Abstract
Tissue salvage of severely ischemic myocardium requires timely reperfusion by thrombolysis, angioplasty, or bypass. However, recovery of left ventricular function is rare. It may be absent or, even worse, reperfusion can induce further damage. Laboratory studies have shown convincingly that reperfusion can increase injury over and above that attributable to the pre-existing ischemia, precipitating arrhythmias, suppressing the recovery of contractile function (“stunning”) and possibly even causing cell death in potentially salvable ischemic tissue. The mechanisms of reperfusion injury have been widely studied and, in the laboratory, it can be attenuated or prevented. Disappointingly, this is not the case in the clinic, particularly after thrombolysis or primary angioplasty. In contrast, excellent results have been achieved by surgeons by means of cardioplegia and hypothermia. For the interventionist, the issue is more complex as, contrary to cardiac surgery where the cardioplegia can be applied before ischemia and the heart can be stopped, during an angioplasty the heart still has to beat to support the circulation. We analyze in detail all these issues.
Medicine has been undergoing constant changes, with cardiology being arguably the fastest changing field of all. The evolution – or should we say the revolution
–
of reducing the progression of coronary atherosclerosis with aspirin, angiotensin enzyme inhibitors and cholesterol-lowering drugs had a remarkable effect on the treatment of coronary artery disease. But perhaps one of the most important changes in cardiology is the ability to reperfuse the ischemic myocardium and consequently to reduce the deleterious effects of acute ischemia. Interestingly, during the second half of the century, 4 independent and different streams of research were initiated and would culminate in the ability to reperfuse the ischemic human heart.
The first line of research related to pathophysiology. The 1960 s witnessed a remarkable increase in the systematic study of (1) the nature of atheroma and thrombosis and (2) the metabolic, contractive, ultrastructural and electrophysiological consequences of myocardial ischemia. Later, in part because of the relative ease of restoring coronary flow, investigators, including the authors of this review, turned their attention to the effects of reperfusion.1–4
The idea of protecting the ischemic myocardium soon attracted a lot of enthusiasm and a cornucopia of compounds such as β-blockers, calcium antagonists, antiinflammatory drugs, and vasodilators were administered to animals with results that appeared dramatically successful. In the 1970 s and 1980 s, however, it became clear that this approach was wrong, an error that was confirmed by a number of completely unsuccessful clinical trials that failed to show the benefits with a range of putative infarct-reducing agents. The main reason for this discrepancy lay in the time of administration of the compounds (i.e., before the onset of ischemia in the experimental setting, after the infarct in the clinical one).
It then became apparent that early reperfusion was a prerequisite for the salvage of ischemic tissue. At the same time, it became also evident that reperfusion per se can be, paradoxically, dangerous, increasing rather than reducing the extension of the infarct.
The second line of research related to cardiac surgery. Quickly, coronary artery bypass became a standard procedure for patients with angina and, with the time, this was extended, although with poor results, to evolving myocardial infarction (MI). No doubt that the surgeons had the monopoly of reperfusion and indeed they managed to develop techniques such as cardioplegia and hypothermia able to protect against the damage induced by reperfusion. Once again, the success of these techniques relies on their application before induction of ischemia.
The third line of research is linked to pharmacologists who, in parallel with surgeons and pathologists, questioned that the thrombus was the result of an infarct. They proved that, actually, it is the cause of the infarct and started to look at measures to dissolve the infarct-initiating thrombi. Early attempts to induce fibrinolysis with streptokinase were, for a variety of reasons, largely ignored until the completion of the Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI) and the Second International Study of Infarct Survival (ISIS2).5,6
These mega trials proved the clinical beneficial effects of thrombolysis, which was accepted as treatment of MI. However, at that time, it was also appreciated that late reperfusion was totally unsuccessful, if not dangerous.
The fourth line of research has led to the concept of percutaneous coronary intervention (PCI) pioneered by Andreas Grüntzig in the 1970s. Interventional cardiologists started to alleviate perfusion deficit and unblock coronary arteries, even in the middle of the development of acute MI (AMI). Timely primary angioplasty is now the gold standard for treating acute ischemia, but again, it was immediately recognized that the time to reperfusion is the key to success, later reperfusion being possibly linked to further extension of ischemic damage. This has led to the establishment of a modern network called “hub and spoke” with the aim of reperfusing as early as possible. Thus, the 1990 s and 2000 s witnessed the development of ingenious catheters armed with cameras, lasers, isotopes, sophisticated stents releasing drugs and/or even disappearing from the coronary arteries, and several drugs aimed at reducing reperfusion injury (RI), avoiding myocardial damage and late further thrombosis.
Thanks to these 4 lines of research we have certainly come a long way since the early days of morphine, β-blockers, and intensive care units. Now we have a better understanding of the nature of ischemia, an appreciation of the importance of early reperfusion and a wide portfolio of different procedures to achieve it. As a result, cardiology has contributed at least 7 years to the 10-year increase in life expectancy that has occurred in the past 30 years.7
This is, by any measure, an incredible record, considering that all the other subspecialties have contributed (at the best) some months!
However, despite modern secondary prevention therapy, the risk of acute coronary events remains high, resulting in acute coronary syndrome (ACS), which is not a single disease entity, but a heterogeneous spectrum of conditions that share key pathophysiological features. The clinical presentation of ACS comprises ST-segment elevation MI (STEMI), non-STEMI (NSTEMI) and unstable angina. The clinical presentation is determined by the extent of coronary obstruction, the temporal pattern of the atherothrombotic disease process and the volume of ischemic myocardium, which, in turn, depends on the degree of collateral circulation, the pre-existing metabolic rate, genetic factors, and the intrinsic survival capacities of the myocytes, making ischemia a rather “personal” conception (Figure 1). Thus, complete coronary occlusion typically underlies STEMI, whereas incomplete coronary occlusion is associated with NSTEMI and ventricular arrhythmias.

The damage inflicted on the myocardium during abrupt occlusion of an epicardial coronary artery is the result of 2 processes: ischemia and, in some instances, subsequent reperfusion (ischemic/reperfusion injury). That reperfusion itself might contribute to the damage during AMI seems a paradox as, unquestionably, timely revascularization either by thrombolytic therapy or primary percutaneous coronary angioplasty (PCA) is, at present, the most effective therapy for limiting infarct size, reducing the healing pattern of the infarcted zone, preserving left ventricular systolic function and delaying the onset of remodeling and heart failure.1–4
In the present review we analyze the “two-faced aspect” of reperfusion itself, we highlight the progress and the failure of each of the 4 lines of research, trying to summarize the most important and probable physiopathologic aspects of RI, as well as the evolving therapies targeting ischemic/reperfusion injury.
