occurrence of typical atherosclerotic lesions only in the area of previous denudation. These experiments made the conclusion unavoidable that the selective dysfunction of regenerated endothelial cells constitutes a locus minoris resistentiae in the coronary arterial wall, which sets the stage for the occurrence of the atherosclerotic process.24–27,30,34–36,44–46

Cumulative concentration-relaxation curves to serotonin (Upper) and bradykinin (Lower) in rings of porcine coronary arteries with endothelium (during sustained contractions to prostaglandin F2α) harvested 4, 8, 16, and 24 weeks after endothelium removal and subsequent regeneration. Relaxation is expressed as percent decrease in tension from the contraction evoked by prostaglandin F2α. Data shown as mean±SEM. Four rings each from control and previously denuded arteries were studied in parallel during each experimental period. The data demonstrate the selective reduction in the response to Gi-protein-dependent responses (serotonin) while that to Gq-dependent activation of eNOS (bradykinin) is unaffected. The preparations of the group at week 4 are the same as those shown in Figure 3, which demonstrated that pertussis toxin inhibits the response to serotonin, but not to bradykinin, in rings with native endothelium, but not in preparations with regenerated endothelium. Note that the selective endothelial dysfunction persisted for a period of at least 6 months. (Modified from Félétou M, et al,6 showing data from Shimokawa H, et al.24 By permission of the American Physiological Society.)
Selective loss of Gi-dependent responses could be caused by either absence of cell membrane receptors or deficiency of the coupling mechanism. The former appears unlikely, at least as regards the endothelial serotoninergic receptors.47 By contrast, the Gi-proteins, although present, appear dysfunctional in regenerated endothelium.48,49 Next, primary cultures were grown from either regenerated or native endothelial cells harvested from the same hearts.49–53 Primary cultures from native endothelium exhibited the expected typical “cobblestone” aspect. However, cultures grown from regenerated endothelial cells contained many large and multinuclear cells with increased presence of β-galactosidase (a prototypical senescence marker), reduced expression and activity of eNOS, greater production of oxygen-derived free radicals, greater uptake of modified LDL and larger generation of oxidized LDL. By contrast, the histochemical presence of Gi-proteins was similar to that of primary native cultures. The changes in mRNA demonstrated in cultures of regenerated endothelial cells were compatible with those phenotypic and functional changes. It had been established by others that exogenously added oxidized LDL blunts NO production and decreases endothelium-dependent relaxation to serotonin in vitro.54,55 Hence it seemed, and still seems logical to conclude that it is the increase in oxidative stress and the resulting augmented presence of oxidized LDL which leads to selective loss in Gi-protein mediated relaxations to serotonin (and thrombin) in regenerated endothelial cells (Figure 5). Obviously, this is but one of the negative effects of oxygen-derived free radicals and oxidized LDL in the endothelial dysfunction that permits the atherosclerotic process.11

G-protein-mediated signal transduction processes in a regenerated endothelial cell. ADP, adenosine diphosphate; α, α-adrenergic receptor; A-FABP, adipocyte-fatty acid binding protein-A B, bradykinin receptor; E, epinephrine; eNOS, endothelial nitric oxide synthase; G, coupling proteins; LDL, low-density lipoproteins; NE, norepinephrine; NO, nitric oxide; oxyLDL, oxidized LDL; PAR, proteinase-activated receptor; P, purinergic receptor; 5-HT, serotonin or serotonin receptor; TX A2, thromboxane A2. Upward arrows indicate increased effect/activity. (Modified with permission from Acta Physiologica.9)
Of the genomic changes observed in primary cultures (Figure 6), the most intriguing ones were the emergence of metalloproteinase-7 (MMP7) and adipocyte-fatty acid binding protein-A (A-FABP). Indeed, the mRNAs of those 2 proteins are expressed in regenerated, but not native endothelial cell cultures.53 The overexpression of both A-FABP and MMP7 is observed only after regeneration in vivo, but is absent if native endothelial cells are made senescent in vitro.53,56 Lipid-induction of A-FABP expression in cultured human endothelial cells reduces the phosphorylation of eNOS and hence the production of NO, effects that can be prevented by the A-FABP inhibitor BMS309403.57 In a rodent model of atherosclerosis (the apolipoprotein E knock-out (ApoE-/-) mouse), the immunochemical presence of A-FABP in the endothelium augments progressively with age, in parallel with endothelial dysfunction and the occurrence of atherosclerotic lesions; chronic administration of BMS309403 delays the atherosclerotic process and prevents the endothelial dysfunction.57,58 These experiments in the ApoE-/- mouse, taken in conjunction with the major overexpression of A-FABP mRNA in primary cultures of porcine regenerated endothelial cells, prompted testing of the effect of chronic administration of BMS309403 in our pig model. Following angioplasty in the coronary artery, chronic treatment with the A-FABP inhibitor prevented the selective reduction in endothelium-dependent responses to serotonin, as well as the intimal thickening caused by endothelial regeneration in vivo, without affecting relaxation responses to either bradykinin or the exogenous NO-donor sodium nitroprusside; a similar protection was obtained with chronic administration of the antioxidant apocynin (Figure 7).59 These observations strongly support the conclusion that the overexpression of A-FABP, the subsequent increase in oxidative stress, and hence the resulting presence of oxidized LDL in the regenerated endothelial cells underlie their dysfunction. At the present time, our findings do not permit us to speculate further as to the molecular link between an increased expression/presence of A-FABP and augmentation in the production of oxygen-derived free radicals. Likewise, we do not know whether or not the augmented expression/presence of MMP7 plays any role at all in the selective blunting of Gi-mediated responses of regenerated endothelial cells, although a role for that particular MMP in cardiovascular disease is conceivable.60

