Abstract
Background: Vericiguat, an oral soluble guanylate cyclase stimulator, is a novel therapeutic agent for patients with heart failure with reduced ejection fraction; however, the detailed cardioprotective mechanism remains unclear. We aimed to explore the mechanism of the effect of vericiguat on the myocardium, particularly focusing on oxidative stress, using in vivo and in vitro experiments.
Methods and Results: Male 8-week-old mice were divided into a control group, angiotensin II (AngII) infusion group, and AngII infusion with low- or high-dose vericiguat treatment group. After 14 days of treatment, vericiguat did not affect the systolic or diastolic blood pressure increase caused by AngII infusion. AngII-induced cardiac hypertrophy and fibrosis in the left ventricle (LV) were significantly ameliorated by high-dose vericiguat treatment. AngII-induced O2−
overproduction and upregulation of messenger RNA levels of Nppa, Nppb, Myh7, Col1a1, Col3A1, and Tgfb1 in the LV were significantly attenuated by vericiguat in a dose-dependent manner. Incubation of neonatal rat cardiomyocytes using vericiguat and AngII revealed that preceding incubation with vericiguat directly reduced AngII-induced cardiomyocyte O2−
production and cardiac hypertrophy-associated gene expression. In addition, AngII-induced phosphorylation of ERK 1/2 or p38 MAPK was significantly attenuated by the incubation with vericiguat.
Conclusions: Our study demonstrated that vericiguat suppresses myocardial oxidative stress via the regulation of ERK 1/2 or p38 MAPK signaling, leading to antihypertrophic/fibrotic effects.
Heart failure (HF) is a major healthcare burden and a life-threatening syndrome characterized by morbidity/mortality and poor functional capacity and quality of life.1 Two of the most critical indicators of HF are overactivity of the β-adrenergic and renin-angiotensin systems2 and cardiac remodeling.3,4 The combination of angiotensin receptor/neprilysin inhibitor, β-blocker, mineralocorticoid receptor antagonist, and sodium-glucose cotransporter 2 inhibitor is a primary intervention for the pathological systems of HF and is termed the “fantastic four” because of the great contribution to improving the poor prognosis of patients with HF with reduced ejection fraction (HFrEF).5
Nevertheless, there are high-risk patients who have had worsening of HF despite treatment with the fantastic four or guideline-based medical therapy. Nitric oxide-soluble guanylate cyclase-cyclic guanosine monophosphate (NO-sGC-cGMP) signaling has been reported to exert cardiovascular protective effects through suppression of reactive oxygen species (ROS)-induced oxidative stress.6,7 However, the signaling is downregulated in patients with HF.8,9 Vericiguat, an oral sGC stimulator, is a novel therapeutic agent for high-risk patients with HFrEF.10 Itt has the potential to enhance NO-sGC-cGMP signaling by stimulating sGC directly with subsequent cardiovascular protective effects.
However, the specific cardioprotective effects of vericiguat remain to be elucidated, so in this study we investigated the novel cardioprotective effect of vericiguat, particularly focusing on oxidative stress and cardiac remodeling using in vivo and in vitro experimental models.
Methods
All experimental procedures were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of the Physiological Society of Oita University, Japan, which follow the guidelines established by the US National Institutes of Health. All animal experiments in this study were approved by Oita University’s ethics review board (243202A and 243201B).
Animals and Treatments
Male C57Bl/6J mice aged 8 weeks (Jackson Laboratory Japan, Yokohama, Japan) were held at Oita University under a 12-h light/dark cycle. Each mouse was randomly assigned to a control (n=6), angiotensin II (Ang II; n=8), Ang II+low-dose vericiguat (n=6), or Ang II+high-dose vericiguat (n=8) group. The study protocol is shown in Figure 1A. An osmotic mini-pump (Alzet 1002, Alzet, Cupertino, CA, USA) was implanted subcutaneously for constant infusion of Ang II (2.0 mg/kg/day; R&D Systems, Minneapolis, MN, USA) for 2 weeks as described previously.11 Mice in the Ang II+vericiguat group received an oral gavage of low- or high-dose vericiguat as previously described12 (1.5 mg/kg/day or 3 mg/kg/day; HY-16774, MCE, Monmouth Junction, NJ, USA) for 3 days prior to infusion of Ang II, and this continued until the end of the Ang II infusion. The mice were monitored daily, and body weight (BW) and tibia length (TL) were measured on days 0 (or pretreatment of vericiguat), 7, and 14. Systolic blood pressure (sBP), diastolic blood pressure (dBP), and heart rate (HR) were also measured at the same time of measuring BW using the tail-cuff method.

