Abstract
Background:
Lipoprotein lipase (LPL) expressed in macrophages plays an important role in promoting the development of atherosclerosis or atherogenesis. MicroRNA-182 (miR-182) is involved in the regulation of lipid metabolism and inflammation. However, it remains unclear how miR-182 regulates LPL and atherogenesis.
Methods and Results:
Using bioinformatics analyses and a dual-luciferase reporter assay, we identified histone deacetylase 9 (HDAC9) as a target gene of miR-182. Moreover, miR-182 upregulated LPL expression by directly targeting
HDAC9
in THP-1 macrophages. Hematoxylin-eosin (H&E), Oil Red O and Masson’s trichrome staining showed that apolipoprotein E (ApoE)-knockout (KO) mice treated with miR-182 exhibited more severe atherosclerotic plaques. Treatment with miR-182 increased CD68 and LPL expression in atherosclerotic lesions in ApoE-KO mice, as indicated by double immunofluorescence staining in the aortic sinus. Increased miR-182-induced increases in LPL expression in ApoE-KO mice was confirmed by real-time quantitative polymerase chain reaction and western blotting analyses. Treatment with miR-182 also increased plasma concentrations of proinflammatory cytokines and lipids in ApoE-KO mice.
Conclusions:
The results of the present study suggest that miR-182 upregulates LPL expression, promotes lipid accumulation in atherosclerotic lesions, and increases proinflammatory cytokine secretion, likely through targeting
HDAC9, leading to an acceleration of atherogenesis in ApoE-KO mice.
Atherosclerosis is the major etiology for cardiovascular complications, such as myocardial infarction and stroke. It is well known that both lipid accumulation and infiltration of inflammatory cells play critical roles in the initiation and development of atherosclerosis.1–3
Lipoprotein lipase (LPL), a key enzyme in lipid metabolism, hydrolyses triglycerides (TG) in circulating TG-rich lipoproteins (TRLs) and promotes the cellular uptake of chylomicron remnants, cholesterol-rich lipoproteins, and free fatty acids by adipose tissue and skeletal muscle.4
The role of LPL in atherogenesis has long been controversial. Yagyu et al showed that overexpression of LPL protects against atherosclerosis in apolipoprotein E (ApoE)-knockout (KO) mice.5
Zhang et al demonstrated spontaneous atherosclerosis in aged LPL-deficient mice that occurred via the oxidation of chylomicrons and activation of vascular endothelial cells.6
However, Qiu et al reported that overexpression of LPL in macrophages markedly promotes proinflammatory cytokine secretion and lipid composition, leading to increased formation of atherosclerotic lesions.7,8
Babaev et al transplanted lethally irradiated female low-density lipoprotein receptor-KO (LDLR-KO) mice with LPL−/−
mouse fetal liver cells and found that, in the arterial wall, macrophage-derived LPL promoted foam cell formation and atherosclerosis in vivo.9
Accumulating evidence demonstrates that macrophage-derived LPL can accelerate the development of atherosclerosis.7–9
Since the pioneering studies performed in
Caenorhabditis elegans,10
microRNAs (miRNAs) have emerged as critical regulators of many biological processes and diseases. Recent studies have shown the roles of miRNAs in lipid metabolism and atherosclerosis.11,12
Our group has recently shown that miR-590, miR-134, and miR-467b can regulate LPL expression in either in vitro or in vivo experiments.13–17
As a polyphenic miRNA, miR-182 is known to be involved in lipid metabolism, inflammation, and angiogenesis.18–20
Interestingly, miR-182 has been connected to the differentiation of T cells and plays an important role in the progression of atherosclerotic plaques.21
It was reported that plasma miR-182 concentrations are significantly increased in patients with coronary artery disease (CAD) compared with healthy control subjects, suggesting that circulating miR-182 may be an important biomarker of CAD.22
However, it remains unclear whether miR-182 affects the development of atherosclerosis. Reversible histone acetylation has been characteristically linked to histone- and chromatin-dependent processes.23,24
It was reported that a genetic variant in the loci corresponding to histone deacetylase 9 (HDAC9) is associated with large vessel stroke.25
HDAC9 expression is upregulated in human atherosclerotic plaques in different arteries.26
Moreover, overexpression of macrophage HDAC9 was reported to be atherogenic via inhibition of cholesterol efflux and the generation of alternatively activated macrophages.27
However, the role of HDAC9 in atherosclerosis has not yet been fully elucidated.
The aim of the present study was to explore the effects of miR-182 on atherosclerosis in ApoE-KO mice and the mechanisms involved. The results indicate that overexpression of miR-182 significantly promotes atherosclerosis in ApoE-KO mice. Mechanistically, miR-182-induced atherogenesis may be associated with upregulation of LPL expression via silencing of
HDAC9
in THP-1 macrophage-derived foam cells, which leads to a marked increase in lipid accumulation and proinflammatory cytokine secretion. Together, the results suggest that miR-182 may act as a novel pharmacologic target for atherosclerosis therapy.
