Abstract
Background:
Plaque erosion can occur quietly without causing clinical symptoms, followed by a healing process resulting in healed plaque. This study aimed to assess culprit and non-culprit plaque characteristics of patients with acute myocardial infarction (AMI) caused by plaque erosion with vs. without healed phenotype at the culprit plaque using optical coherence tomography (OCT).
Methods and Results:
A total of 117 AMI patients caused by plaque erosion who underwent OCT imaging of 3 coronary arteries were included. Patients were divided into 2 groups based on presence or absence of a healed phenotype at the culprit site. Culprit and non-culprit plaque characteristics were compared between the 2 groups. A healed phenotype at the culprit lesion was identified in 47.9% of AMI patients caused by plaque erosion. Patients with a healed phenotype at the culprit site were more frequently with hyperlipidemia, and had a higher prevalence of macrophage infiltration, microchannels, cholesterol crystals, and calcification at the culprit lesion. Moreover, patients with a healed phenotype at the culprit site had more non-culprit plaques and more characteristics of plaque vulnerability at the non-culprit lesion. In addition, patients with a healed phenotype at the culprit site presented with more severe luminal stenosis at both the culprit and non-culprit lesion.
Conclusions:
A healed phenotype was identified in 47.9% of AMI patients caused by plaque erosion at the culprit site. A healed phenotype within eroded culprit plaque was associated with signs of pancoronary vulnerability and advanced atherosclerosis.
Plaque erosion and plaque rupture are the 2 most frequent causes of acute coronary syndrome (ACS), and plaque erosion is responsible for approximately one-third of patients.1,2
Previous pathological and in vivo studies have reported that plaque erosion can occur quietly without causing clinical symptoms followed by a healing process of the erosion site resulting in healed plaque.3–5
Silent plaque instability and the healing process can occur repeatedly, and eventually lead to culprit plaque formation. However, studies regarding the characteristics of plaque erosion with and without healed plaque are limited. Optical coherence tomography (OCT) is an intravascular imaging technique with excellent resolution, which can visualize detailed structures in the coronary arterial plaque.6
On OCT imaging, healed plaques appear as heterogeneous signalrich layers with optical signal intensity that is different from the underlying plaque,3,7
and Shimokado et al demonstrated that OCT-derived healed plaques had good sensitivity and specificity (81% and 98%) to detect histologically defined healed plaques.5
Therefore, in this study, we sought to assess culprit and non-culprit plaque characteristics of culprit plaque erosion with vs. without a healed phenotype in patients with acute myocardial infarction (AMI).
Editorial p ????
Methods
Study Population
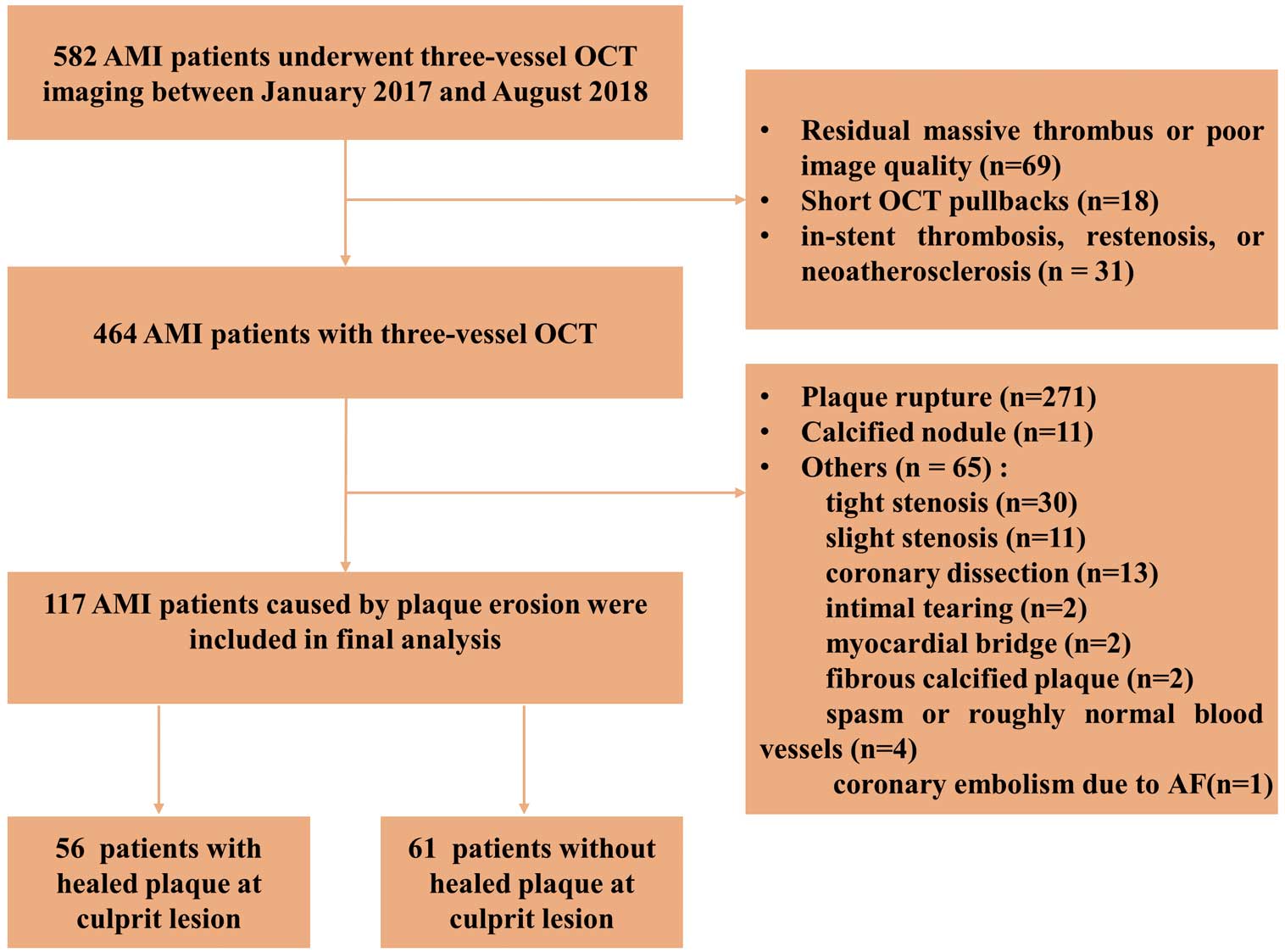
Between January 2017 and August 2018, 582 patients with AMI who underwent pre-intervention OCT imaging of all 3 major epicardial coronary arteries were identified in the Cardiovascular Hospital of the Second Affiliated Hospital of Harbin Medical University (Harbin, China). Among them, 118 patients were excluded for: (1) residual massive thrombus or poor image quality (n=69); (2) short OCT pullbacks less than two-thirds of the lengths of the arteries (n=18); and (3) in-stent thrombosis, restenosis, or neoatherosclerosis (n=31). Therefore, 464 patients with 3-vessel OCT imaging were identified for this study. In addition, patients with plaque rupture (n=271), calcified nodule (n=11) and other lesion types (n=65), including tight stenosis (n=30), slight stenosis (n=11), coronary dissection (n=13), intimal tearing (n=2), myocardial bridge (n=2), fibrous calcified plaque (n=2), spasm or roughly normal blood vessels (n=4), and coronary embolism due to atrial fibrillation (n=1) were also excluded. Finally, 117 patients with AMI diagnosed as plaque erosion were included in the analysis (Figure 1). Pre-intervention OCT of the culprit vessel was carried out before treatment of the culprit plaque, whereas OCT of the non-culprit vessels was performed immediately after the culprit plaque was treated. Diagnosis of AMI included ST-segment elevation myocardial infarction (STEMI) and non-ST-segment elevation myocardial infarction (NSTEMI), as previously described.8,9
The present study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (Harbin, China), and all patients provided written informed consent.
Coronary Angiography Analysis
Quantitative coronary angiography analysis was performed by using a Cardiovascular Angiography Analysis System, version 5.10.1 software (Pie Medical Imaging BV, Maastricht, The Netherlands). Coronary flow was assessed using the Thrombolysis in Myocardial Infarction (TIMI) flow grade classification. The minimal lumen diameter (MLD), reference vessel diameter (RVD), diameter stenosis (DS), and lesion length of the culprit and non-culprit lesions were measured.10
OCT Image Acquisition and Analysis
OCT was performed by using a commercially available frequency domain OCT system (ILUMIEN OPTIS or OPTIS Integrated System; Abbott Vascular, Santa Clara, CA, USA). In patients with TIMI flow grade ≤2 and large thrombus burden, aspiration thrombectomy was allowed before imaging, whereas patients with balloon pre-dilation were not enrolled. All OCT images were analyzed by 2 independent investigators blinded to clinical and angiographic data. In case of discordance, a consensus reading was achieved with a third investigator.
OCT images were analyzed using the previously established criteria.11,12
All plaques were identified by OCT as segments with luminal narrowing and a loss of the normal 3-layered structure of the vessel wall.11
Culprit lesions were identified based on angiographic findings, electrocardiogram changes, and/or left ventricular wall motion abnormalities.11
In patients with multiple stenoses, the plaque with the most severe stenosis or with evidence of acute thrombus on angiography or OCT was considered to be the culprit. Non-culprit lesions were identified by OCT by the presence of a plaque in ≥3 consecutively analyzed frames.11
The reference was the site with the largest lumen area either proximal or distal to the stenosis, and plaque length was determined as the distance between the distal and proximal reference. A distance of at least 5 mm on the longitudinal view was considered 2 separated plaques, otherwise, separate plaques were considered to be one long plaque.11–13
Cross-sectional OCT images were analyzed at 1-mm intervals. Minimal lumen area (MLA) was defined as the smallest lumen area within the length of the plaque. Percent area stenosis (AS%) was calculated as ([Mean Reference Lumen Area − MLA] / Mean Reference Lumen Area) × 100.
Plaque erosion was defined and categorized according to the absence of fibrous cap disruption and the presence of a thrombus. Definite erosion was identified by the presence of an attached thrombus overlying an intact and visualized plaque. Probable erosion was defined by: (1) luminal surface irregularity at the culprit lesion in the absence of thrombus; or (2) attenuation of underlying plaque by a thrombus without superficial lipid or calcification immediately proximal or distal to the site of a thrombus.1
Plaque rupture was identified by the presence of fibrous cap discontinuity with a cavity formed inside the plaque. A calcified nodule was defined by fibrous cap disruption detected over a calcified plaque characterized by protruding calcification, superficial calcium, or the presence of substantive calcium proximal and/or distal to the lesion.