Echocardiography
Transthoracic echocardiography (Hitachi Aloka Medical, Tokyo, Japan) was performed on day 14 under anesthesia, with the body temperature fixed at 37.0℃. The interventricular septal thickness at end-diastole (IVSTd), LV end-diastolic posterior wall thickness (LVPWT), LV end-diastolic diameter (LVDd), LV endsystolic diameter (LVDs), and LV ejection fraction (LVEF) were determined using ImageJ software (version 1.53a, National Institutes of Health, Bethesda, MD, USA).
Histology
Isolated LV tissue samples were immersed in 4% paraformaldehyde, embedded in paraffin, and cut into 5-μm sections. After deparaffinization, sections were stained with hematoxylin-eosin (HE), Alexa Fluor 594-conjugated wheat germ agglutinin (WGA; Life Technologies Corp., Carlsbad, CA, USA) or Masson’s trichrome. Images were acquired and digitized on a BZ-9000 Biolevo epifluorescence microscope (Keyence, Osaka, Japan), and analyzed at ×400 magnification using the associated software (Keyence). The percentage fibrosis was determined by calculating the ratio of fibrotic areas to normal myocardial tissue. To obtain the mean values, 5 images per LV tissue sample from each mouse were analyzed.
Isolation and Culture of Neonatal Rat Myocytes
Cardiomyocytes were isolated from the hearts of 1–3-day-old neonatal Sprague–Dawley rats (Jackson Laboratory) and cultured as described previously.13 Briefly, after death, the hearts were excised and cut into pieces in phosphate-buffered saline. After digestion with trypsin (2 mg/mL), the cardiomyocytes were isolated and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco/Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 mg/mL), vitamin B12, and bromodeoxyuridine (0.1 mmol/L). Cardiomyocytes were attached to the cell culture dishes, started beating after 1–2 days, and were used for the in vitro incubation experiments.
Incubation of Rat Cardiomyocytes With Vericiguat
The rat cardiomyocytes were treated with vericiguat (MCE) for 48 h in DMEM supplemented with 5% FBS, penicillin (100 U/mL), streptomycin (100 mg/mL), vitamin B12, and bromodeoxyuridine (0.1 mmol/L). Next, we changed the cell culture medium to DMEM without FBS and added vericiguat again. The cells were also treated with and without 100 μM ERK or p38 MAPK inhibitor (Cayman Chemical, Michigan, USA). After 24-h incubation, the cardiomyocytes were exposed to 1.0 μM Ang II for 1 h. The dosage of vericiguat was set to 0.1 μM and 1.0 μM using human blood concentration data as mentioned in a phase 1 clinical trial.14
Ribonucleic acid (RNA) Isolation and Quantitative Real-Time Polymerase Chain Reaction (qPCR)
Total RNA was isolated using a spin column-based RNA purification method with the RNeasy Fibrous Tissue Mini Kit or RNeasy Mini Kit (QIAGEN, Hilden, Germany). The RNA concentration and integrity were assessed spectrophotometrically using the NanoDrop 2000 spectrophotometer. RNA was reverse transcribed to complementary deoxyribonucleic acid using SuperScript VILO Master Mix (Thermo Fisher Scientific) with the ezDNaseTM Enzyme Kit (Invitrogen/Thermo Fisher Scientific) according to the manufacturer’s protocols. qPCR was performed with TaqMan chemistry using the standard universal TaqMan protocol, as indicated by the manufacturer, with the LightCycler 96 (Roche Diagnostics, Basel, Switzerland). Glyceraldehyde-3-phosphate dehydrogenase was used as the housekeeping gene. The IDs of the TaqMan probes (Thermo Fisher Scientific) are provided in Supplementary Table 1.