Methods
Cell Culture and Treatments
HEK 293T and human monocyte (THP-1) cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). HEK 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Exposure to 160 nmol/L phorbol-12-myristate acetate (PMA) for 24 h was used to induce the differentiation of THP-1 cells into macrophages. Human monocytes from healthy control subjects were isolated by the OptiPrep procedure (Axis Shield, Dundee, Scotland). Ethical approval for blood donation by healthy participants was obtained from the Faculty of Health Research Ethics Committee, University of South China. Approximately equal numbers of males and females were recruited. The blood was acquired through venipuncture of healthy participants who were enrolled at the Second Affiliated Hospital of University of South China. All donors were not on medication. Whole blood was collected and mixed with OptiPrep in a ratio of 8 : 1, and the mixture covered with 0.5 mL Tris-buffered saline (TBS). Samples were then centrifuged at 1,500 g for 30 min at room temperature, after which monocytes were collected from the upper fraction (at the top of the plasma; >90% monocyte purity) and placed in flasks containing RPMI 1640 medium. Monocytes were allowed to adhere to the flask in RPMI 1640 medium and were differentiated into macrophages by culturing them for 7 days in the presence of macrophage colony-stimulating factor as described previously.28
To induce foam cell formation, THP-1 macrophages and human primary macrophages were cultured in serum-free DMEM medium containing 50 μg/mL oxidized low-density lipoprotein (oxLDL) for 48 h at 37℃.
Real-Time Quantitative Polymerase Chain Reaction (qPCR)
qPCR analysis was performed as described previously.29
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. qPCR with SYBR green detection chemistry was performed using a LightCycler Run 5.32 Real-Time PCR System (Roche). Melting curve analyses of all qPCR products were performed and shown to produce a single DNA duplex. Quantitative measurements were determined using the ∆∆Ct method. In these experiments, β-actin was used as the internal control.
Western Blot Analysis
Peritoneal macrophages and aortic tissues were lysed for the extraction of protein using RIPA buffer (Sigma, St Louis, MO, USA), as described previously.30
Protein lysates from isolated peritoneal macrophages or aortic tissues (20 μg per lane) were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA) and incubated with primary antibodies overnight at 4℃. Next, PVDF membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies at room temperature for 2 h. Finally, protein signals were visualized using a chemiluminescence method and quantified with the Tanon-5500 Chemiluminescent Imaging System (Tanon, Shanghai, China).
ApoE-KO Mice and Treatments
Six-week-old male ApoE-KO mice were purchased from Peking University with Laboratory Animal Center. All procedures were done in accordance with the Institutional Animal Ethics Committee and the University of South China Animal Care Guidelines for the Use of Experimental Animals. ApoE-KO mice were randomized into 4 groups (10 mice in each group): (1) an miR-182 scrambled agomir negative control group (AG-NC); (2) an miR-182 agomir group (AG); (3) an miR-182 scrambled antagomir negative control group (AN-NC); and (4) an miR-182 antagomir group (AN). Mice in the AG-NC, AG, AN-NC and AN groups were injected with miR-182 scrambled agomir negative control, agomir, scrambled antagomir negative control and antagomir. The agomir (2’ OME+5’ chol modified) and antagomir (2’ OME+5’ chol modified) of miR-182 are locked nucleic acids purchased from RiboBio (Guangzhou, China). All sequences are provided as follows: miR-182 agomir: UUUGGCAAUGGUAGAACUCACACU, miR-182 antagomir: UUUGGCAAUGGUAGAACUCACACU. miR-182 agomir negative control: UCACAACCUCCUA GAAAGAGUAGA, and miR-182 antagomir negative controls: UUUGUAC UACACAAAAGUACUG. Each mouse received a tail vein injection of miR-182 agomir, miR-182 antagomir or one of the corresponding controls, respectively, at a dose of 80 mg/kg wt in 0.2 mL saline once every 2 weeks after starting the high-fat/high-cholesterol Western diet (the Paigen diet (Trophic Animal Feed High-tech Co., Ltd, Jiangsu, China)). All mice were bred in a specific pathogen-free environment with unlimited access to food and water. After 8 weeks, and after fasting for 12–14 h, mice were anesthetized (1% Pelltobarbitalum Natricum (Yubo Biologycal, Shanghai, China)) and killed by exsanguination. The heart, aortas, peritoneal macrophages, and blood samples were collected for subsequent experiments.
Statistical Analysis
Data are expressed as the mean±SD. Results were analyzed by Student’s t-test and 1-way analysis of variance (ANOVA; Tukey’s or Dunnett’s tests). Analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA), and statistical significance was set at P≤0.05.