Plaques were classified as fibrous plaques (homogeneous, high backscattering region) or lipid plaques (low signal region with diffuse border).11
For each lipid plaque, the thinnest fibrous cap thickness (FCT) and the maximal arc of lipid were measured. Lipid length was obtained on the longitudinal view, and the lipid index was calculated as the product of mean lipid arc and lipid length. Thin-cap fibroatheroma (TCFA) was defined as a plaque with a maximal lipid arc >90° and thinnest FCT <65 μm. Macrophage accumulation was defined as the presence of highly backscattering focal regions within the fibrous cap. Microchannels were defined as the presence of signal-poor structures with vesicular or tubular shapes. Cholesterol crystals were identified as thin and linear regions of high signal intensity with high backscattering within a plaque. Calcification was defined as a signal-poor or heterogeneous region with a sharply delineated border. Thrombus was defined as an irregular mass with minimum diameter at least 250 μm adherent to the vessel wall or floating within the lumen. Healed plaque was defined as a plaque with heterogeneous signal-rich layered tissue of different optical signal intensity located close to the luminal surface that was clearly demarcated from the underlying tissue.5
The maximum area and thickness of healed tissue was determined from the cross-sectional OCT images.
The inter-observer kappa coefficients for plaque erosion and healed plaque were 0.92 and 0.88, respectively. The intra-observer kappa coefficients for plaque erosion and healed plaque were 0.95 and 0.92, respectively.
Clinical Follow up
Patients were followed for 2 years after discharge at either a hospital visit or via a phone call. Major adverse cardiac events (MACE) were a composite of cardiac death, non-fatal recurrent miocardial infarction (MI), ischemia-driven revascularization, or rehospitalization due to unstable or progressive angina. Detailed definitions of individual outcome measures are provided in
Supplementary Methods.
Statistical Analyses
Statistical analyses were performed by using SPSS version 21.0 (IBM, Armonk, NY, USA). Data distribution was assessed by using the Kolmogorov-Smirnov test. Continuous variables have been shown as mean±SD or as median (25th–75th percentiles) and compared with the independent sample t-test or the Mann-Whitney U-test. Categorical data have been presented as counts (proportions) and compared using the chi-squared or Fisher’s exact test. For plaque level data, a model with generalized estimating equations was used to take into account potential cluster effects of multiple plaques in a single patient. Inter- and intra-observer agreement were assessed by kappa coefficient statistics. A 2-tailed P<0.05 was considered statistically significant.
Results
Patients and Baseline Characteristics
Among 117 AMI patients caused by plaque erosion, 67 (57.3%) patients were with definite plaque erosion and 50 (42.7%) patients were with probable plaque erosion. Moreover, in 117 patients, 56 (47.9%) patients with healed plaque at the culprit lesion had 230 non-culprit plaques and 61 (52.1%) patients without healed plaque at the culprit lesion had 167 non-culprit plaques.
Patient characteristics are listed in
Table 1. Although the difference did not reach statistical significance, patients with healed phenotype at the culprit site had a higher incidence of hyperlipidemia (55.4% vs. 37.7%, P=0.056) than those without. No significant differences were found in other baseline characteristics between the 2 groups.
Table 1.
Baseline Clinical Characteristics of Patients With vs. Without Healed Phenotype at the Culprit Site
| |
Healed phenotype
group (n=56) |
Non-healed phenotype
group (n=61) |
P value |
| Age, years |
53.0±10.3 |
52.4±11.1 |
0.748 |
| Male |
47 (83.9) |
51 (83.6) |
0.962 |
| Risk factors |
| Hypertension |
18 (32.1) |
13 (21.3) |
0.185 |
| Hyperlipidemia |
31 (55.4) |
23 (37.7) |
0.056 |
| Current smoking |
35 (62.5) |
42 (68.9) |
0.469 |
| Diabetes mellitus |
10 (17.9) |
9 (14.8) |
0.649 |
| Chronic kidney disease |
0 (0.0) |
5 (8.2) |
0.083 |
| Clinical presentation |
|
|
0.739 |
| STEMI |
36 (64.3) |
41 (67.2) |
|
| NSTEMI |
20 (35.7) |
20 (32.8) |
|
| Laboratory data |
| Triglyceride, mg/dL |
144.2±78.7 |
121.6±52.3 |
0.08 |
| TC, mg/dL |
179.8±38.1 |
170.6±33.8 |
0.176 |
| LDL-C, mg/dL |
110.2±29.7 |
102.6±27.8 |
0.166 |
| HDL-C, mg/dL |
50.2±10.3 |
52.8±11.0 |
0.202 |
| HbA1c, % |
6.2±1.2 |
5.8±0.9 |
0.086 |
| Hs-CRP, mg/L |
5.5 (1.6–10.3) |
4.6 (2.5–8.3) |
0.736 |
| Creatinine, μmol/L |
75 (63.3–85.3) |
78 (68–94) |
0.062 |
| CK-MB, μg/L |
14.2 (2.7–63.8) |
16 (2.1–96.0) |
0.766 |
| CTnI, μg/L |
2.7 (1.1–13.4) |
2.0 (0.3–24.7) |
0.798 |
| Medication history |
| Antiplatelet therapy |
4 (7.1) |
8 (13.1) |
0.367 |
| Aspirin |
2 (3.6) |
6 (9.8) |
0.275 |
| Clopidogrel |
2 (3.6) |
2 (3.3) |
1.000 |
| Statin |
5 (8.9) |
2 (3.3) |
0.257 |
| ACEI/ARB |
6 (10.7) |
3 (4.9) |
0.308 |
| β-blockers |
1 (1.8) |
2 (3.3) |
1.000 |
Data are presented as n (%), mean±SD, or median (25th–75th percentiles). ACEI, angiotensin-converting enzyme inhibitor; ARB, angiotensin II receptor blocker; β-blockers, β-receptor blockers; CK-MB, creatine kinase isoenzyme MB; CTnI, cardiac troponin I; HbA1c, glycated hemoglobin; HDL-C, high-density lipoprotein-cholesterol; Hs-CRP, high sensitivity C-reactive protein; LDL-C, low-density lipoprotein-cholesterol; SD, standard deviation; STEMI, ST-segment elevation myocardial infarction; NSTEMI, non-ST-segment elevation myocardial infarction; TC, total cholesterol.