Oxidative Stress Quantification
Following the in vivo and in vitro experiments, tissues or cells were scraped and sonicated in ice-cold Krebs-HEPES buffer (pH 7.35) in the presence of a protease inhibitor (Roche Applied Science, Penzberg, Germany). The O2−
production was measured in cell lysates using lucigenin (5 μmol/L)-enhanced chemiluminescence, as previously described.15 The contribution of nicotinamide adenine dinucleotide phosphate (NADPH; 100 μmol/L) to cardiomyocyte O2−
production was quantified, and the chemiluminescence levels of the cardiomyocytes were normalized to the total protein concentration. The production of total ROS and mitochondria-derived O2−
were evaluated using CellROX Green Reagent (Invitrogen/Thermo Fisher Scientific) and MitoSOX Red Superoxide superoxide Indicator indicator (Invitrogen/Thermo Fisher Scientific), respectively, according to the manufacturer’s protocol. The stained specimens were observed under a fluorescence microscope (BZ-9000; KEYENCE) and the fluorescence intensity was measured using ImageJ software.
Western Blotting
Western blotting was performed to determine the expression of extracellular signal-regulated kinase (ERK) 1/2, p-ERK 1/2, p38 mitogen-activated protein kinase (MAPK), and p-p38 MAPK as previously described.13 The catalog numbers of the antibodies (Cell Signaling Technology, Boston, MA, USA) are provided in Supplementary Table 2. Specifically, proteins extracted from mouse LV tissues or rat cardiomyocytes were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred electrophoretically to polyvinylidene fluoride membranes (Merck Millipore, Darmstadt, Germany). After blockade, the membranes were incubated with primary antibodies, and after washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Little Chalfont, UK). Protein bands were visualized using ECLTM Prime (GE Healthcare), and arbitrary optical densities were measured using ImageJ software.
Statistical Analysis
The Shapiro-Wilk test was used to evaluate the normality of continuous variables. Data are presented as mean and standard deviation. Continuous variables were evaluated using analysis of variance, followed by Dunnett’s test correction for multiple comparisons. Statistical significance was set at P<0.05, and all computations were performed using EZR16 for R statistical software (version 25.0; Cary, NC, USA), GraphPad Prism (version 9.5.1; Boston, MA, USA) or JMP (version 13.2.0; SAS, Cary, NC, USA)17 running on Windows 11 (Microsoft, Redmond, WA, USA).
Results
Physiological and Echocardiographic Findings
No significant differences in physiological characteristics were observed in the control, Ang II, and Ang II+vericiguat groups before Ang II or vericiguat treatment (Supplementary Table 3). As shown in Figure 1B,C, the sBP and dBP in the Ang II group were significantly elevated compared with the control group (P<0.001); however, no significant changes were observed, compared with the Ang II+low dose vericiguat group (day 7, P=0.53 and 0.80, respectively; day 14, P=0.98 and 0.06, respectively) and the Ang II+high dose vericiguat group (day 7, P=0.77 and 0.91, respectively; day 14, P=0.72 and 0.35, respectively). HR did not differ among the 3 groups (Figure 1D). The control group had gained more BW than the Ang II group (P=0.002) on day 14, but the Ang II+low- or high-dose vericiguat groups did not (P=0.90 and P=0.71, respectively) (Figure 1E). Representative echocardiographic images of each group are shown in Figure 2A, and the echocardiographic data are summarized in Figure 2B, showing that Ang II caused concentric hypertrophy characterized by increased IVSTd (P=0.001) and LVPWT (P<0.001), and decreased LVDd (P<0.001) and LVDs (P<0.001). High-dose vericiguat treatment suppressed the septal wall thickening (P=0.003) induced by Ang II, but did not affect other parameters obtained by echocardiography. LVEF was not altered by Ang II or vericiguat treatment.

Macroscopic and Microscopic Findings of Cardiac Hypertrophy and Fibrosis
Figure 3A shows representactive images of the macroscopic and microscopic findings (HE, WGA, and Masson’s trichrome staining) in the mouse hearts. We measured the heart weight (HW) and calculated the HW/BW ratio to compensate for body size. As shown in Figure 3B, the HW/BW ratio and HW/TL ratio increased with Ang II infusion (P<0.001). Interestingly, the Ang II-induced increase in HW/BW ratio and HW/TL ratio was significantly suppressed by high-dose vericiguat treatment (P=0.028 and P=0.019, respectively). Analysis of the cardiomyocyte cross-sectional area using WGA staining indicated that Ang II induced the hypertrophy of cardiomyocytes (P<0.001), whereas vericiguat suppressed the hypertrophic changes (P<0.001, Figure 3C). We also evaluated the cardiac fibrotic area using Masson’s trichrome staining, which showed that the fibrotic area was significantly increased by Ang II (P<0.001) and suppressed by vericiguat (P<0.001, Figure 3D).