Pathophysiology of Ischemic Damage
Adult cardiac myocytes are mainly terminal cells, and have no or little replicative capacity with which to replace cell loss.8
It follows that preservation of cardiac structure and function is the ultimate target of every therapeutic intervention, including reperfusion of the ischemic myocardium.
Cell death from coronary artery occlusion starts to occur after approximately 20 min of severe ischemia.9
Collateral coronary blood flow and partial, instead of total, coronary artery occlusion in humans may allow myocyte survival for a longer period of time. Irreversible ischemic injury, cell death and subsequent necrosis begin in the subendocardium and cell death progresses in a wave front from the subendocardium into the subepicardium of the ischemic bed-at-risk.9
Hypoxia and Ischemia
These terms describe a condition of reduced oxygen availability in the cell such that it cannot maintain mitochondrial oxidative processes.10
In ischemia, the lack of oxygen is caused by a reduction in coronary flow. In hypoxia, which is mainly an experimental condition, the flow is normal but it does not provide oxygen. The nearest clinical condition is anemia.
As soon as the supply of energy fails to match the needs of intracellular metabolism, a cascade of increasingly severe metabolic perturbations commences. The cell becomes “metabolically distressed,” and unless interrupted by early reperfusion, ischemia will inevitably progress towards cell death and necrosis and or infarction (Figure 2).

During short periods of ischemia (e.g., in angina), the match between biochemical and mechanical activity is maintained; mechanical activity in the ischemic areas is drastically reduced (hypokinesia and akinesia), and the residual delivery of oxygen may just be sufficient for a short period to maintain cell viability. As shown in
Figure 3, in the isolated and reperfused rabbit heart, complete abolition of coronary flow (global ischemia) results in rapid downregulation of contraction and, eventually, quiescence, caused by intracellular acidosis, which develops within seconds of induction of ischemia and reduces calcium movement within the sarcolemma, sarcoplasmic reticulum (SR), and myofilaments.11
Shortly after this, the energy charge of the myocyte is reduced, and creatine phosphate (CP) levels decline faster and to a greater extent than those of ATP. Anaerobic metabolism develops, leading to lactate production, and contributes to the formation of limited amounts of ATP by oxygen-independent, substrate-level phosphorylation.12
Both contractile downregulation (and therefore decreased ATP consumption) and increased anaerobic ATP production are cellular cardioprotective mechanisms occurring, however, at the expense of contraction.
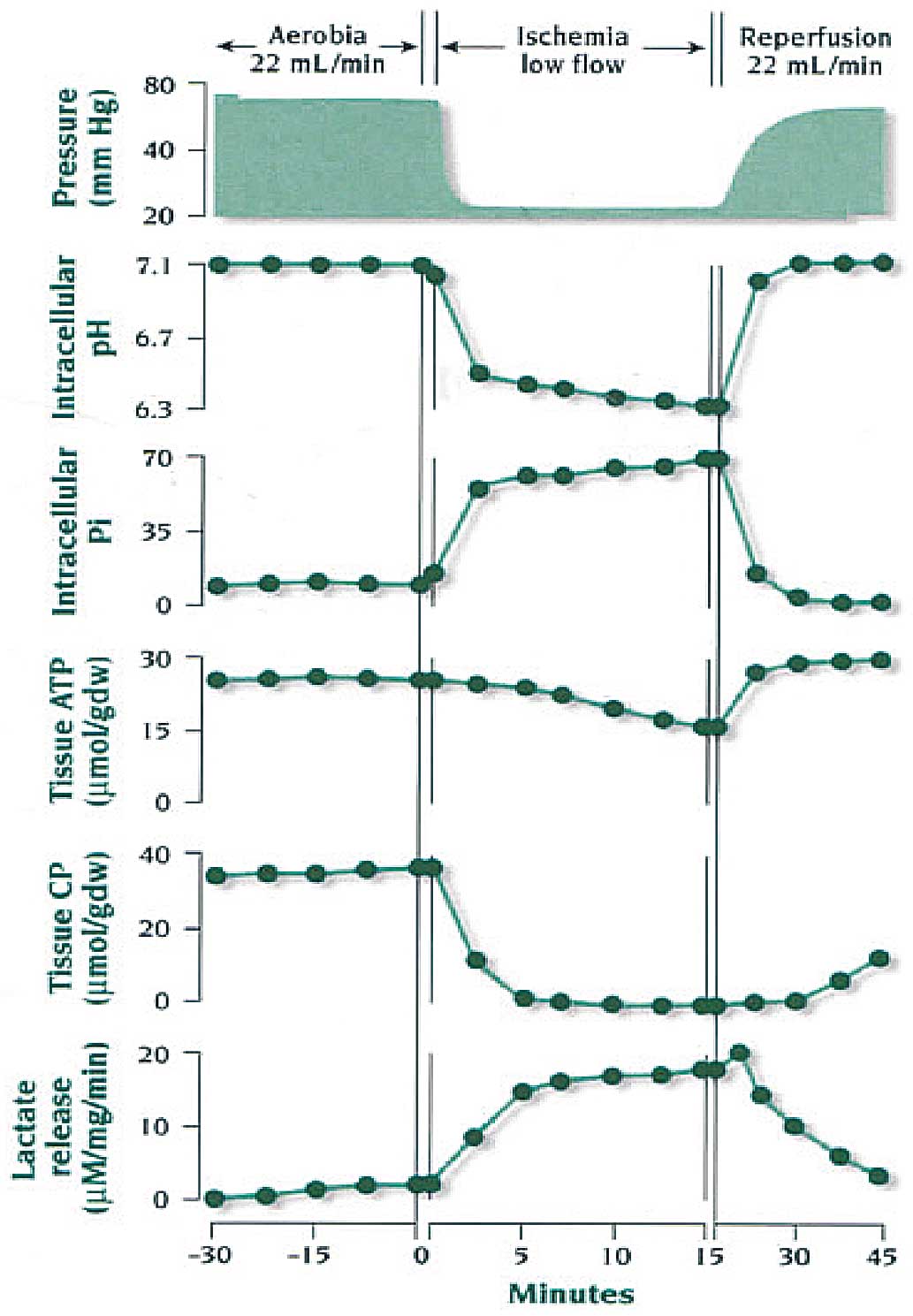
Reperfusion at this stage results in recovery of high-energy phosphate production, which, in turn, indicates that the mitochondria are still functional and capable of normal aerobic metabolism. This is linked to a recovery of mechanical function that can be either immediate or somewhat delayed because of the presence of stunning. Often in this phase of “early reperfusion” after short period of ischemia, several arrhythmias develop independently of the presence of stunning.