Results
Effects of miR-182 on HDAC9
To evaluate the degree to which miR-182 is conserved across various species, the miRDB database (http://mirbase.org/index.shtml) was searched, with the results indicating that miR-182 is evolutionarily conserved in different species. These findings suggest that miR-182 plays a pivotal role in the evolution of species (Figure S1A). Analysis using both microRNA (http://www.microrna.org/microrna/) and TargetScan (http://www.targetscan.org/) software revealed a putative binding site between miR-182 and the 3’ untranslated region (UTR) of
HDAC9
in most animal species, revealing a biological basis for miR-182 to combine with the 3’UTR of
HDAC9
(Figure S1B). Importantly, the miR-182–HDAC9
mRNA hybrid free energy scores tested by RNAhybrid (http://bibiserv.techfak.unibielefeld.de/rnahybrid/) were very low (Figure S1C). Using TargetScan, we found that the 3’UTR of
LPL
had no binding site for miR-182, suggesting that miR-182 had no direct effect on
LPL
(Figure S1D). Finally, we performed a luciferase reporter assay to determine whether
HDAC9
was a direct target of miR-182 in HEK 293T cells. In these experiments, cotransfection of miR-182 mimic and phosphorylated (p-)HDAC9
wild-type (WT) 3’UTR miRNA luciferase reporter vector into HEK 293T cells potently inhibited luciferase activity compared with control transfected cells, but the luciferase activity of the mutant was significantly rescued (Figure S1E). In contrast, cotransfection of miR-182 mimic and p-LPL luciferase reporter vector into HEK 293T cells did not significantly change luciferase activity (Figure S1E).
Effects of miR-182 on HDAC9 Expression and Activity
To further investigate whether miR-182 could negatively regulate
HDAC9
expression, we tested the effects of miR-182 on the expression of HDAC9 in human THP-1 macrophages through transfection of miR-182 mimic or miR-182 inhibitor. As shown in
Figure 1A–F, transfection with miR-182 mimic decreased HDAC9 mRNA and protein levels in both a concentration- and time-dependent manner. However, transfection with miR-182 inhibitor had an opposite effect. Similarly, the results demonstrated that in cells transfected with miR-182, HDAC9 activity was significantly decreased in the presence of miR-182 upregulation (Figure 1G).
Effects of HDAC9 on Endogenous LPL Expression
To determine whether HDAC9 regulates endogenous levels of LPL, human primary macrophages were transfected with 80 nmol/L pAd-Control (recombinant adenoviral vectors), pAd-HDAC9-3’UTR and pAd-shHDAC9- 3’UTR (inhibition of HDAC9 by short hairpin RNAHDAC9 (shHDAC9)) for 24 h. The results showed that overexpression of HDAC9 significantly decreased acetylation of lysine 16 of histone H4 (H4K16 ac) of the
LPL
core protein (Figure 2A) and downregulated LPL mRNA and protein expression (Figure 2B,C). In contrast, HDAC9 knockdown or treatment with 3 μmol/L trichostatin A (TSA; an HDAC9 inhibitor) increased H4K16 ac of the
LPL
core protein, as well as LPL mRNA and protein expression (Figure 2A–C). Similar results were observed in human THP-1 macrophages (Figure S2A–C). Together, these results suggest an inhibitory role for HDAC9 in the regulation of endogenous LPL expression.
Mechanisms of miR-182 on LPL Expression
We first examined the effects of miR-182 on LPL expression. Treatment with miR-182 mimic significantly increased H4K16 ac of the
LPL
core protein (Figure 2D) and upregulated LPL expression in human primary macrophages (Figure 2E,F). Next, we investigated whether downregulation of HDAC9 mediated miR-182 effects by overexpressing HDAC9. As shown in
Figure 2G,H, overexpression of HDAC9 abolished the effects of the miR-182 mimic on LPL expression in human primary macrophages. Consistently, the miR-182 mimic markedly enhanced LPL activity, and this was reversed in the presence of HDAC9 overexpression (Figure 2I). Similar results were obtained in human THP-1 macrophages (Figure S2D–I). Together, these results support the idea that silencing of
HDAC9
induced by miR-182 blocks
LPL
core protein deacetylation and subsequently increases LPL expression and activity.