Expression of mRNAs Related to Hypertrophy and Fibrosis in Mouse LV
Quantitative analysis of the mRNA levels associated with cardiac hypertrophy and fibrosis in the mouse LV are presented in Figure 4. Cardiac hypertrophy-associated mRNA expression of natriuretic peptide type A (Nppa), natriuretic peptide type B (Nppb), and myosin heavy chain 7 (Myh7) were increased by Ang II infusion (P=0.002, P=0.035, and P=0.012, respectively) and suppressed by high-dose vericiguat (P=0.005, P<0.001, and P=0.035, respectively) as shown in Figure 4A. Low-dose vericiguat attenuated Ang II-induced Nppa overexpression (P=0.013). Similarly for the hypertrophy-related genes, Ang II-induced overexpression of cardiac fibrosis-related genes, collagen type I alpha 1 chain (Col1a1; P=0.009), collagen type III alpha 1 chain (Col3a1; P=0.004), and transforming growth factor β1 (Tgfb1; P=0.006), all of which were attenuated by high-dose vericiguat (P=0.032, P<0.001, and P<0.001, respectively, Figure 4B). Low-dose vericiguat attenuated Ang II-induced Col3a1 overexpression (P=0.001).

Quantification of O2−
Overproduction Induced by Ang II in the Mouse LV
To explore vericiguat’s mechanism of regulation of cardiac hypertrophy and fibrosis, we evaluated the O2−
production using luminometry in mouse LV tissue samples. As shown in Figure 4C, basal O2−
production was significantly exacerbated by Ang II (P<0.001) and suppressed by both low- and high-dose vericiguat treatment (P=0.006 and P<0.001, respectively). Furthermore, Ang II-induced NADPH-stimulated O2−
overproduction (P<0.001) was significantly attenuated by both low- and high-dose vericiguat treatment (P=0.002 and P<0.001, respectively, Figure 4D).
Evaluation of Oxidative Stress in Rat Cardiomyocytes
We investigated the direct effects of vericiguat on rat cardiomyocytes. Incubation of neonatal rat cardiomyocytes with vericiguat (0.1 μM and 1.0 μM) for 72 h and subsequent 1 h exposure to Ang II (1.0 μM) revealed that basal (Figure 5A) and NADPH-stimulated (Figure 5B) O2−
production increased with Ang II exposure (P<0.01) and was suppressed by preceding incubation with vericiguat (P<0.05). We also evaluated the total ROS and mitochondria-derived O2−
production in rat cardiomyocytes using live-cell oxidative fluorescence microtopography. Representative images of total ROS production evaluated with CellROX and mitochondria-derived O2−
production evaluated with MitoSOX are shown in Figure 5C. Ang II exposure significantly increased the fluorescent intensity of CellROX (P<0.001, Figure 5D) and MitoSOX (P<0.001, Figure 5E); however, vericiguat treatment significantly decreased the fluorescent intensity in a dose-dependent manner (P<0.05).