Upregulation of genes in primary cultures of regenerated endothelial cells. A-FABP, adipocyte-fatty acid binding protein; MMP7, matrix-metalloproteinase 7; the mRNA for those 2 genes was below detection level in primary cultures of native endothelial cells. Upregulated genes in regenerated compared with native endothelial cells were: HPGD, hydroxyprostaglandin dehydrogenase 15-(NAD); GADD45A&B, growth arrest and DNA-damage-inducible, α and β; F2, coagulation factor II; CXCL12, stromal cell-derived factor-12; MMP23A&B, matrix-metalloproteinase23A&B; COL1A1, collagen α 1; GM-CSF, granulocyte macrophage colony-stimulating factor; TGF-β1, TGF-β 1; PPAR γ 1a, peroxisome proliferator-activated receptor γ 1a; Gp VII PLA2, phospholipase A2. (Modified with permission from Lee MYK, et al of the American Heart Association.53)

(Upper) Responses to serotonin (10–9 to 10–6 mol/L) in porcine coronary arterial rings with native or regenerated (1 month after angioplasty) endothelium harvested from pigs treated with vehicle (Control; Left), the A-FABP inhibitor BMS309403 (1.5 mg·kg–1·day–1; Middle) or the antioxidant apocynin (4 mg·kg–1·day–1; Right). Relaxation is expressed as percentage of the precontraction and shown as mean±SEM. (Middle) Representative cross-sections of arteries of the 3 experimental groups, with native or regenerated endothelium, stained with modified Curtis’s Ponceau. (Lower) Quantification of the cross-sectional area of the intima-medial layer of the arteries with native (black bars) or regenerated (pink bars) endothelium after chronic treatment for 28 days with vehicle BMS309403 or apocynin. Data are shown as mean±SEM; *P<0.05 native vs. regenerated. (Modified with permission Chan C, et al.59)
It is conceivable that, besides angioplasty, any manipulation of a blood vessel will lead to partial disruption of the endothelial lining. This will be followed by rapid regeneration of endothelial cells, which are bound to be dysfunctional in terms of endothelium-dependent responsiveness, in particular as regards relaxation responses to aggregating platelets. This conclusion is comforted by the demonstration that in reversed vein grafts, endothelium-dependent relaxation is absent 4 weeks after implantation.31 Likewise, in the development of the vasculopathy leading to the accelerated atherosclerosis that often accompanies cardiac transplantation, the first sign of vascular dysfunction is the blunting of endothelium-dependent relaxation responses to agonists (serotonin, α2-adrenergic agonists) that are coupled to the activation of eNOS by Gi-proteins; the characteristics of endothelial dysfunction in the coronary arteries of the allografted heart are remarkably similar to those observed in preparations lined with regenerated endothelial cells.61,62 The severity of the vascular lesions in the allografted heart is exacerbated by chronic inhibition of eNOS.63 Hence, one is tempted to conclude that the unavoidable endothelial damage, sustained by the transplanted heart prior to and during the surgical procedure, although it has been followed by regeneration, again sets the stage for the occurrence of the atherosclerotic process.
The activation of eNOS and the resulting increased production of NO constitute a major role played by the endothelial cells in protecting the vascular wall. Some of the products (serotonin and ADP) released by aggregating platelets are among the pathologically most relevant stimuli initiating the protective release of endothelium-derived NO. They are coupled to the activation of eNOS by different Gi-proteins. The efficacy of the platelet products in causing endothelium-dependent relaxation is modulated non-selectively by the diet and is in particular blunted by hypercholesterolemia. When the endothelium is removed mechanically, rapid regeneration of the endothelial lining occurs. However, the regenerated endothelial cells selectively lose their Gi-mediated coupling, rendering them particularly less responsive to serotonin, the major mediator of the endothelium-dependent relaxation/dilatation evoked by aggregating platelets, and thrombin. Hence, the areas of arteries covered with regenerated endothelium cannot produce enough NO in response to these stimuli. This local NO deficiency not only favors the intravascular coagulation process and the occurrence of vasospasm, but also permits the inflammatory reaction leading to atherosclerosis.11,64–66
The author thanks Mrs Ivy Wong most sincerely for her usual, most efficient editorial assistance. He also thanks his former collaborators for their help and inspiration during the studies briefly summarized in this essay, with particular gratitude to a group of outstanding participating Japanese post-doctoral fellows: Hiroaki Shimokawa, Kimihiro Komori, Toshiro Shibano and Phyo Kim.