Reperfusion Arrhythmias
Arrhythmias during or immediately after reperfusion are seen in experimental animal models and in humans.13
Among the animal studies, those of Manning and Hearse using isolated and perfused hearts provided a very clear description of reperfusion-induced arrhythmias and the possibility to reduce their occurrence with different antiarrhythmic drugs.14
In humans, arrhythmias are often present in reperfused STEMI patients, particularly after thrombolysis. Actually, early reperfusion arrhythmias, together with the early blood increase of intracellular biomarkers, are considered evidence of successful reperfusion and vitality of the myocytes.15
Myocardial Stunning
This term refers to reversible post-ischemic contractile dysfunction that can occur in large animal models and in humans.16
It is common knowledge that ischemic myocardium does not necessarily recover its function immediately after reperfusion. It can happen at a later stage, even weeks later in humans. Like arrhythmias, stunning is considered a benign form of reperfusion damage. Normally it does not need any specific treatment because it is unpredictable and a specific anti-stunning treatment is not available. Nonetheless, in the unfortunate cases when reperfusion results in severe stunning, causing low cardiac output, positive inotropes and vasodilators are used in the hope that severe stunning and not irreversible damage is the ultimate cause. Several hypotheses have been taken into consideration to explain stunning, such as extracellular collagen alterations, myofilament malfunction, reduced sympathetic response, reduced energetic reserve, calcium metabolism alterations, and cellular damage mediated by reactive oxygen species.17,18
The most likely and popular explanation is linked to the effects of oxidative stress and calcium overload on the contractile apparatus, but even this hypothesis has never been confirmed.19,20
Myocardial Hibernation
Repetitive episodes of stunning following more prolonged periods of ischemia may, in some patients, result in chronic regional malfunction, which will recover upon reperfusion. This condition is normally referred to as “hibernation”. The term “hibernation” implies an adaptive reduction in energy use in the presence of a reduced energy supply, through reduced activity. In the early 1980 s, Rahimtoola21
reviewed the results of coronary bypass surgery trials and identified a subset of patients with coronary artery disease and chronic left ventricular dysfunction that improved upon revascularization. The rapid amelioration of myocardial function obtained by revascularization ruled out the hypothesis that the reduced dysfunction was caused by histological modification of the myocardium, and left the scientific community with the dilemma of explaining this phenomenon. The concept of chronic, yet reversible, contractile dysfunction in patients with coronary artery disease was not easy to grasp. It challenged traditional view that the extent of chronic contractile dysfunction reflected the amount of infarcted tissue.22
Two hypotheses have been put forward to explain hibernation: (1) the mechanical alteration is caused by “chronic ischemia” or better still, to chronic coronary blood flow reduction with chronic intracellular acidosis, downregulating contraction to allow the maintenance of an energy matching (low or no contraction reduces energy needs) of viability despite reduced ATP production;12,22,23
or (2) hibernation is caused by repetitive ischemic insults that maintain a chronic state of myocardial stunning.24,25
Prolonged Irreversible Ischemia
Experimentally, myocardial reperfusion after prolonged ischemia does not bring about any contractile recovery. Instead, it causes accumulation of tissue and mitochondrial calcium, a reduction in the mitochondrial capacity to utilize oxygen for ATP production, and further deterioration of diastolic function, with reduction in compliance26,27
(Figure 4). Histologically, reperfusion after prolonged ischemia causes explosive edema and swelling of the cytosol, massive hypercontraction bands and sarcolemmal damage, as well as mitochondrial calcium accumulation in the form of phosphate granules.26,27
It is not simple to determine the duration and severity of ischemia capable of producing irreversible damage, as it varies from the experimental setting and in the clinical condition.
Figure 5
shows that in isolated cells it is a question of minutes, whereas in isolated larger preparations, it is a question of hours, and in humans, several hours (unless hibernation develops). If coronary blood flow remains severely reduced, the myocardium will remain quiescent; biochemical ischemia will nonetheless intensify, and proceed towards irreversible damage. In experimental studies, this is indicated by the continuing release of lactate, increases in diastolic pressure (indicating a severe perturbation of ionic calcium homeostasis), and signs of severe sarcolemmal damage (e.g., enzyme and troponin leakage). Under these circumstances, the mitochondria themselves become targets for ischemic damage, as they show reduced function, decreased membrane potential, and decreased activity of NADH dehydrogenase as well as differing degrees of uncoupling.26,27
Some, but not all, investigators believe that reperfusion itself might be detrimental and able to inflict injury over and above the level of that attributable to ischemia.26
Other investigators, however, question the existence of reperfusion-induced injury.1,2
Numerous studies suggest that calcium, oxygen-derived free radicals and pH changes contribute to post-ischemic dysfunction. Electron magnetic resonance spectroscopy has directly demonstrated the formation of free radicals and calcium overload in the stunned myocardium, as well as during reperfusion after prolonged ischemia, leading to the so-called “reperfusion paradox”.

Paradoxes of Reperfusion Injury
Several hypotheses have been proposed to explain why reperfusion should result in further injury. The research has followed different lines of enquiry, depending on the specific time and trend of the moment.