Oxidative Stress-Associated Signaling Cascade and Hypertrophic Gene Expression in Rat Cardiomyocytes
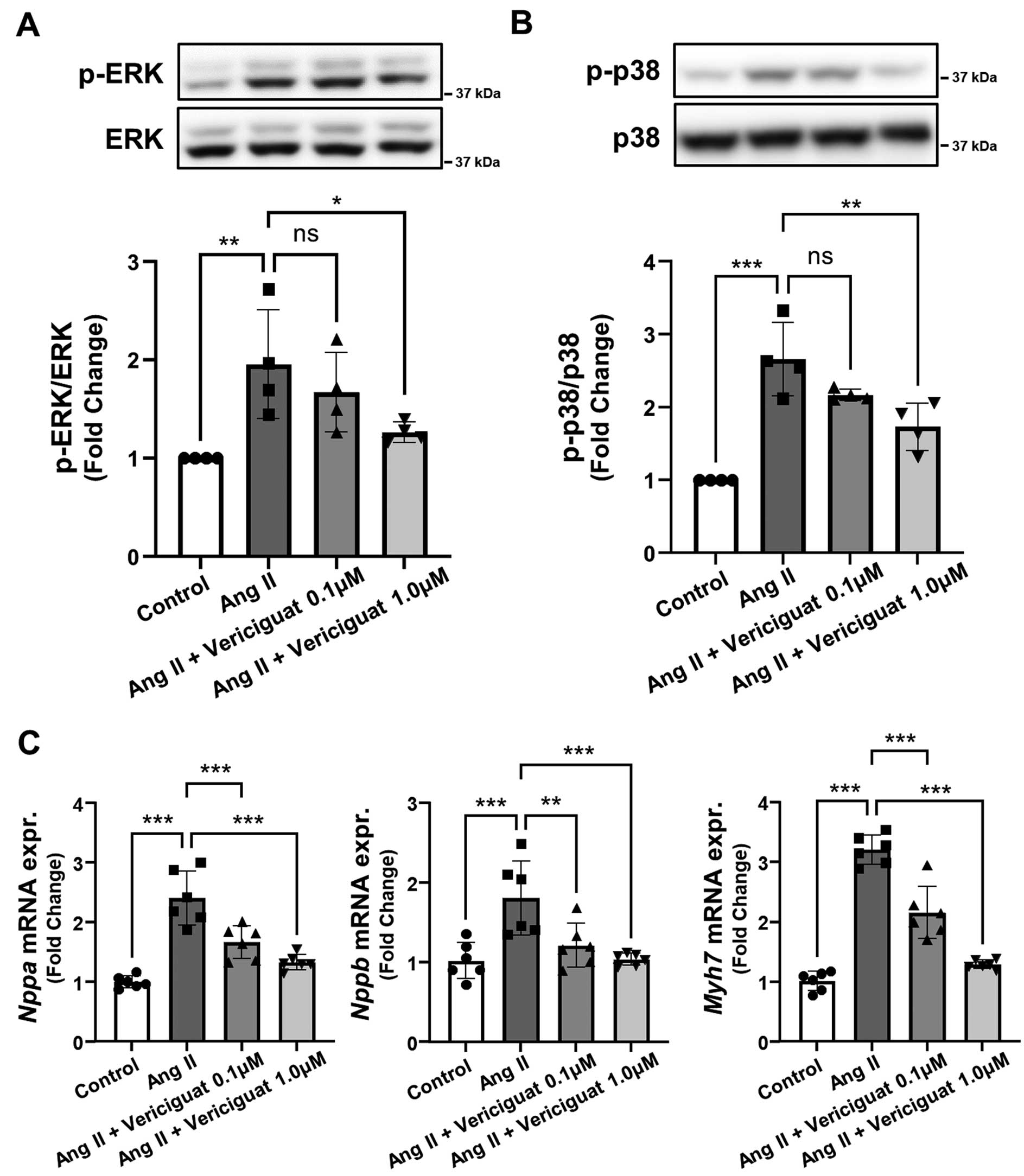
Finally, to investigate the detailed mechanism of the effect of vericiguat on rat cardiomyocytes, we evaluated the phosphorylation of ERK 1/2 and p38 MAPK, which are well-known signaling cascades induced by oxidative stress, followed by hypertrophy-associated gene upregulation. The phosphorylation of ERK 1/2 (Figure 6A) and p38 MAPK (Figure 6B) was enhanced by Ang II exposure (P<0.01), but attenuated by vericiguat treatment (P<0.05). Cardiac hypertrophy-associated gene (Nppa, Nppb, and Myh7) mRNA expression was increased by Ang II, but significantly suppressed by vericiguat in a dose-dependent manner (Figure 6C). We performed an additional in vitro experiment using ERK or p38MAPK inhibitor. The addition of ERK or p38MAPK inhibitor to vericiguat did not reduce the basal and NADPH-stimulated O2−
compared with vericiguat alone (Supplementary Figure A,B), suggesting that vericiguat might improve redox states in cardiomyocytes mainly through inhibition of ERK or p38MAPK signaling. However, we cannot exclude the possibility that the effect of vericiguat on the reduction of O2−
was potent and thus additional effects of ERK or p38MAPK inhibitor were less apparent.