Effects of miR-182 on Lipid Levels in THP-1 Macrophages
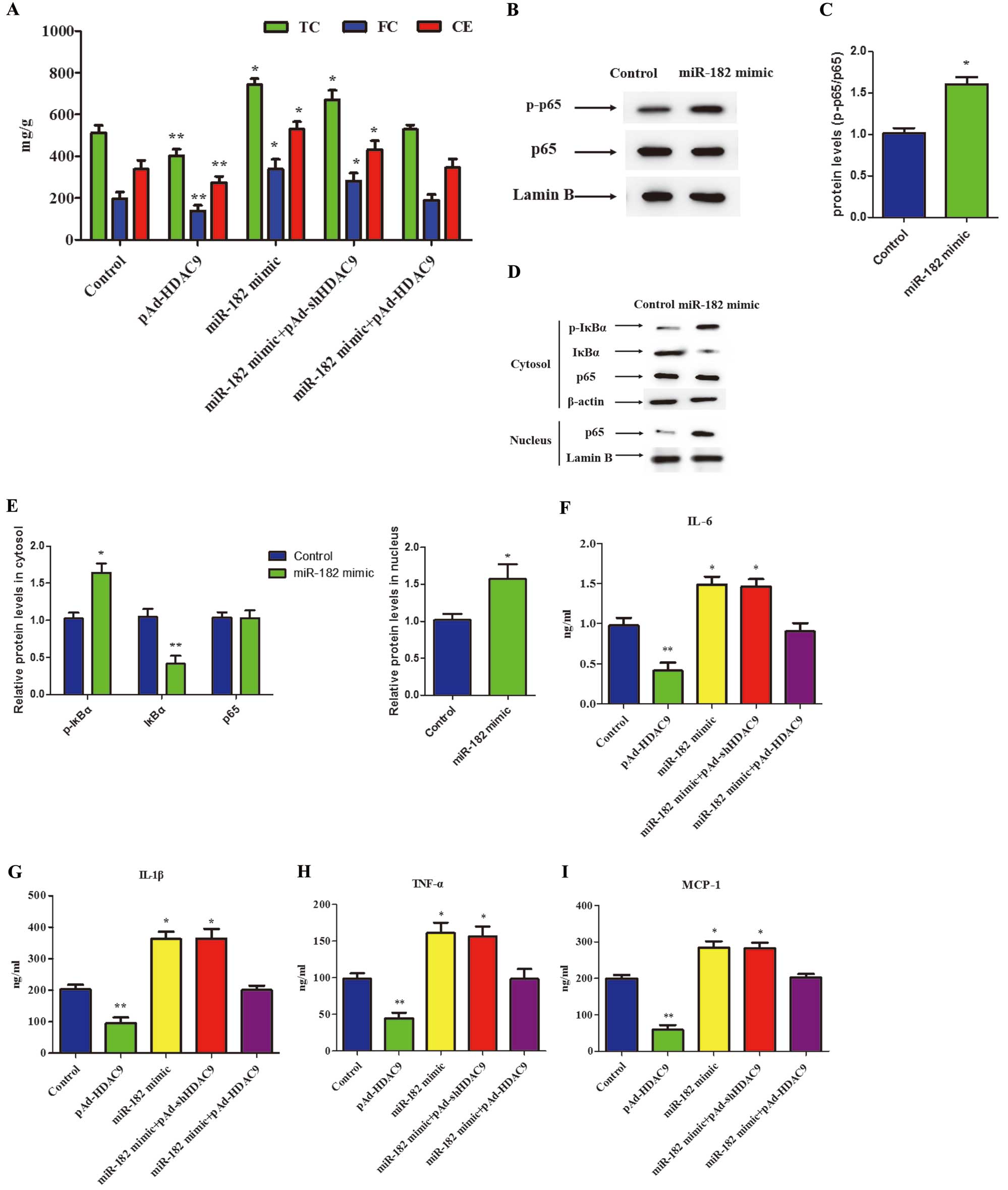
We next investigated whether miR-182 affected lipid content in oxLDL-treated human THP-1 macrophages using HPLC. The results of these experiments showed that miR-182 mimic upregulated total cholesterol (TC), free cholesterol (FC), and cholesteryl ester (CE) levels in human THP-1 macrophages. In addition, pAd-HDAC9 decreased TC, FC, and CE levels in human THP-1 macrophages (Figure 3A). However, there was no significant difference in TC, FC and CE levels in human THP-1 macrophages treated with miR-182 mimic combined with pAd-HDAC9 compared with control cells (Figure 3A). Therefore, the results suggest that miR-182 promotes lipid accumulation by targeting HDAC9 in human THP-1 macrophages.
Effects of miR-182 on Pro-Inflammatory Cytokine Secretion in THP-1 Macrophages
Whether miR-182 affected the secretion of proinflammatory cytokines was investigated in oxLDL-treated human THP-1 macrophages. First, the effects of miR-182 on the nuclear factor (NF)-κB pathway, relative levels of p-IκBα (inhibitor of NF-κB-α), IκBα, and NF-κB in the cytoplasm, and levels of p65 NF-κB in the nucleus were determined by western blot analysis. There was no significant effect of miR-182 on total levels of p65 protein expression, but levels of p-p65 were significantly increased in following treatment with miR-182 mimic (Figure 3B,C). Conversely, treatment with the miR-182 mimic potently induced the phosphorylation and degradation of IκBα in the cytoplasm, thereby reducing p65 levels in the cytoplasm with an increase in nuclear p65 (Figure 3D,E). In addition, treatment of cells with the miR-182 mimic significantly increased interleukin (IL)-6, IL-1β, monocyte chemotactic factor-1 (MCP-1), and tumor necrosis factor (TNF)-α levels, as determined by ELISA. Treatment of cells with pAd-HDAC9 decreased levels of IL-6, IL-1β, MCP-1, and TNF-α in human THP-1 macrophages. However, levels of proinflammatory cytokines did not differ significantly between control group and cells transfected with miR-182 mimic combined with pAd-HDAC9 (Figure 3F–I). These findings suggest that miR-182 promotes proinflammatory cytokine secretion by targeting HDAC9 in human THP-1 macrophages.
