Multiple Arterial Dissections in a Patient With Vascular Ehlers-Danlos Syndrome Caused by a Frameshift Mutation in COL3A1
Article ID: CJ-19-0143

Details
Article ID: CJ-19-0143
A 49-year-old man with a 10-year history of renal infarction complained of acute abdominal pain. Imaging indicated multiple visceral and iliac artery dissections (Figure A–G). Several different diagnoses were considered (Supplementary Table), and genetic testing indicated a frameshift mutation in COL3A1 (Figure H). Based on arterial complications and genetic testing, he was finally diagnosed with vascular Ehlers-Danlos syndrome (vEDS).
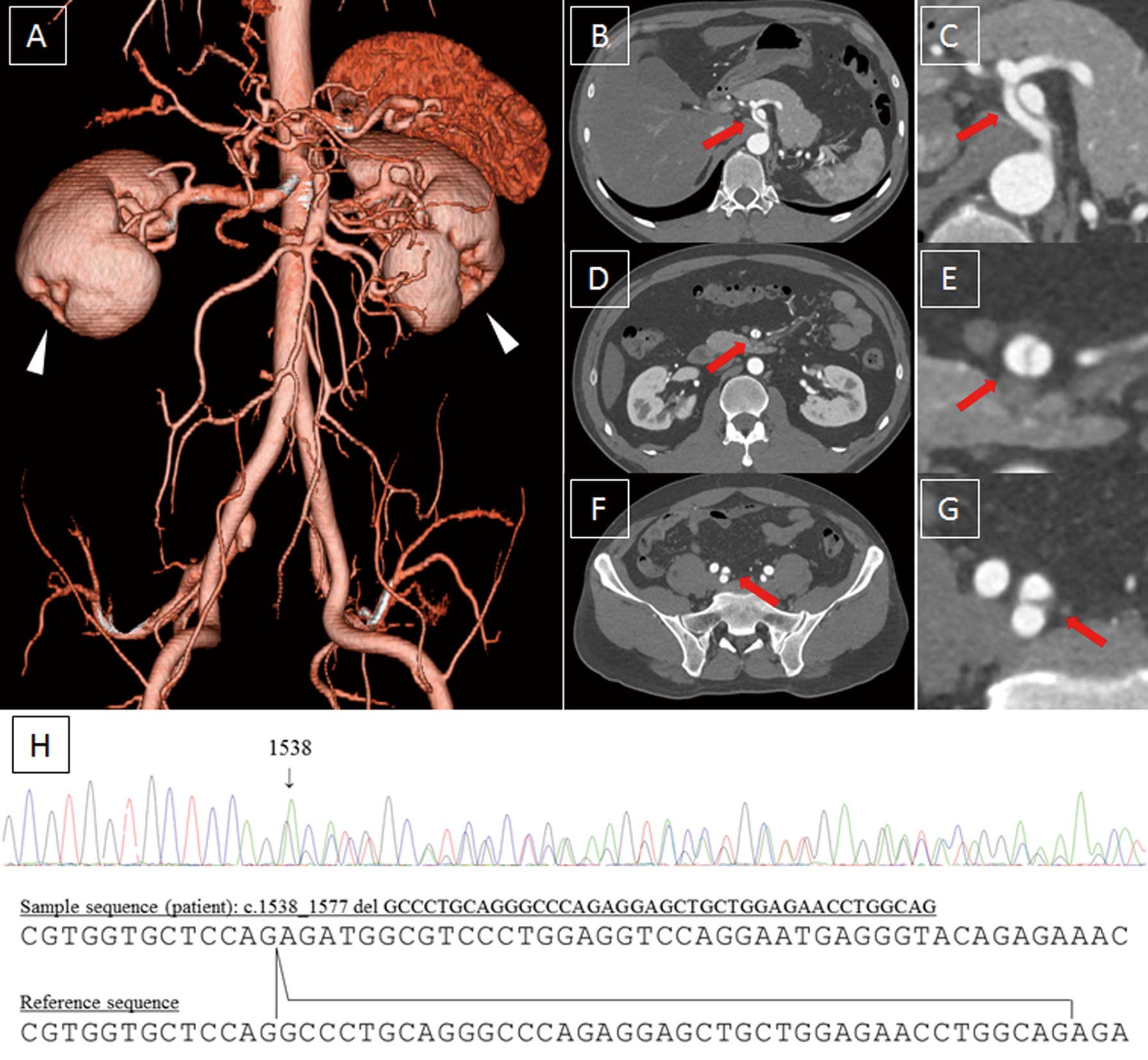
Contrast-enhanced computed tomography shows multiple arterial complications: (A) evidence of bilateral renal infarction on 3-D imaging (arrowheads), (B,C) celiac artery dissection and aneurysm, (D,E) superior mesenteric artery dissection, and (F,G) right internal iliac artery dissection and aneurysm. (H) DNA sequence chromatogram of COL3A1 showing a 40-bp deletion in exon 22 (c.1538_1577 del), causing a frameshift and premature stop codon (p.G513EfsTer10).
vEDS is rare and life-threatening, and can cause spontaneous arterial dissection or rupture. It is caused by heterozygous COL3A1 mutations, predominantly missense mutations altering glycine codons in triple helical domains, or splice-site variants resulting in in-frame exon skipping.1 Although most patients with vEDS have a disease-specific history (spontaneous rupture of the bowel, uterine rupture during pregnancy) and physical features (translucent skin, hypermobility of small joints, easy bruising) other than arterial complications, the present patient did not have any such traits. Frameshift and nonsense mutations of COL3A1 are less frequently reported than the aforementioned major variants, and cause milder phenotypes and later onset of arterial complications.1
In cases wherein patients present with spontaneous arterial complications, one should consider the possibility of vEDS and opt for genetic testing, even if the patients have only a few typical features of the disease.
The authors declare no conflicts of interest.
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-19-0143