Angiographic findings are summarized in
Supplementary Table 1
and
Supplementary Table 2. Lesion distribution of culprit and non-culprit lesions was not significantly different between the 2 groups. Patients with healed phenotype at the culprit site had smaller RVD in culprit (3.0±0.5 mm vs. 3.3±0.6 mm, P=0.017) and non-culprit lesions (3.1±0.7 mm vs. 3.3±0.6 mm, P=0.048) compared with those without healed phenotype.
OCT Findings at the Culprit Lesion
OCT characteristics of culprit lesions are listed in
Table 2
and
Figure 2. Patients with healed phenotype at the culprit site had longer plaque length (22.3 [17.2–25.7] mm vs. 18.0 [16.2–21.5] mm, P=0.003), higher prevalence of lipid plaque (71.4% vs. 52.5%, P=0.035), macrophage infiltration (87.5% vs. 59.0%, P=0.001), microchannel (53.6% vs. 29.5%, P=0.008), cholesterol crystals (25% vs. 9.8%, P=0.03), and calcification (46.4% vs. 11.5%, P<0.001). Patients with healed phenotype at the culprit site also had smaller MLA (1.6 [1.1–2.4] mm2
vs. 2.1 [1.7–3.2] mm2, P=0.007) and greater degree of AS (79.3 [68.4–84.7]% vs. 73.3 [57.4–79.3]%, P=0.035) than those without healed phenotype. The representative OCT image is presented in
Figure 3.
Table 2.
OCT Analysis of Culprit Plaques in Patients With vs. Without Healed Phenotype at the Culprit Site
| |
Healed phenotype
group (n=56) |
Non-healed phenotype
group (n=61) |
P value |
| Plaque length, mm |
22.3 (17.2–25.7) |
18.0 (16.2–21.5) |
0.003 |
| Plaque type |
|
|
0.035 |
| Fibrous |
16 (28.6) |
29 (47.5) |
|
| Lipid |
40 (71.4) |
32 (52.5) |
|
| FCT, μm |
100 (84.2–139.2) |
100 (77.5–121.7) |
0.398 |
| Mean lipid arc, ° |
205.7±43.0 |
203.0±44.5 |
0.801 |
| Max lipid arc, ° |
349.6 (238.8–360) |
287.5 (239.3–360) |
0.355 |
| Lipid length, mm |
13.6±7.3 |
11.8±6.4 |
0.262 |
| Lipid index, mm° |
2,893.7±1,744.4 |
2,422.4±1,383.4 |
0.217 |
| TCFA |
4 (7.1) |
3 (4.9) |
0.907 |
| Macrophage |
49 (87.5) |
36 (59.0) |
0.001 |
| Microchannel |
30 (53.6) |
18 (29.5) |
0.008 |
| Cholesterol crystals |
14 (25.0) |
6 (9.8) |
0.03 |
| Calcification |
26 (46.4) |
7 (11.5) |
<0.001 |
| Thrombus |
56 (100) |
58 (95.1) |
0.273 |
| MLA, mm2 |
1.6 (1.1–2.4) |
2.1 (1.7–3.2) |
0.007 |
| AS, % |
79.3 (68.4–84.7) |
73.3 (57.4–79.3) |
0.035 |
| Mean RLA, mm2 |
8.1 (6.1–9.0) |
8.5 (6.6–10.8) |
0.144 |
Data are presented as n (%), median (interquartile range), or mean±SD. AS, area stenosis; FCT, fibrous cap thickness; MLA, minimal lumen area; OCT, optical coherence tomography; RLA, reference lumen area; SD, standard deviation; TCFA, thin cap fibroatheroma.

OCT characteristics of non-culprit lesions are summarized in
Table 3, Table 4
and
Figure 4. Patients with healed phenotype at the culprit site had more non-culprit plaque (4.1±2.2 vs. 2.7±1.8, P<0.001).