Effects of miR-182 on the Development of Atherosclerosis in ApoE-KO Mice
We further investigated the effects of miR-182 on the development of atherosclerosis in ApoE-KO mice by measuring the surface area of atherosclerotic plaques relative to total surface area of aortas after perfusion with formalin. In ApoE-KO mice treated with AG, there was a significant increase in atherosclerotic plaque area. In contrast, decreased plaque areas were observed in AN-treated mice (Figure 4A,B). These results suggest that miR-182 increases both the number and size of aortic arch plaques.

Plaque area was also examined in hematoxylin and eosin (H&E)-stained cross-sections of the aortic sinus. The aortic root plaque area of AG-treated mice was much greater than that of AG-NC-treated mice, whereas the aortic root plaque area of AN-treated mice was less than that of AN-NC-treated mice (Figure 4C). Moreover, the severity of lipid deposition was significantly increased in the aortic sinus of AG-treated mice. However, lipid deposition was decreased by AN treatment (Figure 4D). Finally, the atherosclerotic plaque collagen content in the aortic sinus was determined using Masson’s trichrome staining in ApoE-KO mice. These studies revealed that AG significantly increased collagen content (Figure 4E). Collectively, the results indicate that miR-182 promotes the development of atherosclerosis.
Effects of miR-182 on LPL Expression in ApoE-KO Mice
Using immunofluorescence, the effects of miR-182 on the expression of macrophage LPL in atherosclerotic lesions in the aortic sinus of ApoE-KO mice were evaluated. The macrophage-rich area identified using CD68 as a marker overlapped with the area staining positive for LPL in aortic atherosclerotic lesions, suggesting are correlation between LPL and macrophages in atherosclerotic lesions in the aortic sinus. In addition, staining of macrophage LPL in atherosclerotic lesions was obviously increased in AG-treated mice, but was significantly decreased in AN-treated mice (Figure 5A,B). Together, these results indicate that miR-182 increases the expression of macrophage LPL in atherosclerotic lesions of ApoE-KO mice.

qPCR and western blot analyses were then used to determine LPL expression in the aortic arch from each group. LPL mRNA and protein expression was significantly increased in AG-treated mice, but significantly decreased in AN-treated mice (Figure 5C,D). Furthermore, expression LPL mRNA and protein was significantly increased in peritoneal macrophages in AG-treated mice, but significantly decreased in AN-treated mice (Figure 5E,F). LPL activity in the aortic arch and peritoneal macrophages was also investigated and found to be significantly increased in the AG-treated group, but reduced in the AN-treated group (Figure 5G,H). These data indicate that miR-182 stimulates LPL expression in ApoE-KO mice.
Effects of miR-182 on Proinflammatory Cytokines and Lipids in ApoE-KO Mice
ELISA was used to determine whether miR-182 affects plasma pro-inflammatory cytokine levels in ApoE-KO mice. Plasma concentrations of IL-6, IL-1β, TNF-α, and MCP-1 were significantly increased in ApoE-KO mice treated with AG. In contrast, AN treatment decreased concentrations of IL-6, IL-1β, TNF-α, and MCP-1 (Figure 5I).
The potential association between miR-182 and lipid levels was explored in ApoE-KO mice. As expected, in AG-treated ApoE-KO mice, higher lipid levels were associated with feeding of the high-fat/high-cholesterol Western diet compared with the AG-NC-treated group, whereas the opposite effects were observed in ApoE-KO mice treated with AN (Table). Plasma TC levels were significantly elevated in ApoE-KO mice treated with AG. Moreover, administration of AG increased plasma low-density lipoprotein cholesterol (LDL-C) concentrations, but decreased plasma TG concentrations. In contrast, AN treatment decreased plasma TC and LDL-C concentrations, but increased plasma TG concentrations in ApoE-KO mice (Table). These findings suggest that miR-182 upregulates plasma concentrations of proinflammatory cytokines and lipids in ApoE-KO mice.
Table.
Effects of miR-182 on BW and the Plasma Lipid Profile in Apolipoprotein E-Knockout Mice
| |
BW (g) |
TG |
TC |
HDL-C |
LDL-C |
| AG-NC |
27.62±1.15 |
2.38±0.36 |
18.44±0.84 |
3.25±0.73 |
15.20±0.61 |
| AG |
29.21±0.68 |
1.37±0.42* |
23.71±1.53* |
3.19±0.53 |
19.45±1.69* |
| AN-NC |
28.43±0.74 |
2.33±0.24 |
18.58±0.86 |
3.05±0.62 |
15.21±0.76 |
| AN |
28.19±1.42 |
3.59±0.57† |
16.59±1.17† |
3.14±0.62 |
12.66±1.58† |
Data are the mean±SEM (n=10). *P<0.05 compared with agomir negative control (AG-NC); †P<0.01 compared with antagomir negative control (AN-NC). Plasma from different experimental groups was measured enzymatically. AG, agomir; AN, antagomir; BW, body weight; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; miR-182, microRNA-182; TC, total cholesterol; TG, triglyceride.