Discussion
The 2 major findings of this study were as follows. (1) Despite the fact that vericiguat did not affect Ang II-induced hypertension in mice, it didt ameliorate Ang II-induced LV hypertrophy, fibrosis, and O2−
overproduction in vivo. (2) Vericiguat directly suppressed Ang II-induced oxidative stress, phosphorylation of ERK 1/2 or p38 MAPK, and upregulation of cardiac hypertrophy-related gene expression levels in a dose-dependent manner in vitro.
In the VICTORIA trial, the composite of cardiovascular morbidity/mortality was significantly lower in the vericiguat arm without affecting blood pressure.10 Consistent with this result, vericiguat did not affect hemodynamic parameters in our study. Taken together, these findings suggest that the cardioprotective effect of vericiguat is not due to an afterload reduction effect of decreasing blood pressure. Vericiguat can directly improve the myocardial redox state, resulting in HF substrate suppression. However, the limitation of this study is that the effects of vericiguat on the vascular endothelium have not been investigated. The NO-sGC-cGMP pathway and subsequent activation of protein kinase G (PKG) in the vascular endothelium exert cardiovascular protective effects. Highly controlled preload regulation due to improved vascular endothelial function by vericiguat, particularly in veins, may potentially contribute to the inhibition of HF substrate progression.
Ang II has been reported to play critical roles in cardiac remodeling, hypertrophy, and fibrosis.2,18,19 Moreover, Ang II induces the overproduction of ROS.18,20 Ang II binds to the angiotensin II type I receptor (AT1R) and activates Gαq proteins.21 AT1R-Gαq signaling activates NADPH oxidase 2 (NOX2) and NOX4, which mediates p47phox- or mitochondria-induced cardiac ROS production.18 On the other hand, activation of the NO-sGC-cGMP-PKG signaling directly modulates p38MAPK or de-activates various signaling pathways via regulator of G protein signaling (RGS2/4), which could inhibit Gαq signaling.22 Based on these previous studies and our findings, vericiguat-induced activation of NO-sGC-cGMP-PKG signaling inhibits ERK or p38MAPK signaling via RGS2/4, leading to inhibition of Gαq signaling, which results in suppression of Ang II-induced ROS overproduction.
In patients with advanced HF, excess oxidative stress disrupts NO-sGC-cGMP signaling and exacerbates HF.9 Notably, vericiguat inhibited the overproduction of myocardial oxidative stress, which was a novel finding of this study. The findings may help us understand the effect of vericiguat in reducing cardiovascular events in patients with HF.
Study Limitation
In this study, we focused on the effect of vericiguat on the redox state of cardiomyocyte. Several pathways other than ROS might contribute to the improvement of cardiac remodeling with vericiguat.
Conclusions
Our study demonstrated that vericiguat plays an important role in attenuating Ang II-induced cardiac remodeling and oxidative stress.
Acknowledgments
We thank Masae Nishibayashi, Tomomi Shuto, Ikuko Morisaki, and Shoi Shiba for their assistance with the manuscript.
Disclosures
N.T. is a member of Circulation Journal’s Editorial Team. The authors declare they have no conflicts of interest.
Funding / IRB Information
None.
Supplementary Files
Please find supplementary file(s);
https://doi.org/10.1253/circj.CJ-24-0659
References
- 1.
Savarese G, Becher PM, Lund LH, Seferovic P, Rosano GMC, Coats AJS. Global burden of heart failure: A comprehensive and updated review of epidemiology. Cardiovasc Res 2023; 118: 3272–3287.
- 2.
Opie LH, Sack MN. Enhanced angiotensin II activity in heart failure: Reevaluation of the counterregulatory hypothesis of receptor subtypes. Circ Res 2001; 88: 654–658.
- 3.
Azevedo PS, Polegato BF, Minicucci MF, Paiva SA, Zornoff LA. Cardiac remodeling: Concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol 2016; 106: 62–69.
- 4.
Takano H, Hasegawa H, Nagai T, Komuro I. Implication of cardiac remodeling in heart failure: Mechanisms and therapeutic strategies. Intern Med 2003; 42: 465–469.
- 5.
Bauersachs J. Heart failure drug treatment: The fantastic four. Eur Heart J 2021; 42: 681–683.