Calcium Paradox
One of the first ideas to explain the reperfusion paradox was linked to the phenomenon named “calcium paradox” (i.e., the readmission of calcium after a short period of calcium-free perfusion causes massive, immediate damage to the myocyte, similar to that seen after late reperfusion, including calcium overload).26
This later event is particularly detrimental because the mitochondria, during reperfusion, may use the restored oxygen for calcium transport instead of for ATP production, as the 2 processes compete for the same energy source (i.e., the ∆Ψ generated across the inner mitochondrial membrane).27
As a consequence, at the time, it was logical to consider calcium antagonists to attenuate calcium overload. These agents, however, despite being highly protective when used before or at the time of ischemia, failed to reduce late reperfusion damage in isolated and perfused heart preparations.28
When tested in larger animal preparations, calcium antagonists decreased infarct size.29
Despite this evidence, the results of the corresponding clinical trials have been unequivocally negative, irrespective of whether the calcium antagonists were administered before, during or after ischemia.30,31
Another possibility of reducing calcium overload is to inhibit the sodium-hydrogen exchange. Again, the literature reports promising experimental results when the inhibitors were used during ischemia,32
but failure when provided during PCI.33
Ranolazine, an inhibitor of late sodium influx, has also been suggested as cardioprotective by reducing the calcium overload, but when tested in 2 large clinical trials, it failed to improve prognosis or reduce the occurrence of MI.34,35
Oxygen Paradox
It has been suggested that the readmission of oxygen causes oxidative stress with consequent membrane peroxidation and immediate damage. This process was considered likely to occur because ischemia causes a time-dependent reduction of the natural myocardial defense mechanism against oxygen free radicals (i.e., mitochondrial superoxide dismutase activity and the cytosolic glutathione-peroxidase and reductase system), as well as a washout of sarcolemmal α-tocopherol.36,37
The readmission of oxygen in the absence of all existing natural scavengers could cause rapid damage to the sarcolemma, which would then become permeable to Ca2+. Unfortunately, despite strong logic, both animal and clinical studies examining the cardioprotective effect of different antioxidants have always been inconclusive.1
As oxidative stress reduces the availability of nitric oxide (NO), it was also thought that the administration of NO donors could be cardioprotective. When tested in the clinic and administered to reperfused STEMI patients, nicorandil, an NO donor, and trimetazidine, a metabolic modulator with antioxidant properties, failed to limit the infarct size.38,39
Today, the damaging effect of oxygen free radicals is recognized by the scientific community in various diseases but, unfortunately, it is not a target for reperfusion therapy.
pH Paradox and Mitochondrial Permeability Transition Pore (mPTP)
During reperfusion there is a rapid washout of lactate and a restoration of the physiological pH. This, in turn, induces opening of the mPTP, which is a non-selective channel of the inner mitochondrial membrane that is closed under physiological conditions (Figure 6).40
During ischemia, the susceptibility of the mPTP is increased, but the pore remains closed when the pH is low. Thus on reperfusion, there is a favorable milieu for mPTP opening and the rapid neutralization of the acid pH triggers the actual opening allowing passage through the inner mitochondrial membrane of all molecules <1.5 kDa. This, obviously, is a major problem for the still viable cells: as the inner mitochondrial membrane becomes freely permeable to protons it effectively uncouples oxidative phosphorylation and disrupts ATP production.41
The abrupt increase in calcium flow into the mitochondria triggers different caspases which, in turn, lead to cell death by apoptosis. It follows that cells that survive the ischemic insult might die from the damage generated by coronary reperfusion. Nowadays, the molecular nature of the mPTP is still unclear.42
Figure 6
shows a schematic model of mPTP and the potential role of the C subunit in its formation. Several agents have been suggested to limit RI by inhibiting the mPTP, including cyclosporine, TRO40303, exenatide and bendavia. Once again, as with all other attempts with different drugs, the clinical results are not encouraging. The compounds that have been most extensively tested are cyclosporine and TRO40303. Unfortunately, a large study with cyclosporine did not show any differences between the placebo and treated groups at 1 year.43
Other trials, testing the role of TRO40303 as an adjunct therapy to PCA (MITOCARE), showed no cardioprotective effects.44
The C subunit of ATP synthase could be an interesting new target.42
Preliminary findings suggested that the C subunit is detectable in serum from patients admitted to hospital for STEMI and undergoing successful primary PCI.45
In addition, elevated levels were significantly related to poor values of surrogate RI endpoints such as ST-segment resolution, TIMI myocardial perfusion grade, TIMI frame count and cardiac marker release.45

Reperfusion Injury: a Series of Paradoxes Solve the Enigma
It seems that reperfusion or re-energization after a prolonged period of ischemia sets in motion a series of paradoxes (Figure 7), which, most likely, all together contributes to RI. The paradoxes are: (1) readmission of oxygen causes oxidative stress, and, at least in some still viable cells, a recovery of energy production that reactivates the SR pump, leading to excess sequestration of calcium, often exceeding the capacity of the SR, thus initiating a continuous release and reuptake of calcium; (2) at the same time the restored oxygen cannot be scavenged, with production of oxygen free radicals that destroy membrane structure, including the sarcolemma, leading to massive calcium entry; (3) interstitial pH is rapidly normalized by proton and lactate washout and a gradient is generated between the extracellular space and the cytosol, which still contains a high concentration of protons. This, in turn, activates proton-extruding mechanisms, namely the sodium/hydrogen exchange, thus causing a net influx of sodium into the cytosol. Depending on the ability of the sodium pump to remove some of the excess sodium, there will likely be further calcium overload (by activation of the sodium/calcium exchanger) and massive cellular uptake of water and subsequent edema; (4) the rapid increase in pH also activates the opening of the mPTP, with immediate entry of calcium into the mitochondria. This dissipates the ∆Ψ and, more importantly, activates the caspase cascade, leading to cell death which, under such a complex scenario, can occur either by necrosis or apoptosis (Figure 8).

Reperfusion Injury in the Clinical Arena
The absolute need for coronary revascularization to save myocardium threatened by ACS arises, to a large extent, from the limitations of currently available pharmacotherapy. Apart from the clear, scientifically proven advantages of reperfusion either by PCI or thrombolysis, there is also a favorable psychological connotation for both patients and practitioners: reperfusion more closely approximates a “cure”! “If I were an ischemic myocardial cell, and someone offered me drugs or blood, I think I would take blood” (W. Paulus, pers. COMM., 1992).