Discussion
Atherosclerosis is associated with imbalanced lipid metabolism and chronic inflammation in the blood vessel wall driven by the accumulation of macrophages in the subendothelium.31,32
There is a large body of evidence supporting the involvement of miRNAs in the development of atherosclerosis.21,33
It has been reported that overexpression of miR-182 induces the production of inflammatory mediators in biliary cells and increase liver damage, bile acid accumulation, and inflammatory responses in mice.34
However, the role of miR-182 in atherogenesis remained unclear. In the present study, we found that miR-182 promoted LPL expression, lipid accumulation, and proinflammatory cytokine secretion by targeting HDAC9 in THP-1 macrophages. Moreover, AG-treated mice showed a significant increase in aortic atherosclerotic lesion area and necrotic core. These findings suggest that miR-182 is an important contributor to atherosclerosis.
It has been well established that macrophage-derived LPL promotes the formation of atherosclerotic lesions by stimulating lipid accumulation.16,35,36
Macrophage-derived LPL acts as a non-enzymatic “molecular bridge” that links the endocytosis receptors, especially LDL receptor-related protein 1 (LRP-1), scavenger receptor A1 (SR-A1), and very low-density lipoprotein receptor (VLDLR), to heparin-like substances.37
In addition, overexpression of macrophage-derived LPL contributes to anchoring some atherogenic lipoproteins, such as LDL and oxLDL, thereby promoting cellular lipid uptake and accumulation. In the present study,
HDAC9
was identified as a direct target of miR-182, which is consistent with a recent report.38
In the present study, silencing of
HDAC9
induced by the miR-182 mimic blocked LPL core protein deacetylation, leading to an increase in LPL expression in both THP-1 macrophages and human primary macrophages. Moreover, LPL expression levels in ApoE-KO mice treated with AG were significantly increased in both the aortic root and peritoneal macrophages. Thus, aggravation of lipid accumulation due to LPL upregulation may be an important mechanism by which miR-182 promotes the development of atherosclerosis.
LPL is an important enzyme that hydrolyses TG in circulating TRLs to liberate fatty acid (FA). Recent studies have shown that accumulation of FC in macrophages leads to the induction and secretion of inflammatory cytokines, such as TNF-α and IL-6, processes that are mediated by FC-induced activation of the IκB kinase/NF-κB pathway.39
Moreover, local high levels of FAs are cytotoxic for FAs can activate toll-like receptor (TLR) 2/4 and FA-binding protein 4 (FABP4) in macrophages,40,41
leading to increased production of proinflammatory cytokines.42
In the present study, miR-182 increased the lipolysis of TRLs by LPL, and then stimulated proinflammatory cytokine production, leading to an acceleration of atherogenesis. Thus, in addition to inducing lipid accumulation, miR-182 accelerates atherosclerosis by amplifying the inflammatory response via upregulation of LPL.
A recent genome-wide association study demonstrated that a genetic variant in the locus corresponding to
HDAC9
is associated with large vessel atherosclerosis, and HDAC9 may have important regulatory roles in atherosclerosis.43
More importantly, overexpression of macrophage HDAC9 markedly decreases the expression of ATP binding cassette (ABC) transporters, such as ABCA1 and ABCG1, which, in turn, inhibits cholesterol efflux from macrophages.44
Macrophage-derived LPL and its associated lipolysis product were shown to decrease expression of ABCA1 and ABCG1, resulting in reduced cholesterol efflux from cells.45
Conversely, downregulation of LPL expression in THP-1 macrophages increased ABCA1 expression and cholesterol efflux from cells.46
Thus, the net result of silencing of
HDAC9
induced by miR-182 is to block cholesterol efflux from macrophages.
In conclusion, the results of the present study show that miR-182 binds to the 3’UTR of
HDAC9
to upregulate LPL expression, which results in increased cellular lipid accumulation and inflammatory cytokine secretion, thereby aggravating atherosclerosis (Figure 6). This finding reveals a novel mechanism by which miR-182 promotes the development of atherosclerosis. Further characterizations may identify miR-182 as a promising target for the prevention and treatment of atherosclerotic diseases.
Acknowledgments
The authors gratefully acknowledge financial support from the National Natural Sciences Foundation of China (81270269, 81570408, 81370377, and 81300224), Aid Program for Science and Technology Department of Hunan Province (2015JC3082, 2015JC3083, and 2015JJ2120), the Construct Program of the Key Discipline in Hunan Province, the Hunan Provincial Natural Science Foundation of China (2017JJ2234), start-up funds for PhDs at the University of South China, Hunan Province Graduate Student Scientific Research Innovation Project (2014SCX02 and 2015XCX03), Graduate Student Innovation Project of Cooperative Innovation Center for Molecular Target New Drug (0223-0002-D0033 and 0223-0002-D0027), and the College Students’ Innovative Project (201510555007). University of South China Innovation Foundation for Postgraduate (2017XCX03).