Table 3.
Plaque-Level OCT Analysis of Non-Culprit Plaques in Patients With vs. Without Healed Phenotype at the Culprit Site
| |
Healed phenotype
group (n=230) |
Non-healed phenotype
group (n=167) |
P value |
| Plaque length, mm |
14.1 (10.6–19.3) |
13.2 (10.2–17.5) |
0.340 |
| Plaque type |
|
|
0.120 |
| Fibrous |
71 (30.9) |
66 (39.5) |
|
| Lipid |
159 (69.1) |
101 (60.5) |
|
| Lipid length, mm |
9.0±6.4 |
9.0±6.7 |
0.992 |
| Mean lipid arc, ° |
146.1±41.3 |
153.4±39.4 |
0.186 |
| Lipid index, mm° |
1,405.8±1,219.7 |
1,429.4±1,172.0 |
0.898 |
| FCT, μm |
103.3 (80.0–150.0) |
116.7 (85.0–165.0) |
0.184 |
| Mean RLA, mm2 |
7.8 (5.7–10.2) |
8.3 (6.2–10.3) |
0.120 |
| MLA, mm2 |
3.6 (2.4–5.5) |
4.3 (2.6–6.2) |
0.037 |
| AS, % |
50.1 (38.7–61.9) |
46.3 (34.3–61.0) |
0.045 |
| TCFA |
20 (8.7) |
11 (6.6) |
0.485 |
| Macrophage |
181 (78.7) |
112 (67.1) |
0.044 |
| Microvessels |
115 (50.0) |
75 (44.9) |
0.604 |
| Cholesterol crystals |
24 (10.4) |
3 (1.8) |
0.003 |
| Thrombus |
5 (2.2) |
6 (3.6) |
0.463 |
| Calcification |
77 (33.5) |
37 (22.2) |
0.043 |
| Healed plaque |
75 (32.6) |
45 (26.9) |
0.284 |
| Healed tissue thickness, μm |
596.9±182.2 |
504.4±170.9 |
0.004 |
| Healed tissue area, mm2 |
1.5±0.9 |
1.1±0.6 |
0.015 |
Data are presented as n (%), median (interquartile range), or mean±SD. Abbreviations as in Table 2.
Table 4.
Patient-Level OCT Analysis of Non-Culprit Plaques in Patients With vs. Without Healed Phenotype at the Culprit Site
| |
Healed phenotype
group (n=56) |
Non-healed phenotype
group (n=61) |
P value |
| Lipid plaque |
51 (91.1) |
46 (75.4) |
0.025 |
| TCFA |
15 (26.8) |
8 (13.1) |
0.063 |
| Macrophage |
49 (87.5) |
48 (78.7) |
0.206 |
| Microchannel |
44 (78.6) |
36 (59.0) |
0.023 |
| Cholesterol crystals |
14 (25.0) |
3 (4.9) |
0.003 |
| Thrombus |
4 (7.1) |
5 (8.2) |
1.000 |
| Calcification |
33 (58.9) |
24 (39.3) |
0.034 |
| Healed plaque |
39 (69.6) |
26 (42.6) |
0.003 |
Data are presented as n (%). OCT, optical coherence tomography; TCFA, thin cap fibroatheroma.

For plaque-based analysis of non-culprit lesions (Table 3, Figure 4), culprit plaque erosion with healed phenotype group had higher incidence of macrophage infiltration (78.7% vs. 67.1%, P=0.044), cholesterol crystals (10.4% vs. 1.8%, P=0.003), calcification (33.5% vs. 22.2%, P=0.043) and had smaller MLA (3.6 [2.4–5.5] mm2
vs. 4.3 [2.6–6.2] mm2, P=0.037) and greater degree of AS% (50.1 [38.7–61.9] % vs. 46.3 [34.3–61.0] %, P=0.045) than culprit plaque erosion without healed phenotype group. The prevalence of non-culprit healed plaque in both groups was similar; however, culprit plaque erosion with healed phenotype had larger non-culprit healed tissue thickness (596.9±182.2 μm vs. 504.4±170.9 μm, P=0.004) and healed tissue area (1.5±0.9 mm2
vs. 1.1±0.6 mm2, P=0.015).
For patient-based analysis of non-culprit lesions (Table 4, Figure 4), patients with healed plaque at the culprit site had higher incidence of lipid plaque (91.1% vs. 75.4%, P=0.025), microchannels (78.6% vs. 59.0%, P=0.023), cholesterol crystals (25.0% vs. 4.9%, P=0.003), calcification (58.9% vs. 39.3%, P=0.034) and healed plaque (69.6% vs. 42.6%, P=0.003) than those without healed plaque at the culprit site.
Clinical Outcomes
Clinical follow-up data were available for 115 (98.3%) patients, and the mean follow-up time was 25.2±3.9 months. The overall incidence of MACE was 16.5%, and there was no significant difference between patients with vs. without healed phenotype at the culprit site (22.2% vs. 11.5%, P=0.138). The incidence of cardiac death, non-fatal MI, ischemia-driven revascularization and rehospitalization were similar between the 2 study groups (Supplementary Table 3).