In the setting of STEMI, randomized clinical trials have consistently demonstrated that emergency PCI is the most effective and safest method of re-canalizing the infarct-related artery and that its use is associated with the lowest mortality and morbidity rates.46,47
There are, however, 2 factors that are the keys to a good outcome in this setting. The first is the timely implementation of PCI. It is essential for patients to be treated within the first 2 or 3 h after symptom onset because recanalization beyond this time will adversely affect infarct size and survival (i.e., RI). The second is the availability of expert and well-trained interventionists and an interventional cardiology team, because higher volume primary PCI centers are associated with improved outcomes. Provided these 2 requirements are met, primary PCI is superior to thrombolysis. However, real-life data from registries have not confirmed the real superiority of primary PCI over intravenous thrombolysis48,49
and this appear to be largely related to RI occurring after longer delays in implementing primary PCI compared with the ideal setting of trials.
Ischemic injury is probably the best demonstration of the reality of lethal RI. Apart from ventricular arrhythmias, myocardial stunning, and further cell death induced by reperfusion, microvascular no-reflow and conditioning are important clinical aspects of this complex phenomenon.
Microvascular No-Reflow
Aside from arrhythmias and stunning, the so-called no-reflow phenomenon has been another suggested form of RI. The coronary microcirculation plays a fundamental role in the success of reperfusion. If blood flow is restored to the epicardial artery but there is an obstruction of the microcirculation, the myocardium will remain ischemic and will never recover. Thus, microvascular obstruction is considered to be an irreversible form of damage that results in both myocyte and endothelial death. The phenomenon was first described in the feline heart,50
and confirmed in dogs,51
as the presence of severe capillary damage, swollen, ruptured endothelial cells, intraluminal thrombosis and irreversibly injured cardiomyocytes.52
Whether the phenomenon itself causes further ischemic damage is doubtful: if reperfusion does not occur, there will inevitably be no-reflow at both the epicardial and microvascular level, in any case resulting in further damage. In the clinic the no-reflow phenomenon is detected angiographically (in the case of PCA) from no or slow flow of contrast media, despite successful recanalization of the epicardial coronary artery.53
It normally develops within minutes after reperfusion and might persist for 1 week.53
As the ischemic zone and the infarct area are not reperfused, the washout of intracellular biomarkers is also reduced or abolished, contributing to underestimation of the final infarct size. This is particularly relevant in the case of thrombolysis, in the absence of angiographic data, and might explain the often detected discrepancy between the estimated (from the biomarkers curve) and actual area of necrosis. It follows that it is difficult to establish the frequency of the no-reflow phenomenon in the clinic, although it has been estimated to occur in 10–30% of patients with reperfused STEMI.54,55
The underlying cause of the microvascular obstruction is unclear. A number of mechanisms have been proposed, including (1) embolization of particulate debris from the spontaneous or induced rupture of the culprit atherosclerotic plaque; (2) vasoconstriction caused by release of vasoconstrictor substances, again from the culprit lesion; (3) microvascular compression by edema of the ischemic myocytes, and (4) platelet and leukocyte aggregates from the culprit lesion or from blood flow as part of the general inflammation associated with STEMI.56
Currently, there is no possibility of predicting the no-reflow phenomenon or an effective specific therapy. Therefore, the slow- or no-reflow phenomenon, unfortunately, remains a neglected therapeutic target.
Preconditioning
Ischemic preconditioning is on the one hand the demonstration of lethal RI and, on the other hand, the prototype of nonpharmacological cardiac protection. The phenomenon was identified by chance by Murry et al,57
who showed that 4 cycles of 5 min of coronary occlusion and 5 min of reperfusion before a sustained coronary occlusion for 40 min remarkably reduced the infarct size in dogs. Importantly, the protection was independent of collateral blood flow, but it is time-limited (i.e., if ischemia is prolonged to 2 or 3 h, ischemic preconditioning has no effect).58
Thus, preconditioning delays but not prevents ischemic damage. The mechanisms that underlie the cardiac protection induced by preconditioning rely on the activation of existing endogenous signaling molecules.59
These molecules trigger intracellular mediator cascades and the activation of different effectors that stabilize myocytes and prevent their death. An abundance of signaling pathways have been discovered in many species and preparations. NO, protein kinase activation and mitochondria with the opening of the mPTP are considered indispensable elements in the signal pathways of preconditioning.60
The unpredictability of STEMI makes the application of preconditioning impossible in this clinical setting, although it could have a role in planned cardiac surgery.61,62
Post- and Remote Conditioning
Zhao et al showed (in dogs) that brief episodes of ischemia and reperfusion, performed immediately after reperfusion following prolonged ischemia reduce the final infarct size and they named this phenomenon ‘post-conditioning’.63
Staat et al64
applied a similar protocol in men with STEMI, within 1 min after primary angioplasty, by inflating/deflating the balloon in a 1-min cycle. This resulted in a 34% reduction of the area under the curve for creatine phosphokinase. However, a larger randomized clinical trial of post-conditioning in STEMI was neutral.65
Another very attractive form of myocardial conditioning that does not involve manipulation of the culprit coronary lesion is the so-called “remote conditioning”: conditioning performed in a distant organ.66
The mechanism of transfer of the cardioprotective signal from a distance is unclear. Both neuronal and humoral hypotheses have been suggested.67,68
Interestingly, a few groups have routinely applied remote preconditioning protocols in the ambulance during transfer to the PCI center, using 4- to 5-min brachial cuff inflation, with encouraging results.18
Reperfusion Injury in the Surgical Arena
Like PCI, revascularization of ischemic patients by means of coronary artery bypass grafting (CABG) has developed very rapidly, largely because of the heroic nature of adventurous surgeons, the development of the heart-lung machine and of techniques able to protect against surgically induced ischemia, allowing the human heart to undergo total ischemic arrest for longer than 3 h without inducing any RI. Despite this, PCI is increasingly taking center stage with respect to CABG as the therapeutic option for revascularization and it is steadily gaining new indications. Whether the ongoing replacement of CABG with PCI is justified or not is not the scope of this review. Instead, we are interested in CABG as a demonstration that RI does exist in the clinic and, more importantly, can be prevented. It is also interesting to elaborate the reasons why it is possible to avoid post-RI during CABG but not during PCI.