Conflict of Interest
The authors declare no actual or potential conflicts of interest.
Supplementary Files
Supplementary File 1
Supplementary Materials and Methods
Figure S1.
Predicted annealing of microRNA-182 (miR-182) to the 3’ untranslated region (UTR) of histone deacetylase 9 (HDAC9).
Figure S2.
MicroRNA-182 (miR-182) increases lipoprotein lipase (LPL) expression by targeting histone deacetylase 9 (HDAC9) in human THP-1 macrophages.
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-16-1165
References
- 1.
Ouimet M, Marcel YL. Regulation of lipid droplet cholesterol efflux from macrophage foam cells. Arterioscler Thromb Vasc Biol 2012; 32: 575–581.
- 2.
Bornfeldt KE. 2013 Russell Ross Memorial Lecture in Vascular Biology: Cellular and molecular mechanisms of diabetes mellitus-accelerated atherosclerosis. Arterioscler Thromb Vasc Biol 2014; 34: 705–714.
- 3.
Stachon P, Peikert A, Michel NA, Hergeth S, Marchini T, Wolf D, et al. P2Y6 deficiency limits vascular inflammation and atherosclerosis in mice. Arterioscler Thromb Vasc Biol 2014; 34: 2237–2245.
- 4.
Kim HK, Shin MS, Youn BS, Kang GM, Gil SY, Lee CH, et al. Regulation of energy balance by the hypothalamic lipoprotein lipase regulator angptl3. Diabetes 2015; 64: 1142–1153.
- 5.
Yagyu H, Ishibashi S, Chen Z, Osuga J, Okazaki M, Perrey S, et al. Overexpressed lipoprotein lipase protects against atherosclerosis in apolipoprotein E knockout mice. J Lipid Res 1999; 40: 1677–1685.
- 6.
Zhang X, Qi R, Xian X, Yang F, Blackstein M, Deng X, et al. Spontaneous atherosclerosis in aged lipoprotein lipase-deficient mice with severe hypertriglyceridemia on a normal chow diet. Circ Res 2008; 102: 250–256.
- 7.
Qiu G, Hill JS. Atorvastatin decreases lipoprotein lipase and endothelial lipase expression in human THP-1 macrophages. J Lipid Res 2007; 48: 2112–2122.
- 8.
Qiu G, Ho AC, Yu W, Hill JS. Suppression of endothelial or lipoprotein lipase in THP-1 macrophages attenuates proinflammatory cytokine secretion. J Lipid Res 2007; 48: 385–394.
- 9.
Babaev VR, Patel MB, Semenkovich CF, Fazio S, Linton MF. Macrophage lipoprotein lipase promotes foam cell formation and atherosclerosis in low density lipoprotein receptor-deficient mice. J Biol Chem 2000; 275: 26293–26299.
- 10.
Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993; 75: 855–862.
- 11.
Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res 2012; 93: 583–593.
- 12.
Hulsmans M, Holvoet P. MicroRNAs as early biomarkers in obesity and related metabolic and cardiovascular diseases. Curr Pharm Des 2013; 19: 5704–5717.
- 13.
Yin W, Tsutsumi K. Lipoprotein lipase activator NO-1886. Cardiovasc Drug Rev 2003; 21: 133–142.
- 14.
He PP, Ouyang XP, Tang YY, Liao L, Wang ZB, Lv YC, et al. MicroRNA-590 attenuates lipid accumulation and pro-inflammatory cytokine secretion by targeting lipoprotein lipase gene in human THP-1 macrophages. Biochimie 2014; 106: 81–90.
- 15.
Tian GP, Tang YY, He PP, Lv YC, Ouyang XP, Zhao GJ, et al. The effects of miR-467b on lipoprotein lipase (LPL) expression, pro-inflammatory cytokine, lipid levels and atherosclerotic lesions in apolipoprotein E knockout mice. Biochem Biophys Res Commun 2014; 443: 428–434.
- 16.
Li Y, He PP, Zhang DW, Zheng XL, Cayabyab FS, Yin WD, et al. Lipoprotein lipase: From gene to atherosclerosis. Atherosclerosis 2014; 237: 597–608.
- 17.
Lan G, Xie W, Li L, Zhang M, Liu D, Tan YL, et al. MicroRNA-134 actives lipoprotein lipase-mediated lipid accumulation and inflammatory response by targeting angiopoietin-like 4 in THP-1 macrophages. Biochem Biophys Res Commun 2015; 472: 410–417.
- 18.
Raitoharju E, Oksala N, Lehtimaki T. MicroRNAs in the atherosclerotic plaque. Clin Chem 2013; 59: 1708–1721.
- 19.
Zhou J, Meng Y, Tian S, Chen J. Comparative MicroRNA expression profiles of cynomolgus monkeys, rat, and human reveal that miR-182 is involved in T2D pathogenic processes. J Diabetes Res 2014; 2014: 760397.
- 20.
Rotllan N, Price N, Pati P, Goedeke L, Fernandez-Hernando C. microRNAs in lipoprotein metabolism and cardiometabolic disorders. Atherosclerosis 2016; 246: 352–360.