- 6.
Butler J, Usman MS, Anstrom KJ, Blaustein RO, Bonaca MP, Ezekowitz JA, et al. Soluble guanylate cyclase stimulators in patients with heart failure with reduced ejection fraction across the risk spectrum. Eur J Heart Fail 2022; 24: 2029–2036.
- 7.
Masuyama H, Tsuruda T, Sekita Y, Hatakeyama K, Imamura T, Kato J, et al. Pressure-independent effects of pharmacological stimulation of soluble guanylate cyclase on fibrosis in pressure-overloaded rat heart. Hypertens Res 2009; 32: 597–603.
- 8.
Emdin M, Aimo A, Castiglione V, Vergaro G, Georgiopoulos G, Saccaro LF, et al. Targeting cyclic guanosine monophosphate to treat heart failure: JACC Review Topic of the Week. J Am Coll Cardiol 2020; 76: 1795–1807.
- 9.
Breitenstein S, Roessig L, Sandner P, Lewis KS. Novel sGC stimulators and sGC activators for the treatment of heart failure. Handb Exp Pharmacol 2017; 243: 225–247.
- 10.
Armstrong PW, Pieske B, Anstrom KJ, Ezekowitz J, Hernandez AF, Butler J, et al. Vericiguat in patients with heart failure and reduced ejection fraction. N Engl J Med 2020; 382: 1883–1893.
- 11.
Fukui A, Takahashi N, Nakada C, Masaki T, Kume O, Shinohara T, et al. Role of leptin signaling in the pathogenesis of angiotensin II-mediated atrial fibrosis and fibrillation. Circ Arrhythm Electrophysiol 2013; 6: 402–409.
- 12.
Follmann M, Ackerstaff J, Redlich G, Wunder F, Lang D, Kern A, et al. Discovery of the soluble guanylate cyclase stimulator vericiguat (BAY 1021189) for the treatment of chronic heart failure. J Med Chem 2017; 60: 5146–5161.
- 13.
Kondo H, Kira S, Oniki T, Gotoh K, Fukui A, Abe I, et al. Interleukin-10 treatment attenuates sinus node dysfunction caused by streptozotocin-induced hyperglycaemia in mice. Cardiovasc Res 2019; 115: 57–70.
- 14.
Boettcher M, Thomas D, Mueck W, Loewen S, Arens E, Yoshikawa K, et al. Safety, pharmacodynamic, and pharmacokinetic characterization of vericiguat: Results from six phase I studies in healthy subjects. Eur J Clin Pharmacol 2021; 77: 527–537.
- 15.
Kondo H, Akoumianakis I, Badi I, Akawi N, Kotanidis CP, Polkinghorne M, et al. Effects of canagliflozin on human myocardial redox signalling: Clinical implications. Eur Heart J 2021; 42: 4947–4960.
- 16.
Yamasaki H, Kondo H, Shiroo T, Iwata N, Masuda T, Makita T, et al. Efficacy of computed tomography-based evaluation of myocardial extracellular volume combined with red flags for early screening of concealed cardiac amyloidosis in patients with atrial fibrillation. Circ J 2024; 88: 1167–1175.
- 17.
Yufu K, Shimomura T, Kawano K, Sato H, Yonezu K, Saito S, et al. Usefulness of prehospital 12-lead electrocardiography system in ST-segment elevation myocardial infarction patients in Oita: Comparison between urban and rural areas, weekday daytime and weekday nighttime/holidays. Circ J 2024; 88: 1293–1301.
- 18.
Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 2011; 108: 837–846.
- 19.
Schnee JM, Hsueh WA. Angiotensin II, adhesion, and cardiac fibrosis. Cardiovasc Res 2000; 46: 264–268.
- 20.
Zablocki D, Sadoshima J. Angiotensin II and oxidative stress in the failing heart. Antioxid Redox Signal 2013; 19: 1095–1109.
- 21.
Ikeda Y, Kumagai H, Motozawa Y, Suzuki J, Komuro I. Biased agonism of the angiotensin II type I receptor. Int Heart J 2015; 56: 485–488.
- 22.
Numata G, Takimoto E. Cyclic GMP and PKG signaling in heart failure. Front Pharmacol 2022; 13: 792798.