The Stone Heart Syndrome
During the early days, cardiac surgery was aggravated by a troublesome syndrome delineated by Cooley and his associates as “the stone heart”.69
This condition was observed in patients undergoing open heart surgery with anoxic arrest of the myocardium. Despite the operation proceeding uneventfully, cardiac function failed to resume on reperfusion, when cardiopulmonary bypass was discontinued. Typically, the heart remained firm, the left ventricle became hyper contracted, like a “stone”, prone to fibrillate and unable to sustain the circulation despite patent coronary arteries. When examined at autopsy, the myocardium of both ventricles displayed severe subendocardial contraction band necrosis similar, if not identical, to that occurring during experimental RI.70
The possible explanation at that time was that the period of non-perfusion of the myocardium depleted the energy reserves of the myocyte and therefore damaged the cell membrane, making it abnormally permeable to calcium. Under these conditions, reflow of blood at the discontinuation of cardiopulmonary bypass resulted in a massive entry of calcium through the damaged sarcolemma and SR.70
Of course, at that time, the existence of the mitochondrial calcium pore was unknown, but the explanation is rather similar to the current one and, in any case, the stone heart is a clear demonstration that RI also exists in humans.
Cardioplegia and Hypothermia
Cold chemical cardioplegia is currently the method of choice for providing a still, relaxed and unobstructed operating field, as well as providing substantial protection from ischemic/reperfusion injury. This procedure involves aortic cross-clamping, with the resulting tissue ischemia being protected by infusion of cold chemical solutions together with the application of topical hypothermia. The use of hypothermia for clinical purposes dates back many years and has completely avoided the occurrence of the “stone heart”. For example, Hippocrates advocated packing wounded patients in snow and ice to reduce hemorrhage and other damage.71
Infants abandoned and exposed to cold remained viable for prolonged periods, but the first clinical trials started in the 1960 s.72
No doubt that the introduction of these techniques to routine cardiac surgery has allowed the tolerable duration of ischemia to be extended from less than 1 h to 3 h, and in experimental studies up to 24 h, before ischemia become irreversible. Three components of effective protection has been identified: (1) conservation of energy by inducing instantaneous arrest with agents such as potassium and by hypothermia; (2) slowing the metabolic rate of energy consumption and degenerative reaction; and (3) combating deleterious ischemia-induced changes with specific protective agents. Recent attempts to further protect the ischemic heart during cardiac surgery include inhibitors of the PARP-pathway, inhibitors of the Na+/H+
exchanger and of the mPTP, and antioxidants.73
So it seems that surgeons and interventionists are mainly following the same paths in terms of avoiding ischemic/reperfusion injury, with 1 major difference: the surgeons have been successful but not the interventionists.1
Why such a consolidated success in comparison with failure in the other clinical setting such as PCI? Very simply, the surgeons apply cardiac protection before (and not after) ischemia and to a quiescent heart that does not need to contract. Administration of cardioplegia before clamping cause immediate arrest without depleting ATP or CP, and the non-beating heart requires little energy to maintain structural integrity. These 2 important issues make the difference. During PCI, ischemia occurs in a working heart and reperfusion often occurs too late, when the damage is likely already occurred.
Conclusions
Despite improvements in its prevention and acute treatment, CAD and MI remain major causes of death and their incidence is rising. Reperfusion, by any means, is the most effective strategy for reducing infarct size and improving clinical outcome. The process of myocardial reperfusion itself, however, can induce injury to the myocardium, thereby reducing the beneficial effects of the reperfusion. Nonetheless, the existence of RI has been and still is a matter of debate. We are lacking a clinical demonstration that post-ischemic recovery can be improved by an intervention applied at the time of reperfusion. This is partly related to the difficulty in the timing of the intervention and of properly designed phase III clinical trials in humans subjected to revascularization for STEMI and to the lack of clearly defined targets for therapy.
It follows that today the only realistic strategy to reduce RI in STEMI patients is to prevent it from occurring by reperfusing as early as possible. This can be achieved by early diagnosis and immediate intervention using a network allowing ambulance diagnosis and direct access to intervention facilities. For those who are reperfused later, unfortunately the problem exists. This is the reason why the drug industry and academia should maintain support and investment aimed at providing a clear understanding of ischemic/reperfusion injury, which will allow the development of specific drugs and interventions to counteract the damage of AMI, which today is the most relevant killer.
The 4 lines of research described in this review should join hands to find out, together, the ultimate solution.
Acknowledgments
This work was supported by a grant from Fondazione Anna Maria Sechi per il Cuore (FASC), Italy. The funders had no role in the preparation and decision to publish or the preparation of the manuscript.
Conflict of Interests
None for this work.
References
- 1.
Ferrari R, Biscaglia S, Malagù M, Bertini M, Campo G. Can we improve myocardial protection during ischaemic injury? Cardiology 2016; 135: 14–26.
- 2.
Ferrari R, Ceconi C, Curello S, Cargnoni A, Pasini E, Visioli O. The occurrence of oxidative stress during reperfusion in experimental animals and men. Cardiovasc Drugs Ther 1991; 5: 277–288.
- 3.
Yellon DM, Pasini E, Cargnoni A, Marber M, Latchman DS, Ferrari R. The protective role of heat stress in the ischaemic and reperfused rabbit myocardium. J Mol Cell Cardiol 1992; 24: 895–907.
- 4.
Ferrari R, Ceconi C, Curello S, Alfieri O, Visioli O. Myocardial damage during ischaemia and reperfusion. Eur Heart J 1993; 14: 25–30.
- 5.
Gruppo Italiano per lo Studio della Streptochinasi nell’Infarto Miocardico (GISSI). Effectiveness of intravenous thrombolytic treatment in acute myocardial infarction. Lancet 1986; 1: 397–402.
- 6.
The GUSTO Investigators. An international randomized trial comparing four thrombolytic strategies for acute myocardial infarction. N Engl J Med 1993; 329: 673–682.
- 7.
Cutler DM, McClellan M. Is technological change in medicine worth it? Health Aff (Millwood) 2001; 20: 11–29.
- 8.
Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res 1998; 83: 15–26.
- 9.
Jennings RB. Early phase of myocardial ischemic injury and infarction. Am J Cardiol 1969; 24: 753–765.
- 10.
Ferrari R, Opie L. Atlas of the myocardium. New York, NY: Raven Press; 1992.
- 11.
Ferrari R, Cargnoni A, Bernocchi P, Pasini E, Curello S, Ceconi C, et al. Metabolic adaptation during a sequence of no-flow and low-flow ischemia: A possible trigger for hibernation. Circulation 1996; 94: 2587–2596.