Discussion
In the current study, we compared the characteristics of culprit and non-culprit plaques in AMI patients caused by plaque erosion with and without healed plaque at the culprit site. Our study demonstrated that patients with healed phenotype at the culprit site: (1) were identified in 47.9% of AMI patients caused by plaque erosion and had a higher incidence of hyperlipidemia; (2) had higher levels of local inflammation and plaque vulnerability at both the culprit and non-culprit lesions; and (3) presented with more severe luminal stenosis at both culprit and non-culprit lesions than patients without healed phenotype at the culprit site.
Prevalence and Baseline Characteristics of Patients With Healed Phenotype
Healed plaques represent signs of previous plaque instability, morphologically characterized by distinct layers of collagen in histopathology14,15
and appear as heterogeneous signalrich layers with optical signal intensity that is different from the underlying plaque on OCT.5
In an autopsy study, Burke et al reported that healed plaque had been found in 53% of subjects who suffered from sudden coronary death, who had no evidence of previous myocardial infarction.15
Recently, Dai et al found 40.3% of AMI patients had healed culprit plaques in an OCT study.16
In the present study, we reported that the prevalence of healed plaques at the culprit lesion was 47.9% in AMI patients caused by plaque erosion, which was similar to the findings of previous studies. In another OCT study, however, Fracassi et al reported that 29% of patients had healed phenotype at the culprit lesion in 376 ACS patients, which was lower than our study.7
This difference may be due to the patients enrolled being identified from 21 sites over 6 years, which indicates a highly selected cohort. In our study, patients with healed phenotype at the culprit lesion were more frequent with hyperlipidemia. Previous studies revealed that hyperlipidemia was associated with a higher risk of thrombosis.17
Under a hypercoagulable state, thrombus stimulation will exceed the endogenous thrombolytic activity, leading to mural thrombosis, followed by the healing process, resulting in healed plaques.
Culprit Plaque Characteristics in AMI Patients Caused by Plaque Erosion With or Without Healed Phenotype at the Culprit Site
In the present study, patients with healed phenotype at the culprit site had higher prevalence of lipid plaque, macrophage infiltration, microchannel, and cholesterol crystals than patients without healed phenotype, indicating higher plaque vulnerability at the culprit lesion. Previous OCT studies have found similar findings.7
Russo et al had reported that plaques with healed phenotype at the culprit site showed more signs of vulnerability (more lipid plaque, greater lipid burden, and more macrophage infiltration) than non-healed ones.18
Shimokado et al also found that microvessels and macrophages were more frequently identified in OCT-derived healed plaque.5
Macrophages indicating higher local inflammation are the sign of plaque activity and play an important role in extracellular matrix degradation and fibrous cap disruption, as well as plaque progress.19
The formation of microvessels is a common feature during thrombus organization and is observed more frequently in healed plaques than in non-healed plaques, and plays an important role in thrombus organization for providing oxygen and nutrients to the healing tissue.20
Cholesterol crystallization is thought to be the result of accumulation of free cholesterol in macrophages and destruction of cholesterol homeostasis. Excess esterified cholesterol and crystals are released from the dead foam cells into the extracellular space, and then identified as cholesterol crystals in coronary plaques.21,22
Sugane et al found that cholesterol-crystalized coronary atheroma was a potential precursor lesion causing plaque instable, leading to obstructive and non-obstructive thrombosis.23
In addition, patients with healed phenotype at the culprit site had a higher incidence of calcification than those without. Russo et al18
also showed a higher prevalence of calcifications in healed culprit plaques compared with non-healed ones. As we know, in the process of plaque healing, especially in the late stage, type 2 macrophages can promote the calcification of plaque by promoting the differentiation of osteoblasts and maturation of vascular smooth muscle cells.3
Moreover, Burke et al suggested that previous plaque instability, especially plaque rupture, via hemorrhage and cellular breakdown, resulted in coronary calcification.24
Non-Culprit Plaque Characteristics in AMI Patients Caused by Plaque Erosion With or Without Healed Phenotype at the Culprit Site
In the current study, we found that patients with healed phenotype at the culprit site had more non-culprit plaques and a higher incidence of macrophages, cholesterol crystals and calcification at plaque level, and higher prevalence of lipid plaque, microchannels, cholesterol crystals, calcification and healed plaque at the patient level, all of which indicated that patients with healed phenotype at the culprit site also had higher plaque vulnerability in non-culprit lesions. Russo et al reported that patients with healed culprit plaque had more non-culprit plaque, and had higher prevalence of lipid plaque and macrophage infiltration than those without in patients with ACS or stable angina pectoris (SAP),18,25
which was similar with our results. Moreover, multiple studies demonstrated that the vulnerability of non-culprit plaque was consistent with the vulnerability of culprit plaque in the same patients.26,27
These results revealed that the presence of healed phenotype at the culprit site in patients with ACS or SAP represented a higher level of pan-coronary plaque vulnerability through previous plaque destabilization, indicating higher instability in the future.