- 21.
Romaine SP, Tomaszewski M, Condorelli G, Samani NJ. MicroRNAs in cardiovascular disease: An introduction for clinicians. Heart 2015; 101: 921–928.
- 22.
Taurino C, Miller WH, McBride MW, McClure JD, Khanin R, Moreno MU, et al. Gene expression profiling in whole blood of patients with coronary artery disease. Clin Sci (Lond) 2010; 119: 335–343.
- 23.
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, et al. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 2005; 18: 601–607.
- 24.
Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat Rev Genet 2009; 10: 32–42.
- 25.
Dichgans M, Malik R, Konig IR, Rosand J, Clarke R, Gretarsdottir S, et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome-wide analysis of common variants. Stroke 2014; 45: 24–36.
- 26.
Markus HS, Makela KM, Bevan S, Raitoharju E, Oksala N, Bis JC, et al. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke 2013; 44: 1220–1225.
- 27.
Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol 2014; 34: 1871–1879.
- 28.
Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, et al. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood 2002; 100: 4446–4453.
- 29.
Yin K, Chen WJ, Zhou ZG, Zhao GJ, Lv YC, Ouyang XP, et al. Apolipoprotein A-I inhibits CD40 proinflammatory signaling via ATP-binding cassette transporter A1-mediated modulation of lipid raft in macrophages. J Atheroscler Thromb 2012; 19: 823–836.
- 30.
Chi X, Shetty SK, Shows HW, Hjelmaas AJ, Malcolm EK, Davies BS. Angiopoietin-like 4 modifies the interactions between lipoprotein lipase and its endothelial cell transporter GPIHBP1. J Biol Chem 2015; 290: 11865–11877.
- 31.
Yu XH, Fu YC, Zhang DW, Yin K, Tang CK. Foam cells in atherosclerosis. Clin Chim Acta 2013; 424: 245–252.
- 32.
Son SH, Goo YH, Choi M, Saha PK, Oka K, Chan LC, et al. Enhanced atheroprotection and lesion remodelling by targeting the foam cell and increasing plasma cholesterol acceptors. Cardiovasc Res 2015; 109: 294–304.
- 33.
Hosin AA, Prasad A, Viiri LE, Davies AH, Shalhoub J. MicroRNAs in Atherosclerosis. J Vasc Res 2014; 51: 338–349.
- 34.
Blaya D, Coll M, Rodrigo-Torres D, Vila-Casadesus M, Altamirano J, Llopis M, et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut 2016; 65: 1535–1545.
- 35.
Olivier M, Tanck MW, Out R, Villard EF, Lammers B, Bouchareychas L, et al. Human ATP-binding cassette G1 controls macrophage lipoprotein lipase bioavailability and promotes foam cell formation. Arterioscler Thromb Vasc Biol 2012; 32: 2223–2231.
- 36.
Jiang Q, Wang D, Han Y, Han Z, Zhong W, Wang C. Modulation of oxidized-LDL receptor-1 (LOX1) contributes to the antiatherosclerosis effect of oleanolic acid. Int J Biochem Cell Biol 2015; 69: 142–152.
- 37.
Sato K, Okajima F, Miyashita K, Imamura S, Kobayashi J, Stanhope KL, et al. The majority of lipoprotein lipase in plasma is bound to remnant lipoproteins: A new definition of remnant lipoproteins. Clin Chim Acta 2016; 461: 114–125.
- 38.
Woldemichael BT, Jawaid A, Kremer EA, Gaur N, Krol J, Marchais A, et al. The microRNA cluster miR-183/96/182 contributes to long-term memory in a protein phosphatase 1-dependent manner. Nat Commun 2016; 7: 12594.
- 39.
Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and MAP kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem 2005; 280: 21763–21772.
- 40.
Erbay E, Babaev VR, Mayers JR, Makowski L, Charles KN, Snitow ME, et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat Med 2009; 15: 1383–1391.
- 41.
Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr Opin Lipidol 2009; 20: 379–385.
- 42.
Suganami T, Nishida J, Ogawa Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler Thromb Vasc Biol 2005; 25: 2062–2068.
- 43.
Azghandi S, Prell C, van der Laan SW, Schneider M, Malik R, Berer K, et al. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke 2015; 46: 197–202.
- 44.
Smith JD. New role for histone deacetylase 9 in atherosclerosis and inflammation. Arterioscler Thromb Vasc Biol 2014; 34: 1798–1799.
- 45.
Yang Y, Thyagarajan N, Coady BM, Brown RJ. Cholesterol efflux from THP-1 macrophages is impaired by the fatty acid component from lipoprotein hydrolysis by lipoprotein lipase. Biochem Biophys Res Commun 2014; 451: 632–636.
- 46.
Kawashima RL, Medh JD. Down-regulation of lipoprotein lipase increases ABCA1-mediated cholesterol efflux in THP-1 macrophages. Biochem Biophys Res Commun 2014; 450: 1416–1421.