- 12.
Ferrari R, Ceconi C, Curello S, Benigno M, La Canna G, Visioli O. Left ventricular dysfunction due to the new ischaemic outcomes: Stunning and hibernation. J Cardiovasc Pharmacol 1996; 28(Suppl 1): S18–S26.
- 13.
Fröhlich GM, Meier P, White SK, Yellon DM, Hausenloy DJ. Myocardial reperfusion injury: Looking beyond primary PCI. Eur Heart J 2013; 34: 1714–1724.
- 14.
Manning AS, Hearse DJ. Reperfusion-induced arrhythmias: Mechanisms and prevention. J Mol Cell Cardiol 1984; 16: 497–518.
- 15.
Kukreja RC, Janin Y. Reperfusion injury: Basic concepts and protection strategies. J Thromb Thrombolysis 1997; 4: 7–24.
- 16.
Van de Werf F. The history of coronary reperfusion. Eur Heart J 2014; 35: 2510–2515.
- 17.
Krause SM, Jacobus WE, Becker LC. Alterations in cardiac sarcoplasmic reticulum calcium transport in the postischemic “stunned” myocardium. Circ Res 1989; 65: 526–530.
- 18.
Bolli R, Patel BS, Jeroudi MO, Lai EK, McCay PB. Demonstration of free radical generation in “stunned” myocardium of intact dogs with the use of the spin trap alpha-phenyl N-tert-butyl nitrone. J Clin Invest 1988; 82: 476–485(erratum J Clin Invest 1988; 82: following 1807).
- 19.
Bolli R, Marban E. Molecular and cellular mechanisms of myocardial stunning. Physiol Rev 1999; 79: 609–634.
- 20.
Ferrari R. Commentary on myocardial stunning and its clinical relevance. Basic Res Cardiol 1995; 90: 300–302.
- 21.
Rahimtoola SH. A perspective on the three large multicenter randomized clinical trials of coronary bypass surgery for chronic stable angina. Circulation 1985; 72: V123–V135.
- 22.
Braunwald E, Rutherford JD. Reversible ischemic left ventricular dysfunction: Evidence for the “hibernating myocardium. J Am Coll Cardiol 1986; 8: 1467–1470.
- 23.
Ferrari R, Williams AJ. The role of mitochondria in myocardial damage occurring on post-ischaemic reperfusion. J Appl Cardiol 1986; 1: 501–519.
- 24.
Ferrari R, Hearse DJ. Reperfusion injury: Does it exist and does it have clinical relevance? J Thromb Thrombolysis 1997; 4: 25–34.
- 25.
Rahimtoola SH. The hibernating myocardium. Am Heart J 1989; 117: 211–221.
- 26.
Ferrari R, Ceconi C, Curello S, Alfieri O, Visioli O. Myocardial damage during ischaemia and reperfusion. Eur Heart J 1993; 14: 25–30.
- 27.
Ferrari R, Williams A. The role of mitochondria in myocardial damage occurring on post-ischaemic reperfusion. J Appl Cardiol 1986; 1: 501–519.
- 28.
Ferrari R, Boffa GM, Ceconi C, Curello S, Boraso A, Ghielmi S, et al. Effect of D-600 on ischaemic and reperfused rabbit myocardium: Relation with timing modality of administration. Basic Res Cardiol 1989; 84: 606–622.
- 29.
Klein HH, Pich S, Lindert S, Nebendahl K, Warneke G, Kreuzer H. Treatment of reperfusion injury with intracoronary calcium channel antagonists and reduced coronary free calcium concentration in regionally ischemic, reperfused porcine hearts. J Am Coll Cardiol 1989; 13: 1395–1401.
- 30.
Beltrame JF. Ivabradine and the SIGNIFY conundrum. Heart J 2015; 36: 3297–3299.
- 31.
Cocco G, Jerie P. Torsades de pointes induced by the concomitant use of ivabradine and azithromycin: An unexpected dangerous interaction. Cardiovasc Toxicol 2015; 15: 104–106.
- 32.
Avkiran M, Marber MS. Na+/H+ exchange inhibitors for cardioprotective therapy: Progress, problems and prospects. J Am Coll Cardiol 2002; 39: 747–753.
- 33.
Théroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, et al. Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations: Main results of the GUARDIAN trial. Circulation 2000; 102: 3032–3038.
- 34.
Morrow DA, Scirica BM, Karwatowska-Prokopczuk E, Murphy SA, Budaj A, Varshavsky S, et al. Effects of ranolazine on recurrent cardiovascular events in patients with non-ST-elevation acute coronary syndromes: The MERLIN-TIMI 36 randomized trial. JAMA Cardiol 2007; 297: 1775–1783.
- 35.
Weisz G, Généreux P, Iñiguez A, Zurakowski A, Shechter M, Alexander KP, et al. Ranolazine in patients with incomplete revascularisation after percutaneous coronary intervention (RIVER-PCI): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2016; 387: 136–145.
- 36.
Ferrari R, Ceconi C, Curello S, Cargnoni A, de Giuli F, Visioli O. Occurrence of oxidative stress during myocardial reperfusion. Mol Cell Biochem 1992; 111: 61–69.
- 37.
Curello S, Ceconi C, de Giuli F, Panzali AF, Milanesi B, Calarco M, et al. Oxidative stress during reperfusion of human hearts: Potential sources of oxygen free radicals. Cardiovasc Res 1995; 29: 118–125.
- 38.
The European Myocardial Infarction Project-Free Radicals Group. Effect of 48-h intravenous trimetazidine on short- and long-term outcomes of patients with acute myocardial infarction, with and without thrombolytic therapy; a double-blind, placebo-controlled, randomized trial. Eur Heart J 2000; 21: 1537–1546.
- 39.
Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): Two randomised trials. Lancet 2007; 370: 1483–1493.
- 40.
He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: A new paradigm of pore structure and function? FEBS Lett 2002; 512: 1–7.
- 41.
Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 1995; 307: 93–98.
- 42.
Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, et al. Molecular identity of the mitochondrial permeability transition pore and its role in ischemia-reperfusion injury. J Mol Cell Cardiol 2015; 78: 142–153.
- 43.
Cung TT, Morel O, Cayla G, Rioufol G, Garcia Dorado D, Angoulvant D, et al. Cyclosporine before PCI in patients with acute myocardial infarction. N Engl J Med 2015; 373: 1021–1031.