Lumen Stenosis in AMI Patients Caused by Plaque Erosion With or Without a Healed Phenotype at the Culprit Site
In our study, patients with healed phenotype at the culprit site had a more severe degree of lumen stenosis in the culprit and non-culprit lesions, indicating greater plaque burden and a more advanced atherosclerosis process. Mann and Davies reported that >70% of plaques with a diameter stenosis ≥51% had a healed pattern in men who died from ischemic heart disease.14
Burke et al found that the mean percent luminal narrowing increased in both acute and stable plaque sites with increased numbers of healed plaques.15
Yamamoto et al demonstrated that healed non-culprit plaques represented rapid progression with a greater increase in plaque volume compared with non-healed phenotype.4
These findings suggested the potential contribution of plaque healing to progression of coronary atherosclerosis. In addition, we also found the healed tissue thickness and healed tissue area in non-culprit healed plaques was greater in patients with healed phenotype at the culprit site than those without. We considered this phenomenon may attribute to higher plaque vulnerability in non-culprit plaques, and there may also be multiple healing processes in single non-culprit coronary lesions, which might not be visible due to the limited penetrability of OCT.
Clinical Significance
In our study, although not statistically significant, the overall incidence of MACE in patients with healed phenotype was nearly twice as high as that of patients without healed phenotype. We considered that the reason for this phenomenon may come from patients with healed phenotype at the culprit site having higher levels of local inflammation and plaque vulnerability at both culprit and non-culprit lesions. Moreover, patients with healed phenotype at the culprit site had more severe luminal stenosis at both culprit and non-culprit lesions than for those without healed phenotype, indicating greater plaque burden and a more advanced atherosclerosis process. Therefore, for AMI patients caused by plaque erosion with a healed phenotype, stronger antithrombotic and lipid-lowering therapy might have more clinical benefits. However, as the sample size of our study is small and follow-up time is relatively short, studies with larger samples and longer follow-up time are needed.
Study Limitations
This study had several limitations. First, this study was a retrospective single-center study and patients with highly unstable clinical conditions and cases with massive thrombus or poor image quality were excluded. Therefore, selection bias cannot be excluded. Second, this study lacked histopathologic correlation, and we cannot exclude other potential mechanisms of atherosclerosis progression (i.e., neointimal hyperplasia, lipid, or calcification) that may lead to intraplaque heterogeneity with a healed appearance on OCT. Third, the OCT definition of plaque erosion was a diagnosis of exclusion requiring the absence of fibrous cap disruption. Fourth, manual thrombectomy may have altered the underlying plaque morphology, although extreme care was taken to avoid excess trauma. Finally, despite the high specificity of OCT-defined healed plaques to detect histologically defined healed plaques, sensitivity was slightly lower. Moreover, in the presence of lipid plaques, OCT showed lower concordance with histology.
Conclusions
In patients with AMI caused by plaque erosion, healed phenotype was identified in 47.9% of patients at the culprit site. Healed phenotype within eroded culprit plaque was associated with signs of pancoronary vulnerability and advanced atherosclerosis.
Acknowledgments
The authors sincerely thank all colleagues and patients who participated in this study.
Sources of Funding
This work was supported by the National Natural Science Foundation of China (grant No. 81801861 to J.D. and No. 81827806 to B.Y.), and Natural Science Foundation of Heilongjiang Province (YQ2020H017 to J.D.).
Disclosures
All authors declared that they had nothing to disclose regarding conflict of interest with respect to this manuscript. B.Y. is an International Associate Editor of
Circulation Journal.
Author Contributions
Yanwei Yin, Chao Fang, Senqing Jiang: the design of research, data acquisition analysis, and manuscript drafting; Jifei Wang, Yidan Wang, Junchen Guo, Fangmeng Lei, Sibo Sun, Xueying Pei, Ruyi Jia, Caiying Tang, Yini Wang: data acquisition; Lulu Li: statistical analysis; Huai Yu: patient enrollment and performing cardiac intervention; Jiannan Dai, Bo Yu: substantial contribution to the design of the research and critical manuscript revision.
IRB Information
The original study was approved by the Ethics Committee of the Second Affiliated Hospital of Harbin Medical University (Reference number: No. KY2017-249).
Data Availability
The deidentified participant data will not be shared.
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circj.CJ-21-0635
References
- 1.
Jia H, Abtahian F, Aguirre AD, Lee S, Chia S, Lowe H, et al. In vivo diagnosis of plaque erosion and calcified nodule in patients with acute coronary syndrome by intravascular optical coherence tomography. J Am Coll Cardiol 2013; 62: 1748–1758.
- 2.
Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: The pathologists’ view. Eur Heart J 2013; 34: 719–728.
- 3.
Vergallo R, Crea F. Atherosclerotic plaque healing. N Engl J Med 2020; 383: 846–857.
- 4.
Yamamoto MH, Yamashita K, Matsumura M, Fujino A, Ishida M, Ebara S, et al. Serial 3-vessel optical coherence tomography and intravascular ultrasound analysis of changing morphologies associated with lesion progression in patients with stable angina pectoris. Circ Cardiovasc Imaging 2017; 10: e006347.
- 5.
Shimokado A, Matsuo Y, Kubo T, Nishiguchi T, Taruya A, Teraguchi I, et al. In vivo optical coherence tomography imaging and histopathology of healed coronary plaques. Atherosclerosis 2018; 275: 35–42.
- 6.
Higuma T, Soeda T, Abe N, Yamada M, Yokoyama H, Shibutani S, et al. A combined optical coherence tomography and intravascular ultrasound study on plaque rupture, plaque erosion, and calcified nodule in patients with ST-segment elevation myocardial infarction: Incidence, morphologic characteristics, and outcomes after percutaneous coronary intervention. JACC Cardiovasc Interv 2015; 8: 1166–1176.