- 44.
Atar D, Arheden H, Berdeaux A, Bonnet JL, Carlsson M, Clemmensen P, et al. Effect of intravenous TR040303 as an adjunct to primary percutaneous coronary intervention for acute ST-elevation myocardial infarction: MITOCARE study results. Eur Heart J 2015; 36: 112–119.
- 45.
Campo G, Morciano G, Pavasini R, Bonora M, Sbano L, Biscaglia S, et al. Fo ATP synthase C subunit serum levels in patients with ST-segment elevation myocardial infarction: Preliminary findings. Int J Cardiol 2016; 221: 993–997.
- 46.
Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: A quantitative review of 23 randomised trials. Lancet 2003; 361: 13–20.
- 47.
De Boer SP, Westerhout CM, Simes RJ, Granger CB, Zijlstra F, Boersma E; Primary Coronary Angioplasty Versus Thrombolysis-2 (PCAT-2) Trialists Collaborators Group. Mortality and morbidity reduction by primary percutaneous coronary intervention is independent of the patient’s age. JACC Cardiovasc Interv 2010; 3: 324–331.
- 48.
Every NR, Parsons LS, Hlatky M, Martin JS, Weaver WD; Myocardial Infarction Triage and Intervention Investigators. A comparison of thrombolytic therapy with primary coronary angioplasty for acute myocardial infarction. N Engl J Med 1996; 335: 1253–1260.
- 49.
Danchin N, Blanchard D, Steg PG, Sauval P, Hanania G, Goldstein P, et al; USIC 2000 Investigators. Impact of prehospital thrombolysis for acute myocardial infarction on 1-year outcome: Results from the French Nationwide USIC 2000 Registry. Circulation 2004; 110: 1909–1915.
- 50.
Krug A, du Mesnil de Rochemont R, Korb G. Blood supply of the myocardium after temporary coronary occlusion. Circ Res 1966; 19: 57–62.
- 51.
Kloner RA, Rude RE, Carlson N, Maroko PR, DeBoer LWV, Braunwald E. Ultrastructural evidence of microvascular damage and myocardial cell injury after coronary artery occlusion: Which comes first? Circulation 1980; 62: 945–952.
- 52.
Camici PG, Wijns W, Borgers M, De Silva R, Ferrari R, Knuuti J, et al. Pathophysiological mechanisms of chronic reversible left ventricular dysfunction due to coronary artery disease (hibernating myocardium). Circulation 1997; 96: 3205–3214.
- 53.
Kloner RA, Ganote CE, Jennings RB. The ‘no-reflow’ phenomenon after temporary coronary occlusion in the dog. J Clin Invest 1974; 54: 1496–1508.
- 54.
Niccoli G, Burzotta F, Galiuto L, Crea F. Myocardial no-reflow in humans. J Am Coll Cardiol 2009; 54: 281–292.
- 55.
Bekkers SC, Yazdani SK, Virmani R, Waltenberger J. Microvascular obstruction: Underlying pathophysiology and clinical diagnosis. J Am Coll Cardiol 2010; 55: 1649–1660.
- 56.
Galinanes M, Lawson CS, Ferrari R, Limb GA, Derias NW, Hearse DJ. Early and late effects of leukopenic reperfusion on the recovery of cardiac contractile function: Studies in the transplanted and isolated blood-perfused rat heart. Circulation 1993; 88: 673–683.
- 57.
Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124–1136.
- 58.
Heusch G. Cardioprotection: Chances and challenges of its translation to the clinic. Lancet 2013; 381: 166–175.
- 59.
Przyklenk K, Whittaker P. Remote ischemic preconditioning: Current knowledge, unresolved questions, and future priorities. J Cardiovasc Pharmacol Ther 2011; 16: 255–259.
- 60.
Heusch G, Boengler K, Schulz R. Cardioprotection: Nitric oxide, protein kinases, and mitochondria. Circulation 2008; 118: 1915–1919.
- 61.
Kloner RA. Current state of clinical translation of cardioprotective agents for acute myocardial infarction. Circ Res 2013; 113: 451–463.
- 62.
Yellon DM, Opie LH. Postconditioning for protection of the infarcting heart. Lancet 2006; 367: 456–458.
- 63.
Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, et al. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: Comparison with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003; 285: H579–H588.
- 64.
Staat P, Rioufol G, Piot C, Cottin Y, Cung TT, L’Huillier I, et al. Postconditioning the human heart. Circulation 2005; 112: 2143–2148.
- 65.
Hahn JY, Song YB, Kim EK, Yu CW, Bae JW, Chung WY, et al. Ischemic postconditioning during primary percutaneous coronary intervention: The effects of postconditioning on myocardial reperfusion in patients with ST-segment elevation myocardial infarction (POST) randomized trial. Circulation 2013; 128: 1889–1896.
- 66.
Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D. Remote ischemic conditioning. J Am Coll Cardiol 2015; 65: 177–195.
- 67.
Bøtker HE, Kharbanda R, Schmidt MR, Bøttcher M, Kaltoft AK, Terkelsen CJ, et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: A randomised trial. Lancet 2010; 375: 727–734.
- 68.
Sloth AD, Schmidt MR, Munk K, Schmidt M, Pedersen L, Sørensen HT, et al. Improved long-term clinical outcomes in patients with ST-elevation myocardial infarction undergoing remote ischaemic conditioning as an adjunct to primary percutaneous coronary intervention. Eur Heart J 2013; 35: 168–175.
- 69.
Cooley DA, Reul GJ, Wucasch DC. Ischemic contracture of the heart: “Stone heart”. Am J Cardiol 1972; 29: 575–577.
- 70.
Hutchins GM, Silverman KJ. Pathology of the stone heart syndrome. Am J Pathol 1979; 95: 745–752.
- 71.
Hippocrates (460–375 BC). De Vetere Medicina.
- 72.
Vaity C, Al-Subaie N, Cecconi M. Cooling techniques for targeted temperature management post-cardiac arrest. Crit Care 2015; 19: 103.
- 73.
Murphy GJ, Ascione R, Angelini GD. Coronary artery bypass grafting on the beating heart: Surgical revascularization for the next decade? (Review) Eur Heart J 2004; 25: 2077–2085.