- 7.
Fracassi F, Crea F, Sugiyama T, Yamamoto E, Uemura S, Vergallo R, et al. Healed culprit plaques in patients with acute coronary syndromes. J Am Coll Cardiol 2019; 73: 2253–2263.
- 8.
O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 61: e78–e140.
- 9.
Amsterdam EA, Wenger NK, Brindis RG, Casey DE Jr, Ganiats TG, Holmes DR Jr, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2014; 64: e139–e228.
- 10.
Garrone P, Biondi-Zoccai G, Salvetti I, Sina N, Sheiban I, Stella PR, et al. Quantitative coronary angiography in the current era: Principles and applications. J Interv Cardiol 2009; 22: 527–536.
- 11.
Tearney GJ, Regar E, Akasaka T, Adriaenssens T, Barlis P, Bezerra HG, et al. Consensus standards for acquisition, measurement, and reporting of intravascular optical coherence tomography studies: A report from the International Working Group for Intravascular Optical Coherence Tomography Standardization and Validation. J Am Coll Cardiol 2012; 59: 1058–1072.
- 12.
Sugiyama T, Yamamoto E, Bryniarski K, Xing L, Lee H, Isobe M, et al. Nonculprit plaque characteristics in patients with acute coronary syndrome caused by plaque erosion vs plaque rupture: A 3-vessel optical coherence tomography study. JAMA Cardiol 2018; 3: 207–214.
- 13.
Cao M, Zhao L, Ren X, Wu T, Yang G, Du Z, et al. Pancoronary plaque characteristics in STEMI caused by culprit plaque erosion versus rupture: 3-vessel OCT study. JACC Cardiovasc Imaging 2021; 14: 1235–1245.
- 14.
Mann J, Davies MJ. Mechanisms of progression in native coronary artery disease: Role of healed plaque disruption. Heart 1999; 82: 265–268.
- 15.
Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, et al. Healed plaque ruptures and sudden coronary death: Evidence that subclinical rupture has a role in plaque progression. Circulation 2001; 103: 934–940.
- 16.
Dai J, Fang C, Zhang S, Li L, Wang Y, Xing L, et al. Frequency, predictors, distribution, and morphological characteristics of layered culprit and nonculprit plaques of patients with acute myocardial infarction: In vivo 3-vessel optical coherence tomography. Circ Cardiovasc Interv 2020; 13: e009125.
- 17.
Owens AP, Byrnes JR, Mackman N. Hyperlipidemia, tissue factor, coagulation, and simvastatin. Trends Cardiovasc Med 2014; 24: 95–98.
- 18.
Russo M, Fracassi F, Kurihara O, Kim HO, Thondapu V, Araki M, et al. Healed plaques in patients with stable angina pectoris. Arterioscler Thromb Vasc Biol 2020; 40: 1587–1597.
- 19.
Otsuka F, Joner M, Prati F, Virmani R, Narula J. Clinical classification of plaque morphology in coronary disease. Nat Rev Cardiol 2014; 11: 379–389.
- 20.
Wakefield TW, Linn MJ, Henke PK, Kadell AM, Wilke CA, Wrobleski SK, et al. Neovascularization during venous thrombosis organization: A preliminary study. J Vasc Surg 1999; 30: 885–892.
- 21.
Fujiyoshi K, Minami Y, Ishida K, Kato A, Katsura A, Muramatsu Y, et al. Incidence, factors, and clinical significance of cholesterol crystals in coronary plaque: An optical coherence tomography study. Atherosclerosis 2019; 283: 79–84.
- 22.
Kellner-Weibel G, Jerome WG, Small DM, Warner GJ, Stoltenborg JK, Kearney MA, et al. Effects of intracellular free cholesterol accumulation on macrophage viability. Arterioscler Thromb Vasc Biol 1998; 18: 423–431.
- 23.
Sugane H, Kataoka Y, Otsuka F, Yasuda S. Cholesterol-crystalized coronary atheroma as a potential precursor lesion causing acute coronary syndrome: A case report. Eur Heart J Case Rep 2019; 3: ytz128.
- 24.
Burke AP, Weber DK, Kolodgie FD, Farb A, Taylor AJ, Virmani R. Pathophysiology of calcium deposition in coronary arteries. Herz 2001; 26: 239–244.
- 25.
Russo M, Kim HO, Kurihara O, Araki M, Shinohara H, Thondapu V, et al. Characteristics of non-culprit plaques in acute coronary syndrome patients with layered culprit plaque. Eur Heart J Cardiovasc Imaging 2020; 21: 1421–1430.
- 26.
Vergallo R, Uemura S, Soeda T, Minami Y, Cho JM, Ong DS, et al. Prevalence and predictors of multiple coronary plaque ruptures. Arterioscler Thromb Vasc Biol 2016; 36: 2229–2238.
- 27.
Kato K, Yonetsu T, Kim SJ, Xing L, Lee H, McNulty I, et al. Nonculprit plaques in patients with acute coronary syndromes have more vulnerable features compared with those with non–acute coronary syndromes. Circ Cardiovasc Imaging 2012; 5: 433–440.

