Abstract
Variant types and sites in a single gene could influence the age of onset, severity, and pattern of affected organs of the genetic disease, such as in Marfan syndrome (MFS)-causing
FBN1, and understanding the genotype-phenotype relationship could aid in determining the treatment strategy. In contrast, completely distinct system and/or organ diseases induced by 1 gene mutation have been rarely reported. Transforming growth factor-β (TGF-β) type I receptor-encoding
TGFBR1
is such a gene, causing Loeys-Dietz syndrome (LDS) closely related to MFS, and also multiple self-healing squamous epithelioma (MSSE) without clinical overlap. The detailed mechanisms underlying this effect, however, remain elusive. We recently reported the significance of 2 distinct intronic variants (c.973+1G>A and c.806-2A>C) of
TGFBR1, which were both predicted to mediate in-frame exon 5 skipping but caused LDS and MSSE, respectively. On ex vivo minigene splicing assay analysis we demonstrated that 2 different cryptic splice sites were activated, and in-frame and out-of-frame transcripts were produced in LDS and MSSE, respectively, supporting the previously proposed but not yet approved mechanism that loss-of-function and haploinsufficiency-causing variants in serine/threonine kinase domains induce LDS and MSSE, respectively. In this review, we briefly summarize the recent findings and unresolved problems for the pathogenesis of LDS, including the TGF-β signaling paradox: most variants have been verified or predicted to be loss of function in vitro, but these variants enhanced TGF-β signaling in vivo.
Loeys-Dietz syndrome (LDS) is an autosomal dominant heritable disorder of the connective tissue closely related to Marfan syndrome (MFS), which is characterized by a triad of arterial tortuosity and aneurysm, widely spaced eyes (hypertelorism), and bifid uvula. Patients with LDS are more likely to exhibit rapidly progressive aortopathy with a tendency to rupture and dissection in the aorta/arteries at a young age and at smaller dimensions compared with MFS.1–3
LDS is caused by a pathogenic variant in transforming growth factor-β (TGF-β) signaling-related genes and classified according to the pathogenic genes:
TGFBR1
(LDS1),
TGFBR2
(LDS2),
SMAD3
(LDS3),
TGFB2
(LDS4),
TGFB3
(LDS5), and
SMAD2
(LDS6). A large proportion of LDS patients have pathogenic variants in the genes encoding TGF-β types I and II receptors,
TGFBR1
(20–25%) and
TGFBR2
(55–60%), especially in the serine/threonine kinase (STK)-encoding regions.4,5
Most variants have been verified or predicted to cause STK loss of function in vitro, but these variants enhanced TGF-β signaling in the in vivo aortic regions, referred to as the TGF-β signaling paradox.
In contrast, pathogenic variants in
TGFBR1
also induce a distinct skin disease called multiple self-healing squamous epithelioma (MSSE).6
MSSE is an autosomal dominant skin cancer syndrome characterized by the development of multiple rapidly growing invasive skin tumors that emerge for a few weeks only to subsequently spontaneously regress and heal with scarring, and there is no clinical overlap between LDS and MSSE.
We recently reported a familial Japanese case of LDS with a novel splice donor site variant in intron 5 in
TGFBR1
(c.973+1G>A).7
The in-frame deletion of the whole exon 5 was predicted to be elicited by this variant, whereas a variant of MSSE in a British family (c.806-2A>C) was also predicted to induce the same deletion of exon 5.6
To clarify this, we performed minigene-based splicing assays of both variants and found that these 2 different diseases were caused by differences in the splicing patterns.
In this mini review, we present and discuss the recent understanding of the molecular mechanism of LDS and the unresolved problems, including the TGF-β signaling paradox and the mechanism by which
TGFBR1
variants elicit 2 completely distinct diseases, LDS and MSSE.7
TGFBR1/TGFBR2 STK Domain and Cause of LDS1 and LDS2
TGFBR1 and TGFBR2 are transmembrane STK receptors consisting of 9 and 7 exons, respectively. The activation of TGFBR2 by TGF-β ligands induces TGFBR2 dimerization, and the TGFBR2 homodimer forms a stable receptor complex with TGFBR1 homodimer and phosphorylates TGFBR1, leading to the subsequent activation of the SMAD signaling pathway. Phosphorylated SMAD2 and SMAD3 form stable complexes with SMAD4, which translocate to the nucleus and regulate the transcription of target genes (Figure 1A).8
In LDS, most variants in
TGFBR1/TGFBR2
are missense and are located in or immediately flanking the evolutionarily conserved STK domain.9
Recently reported knock-in mice with missense mutations (Tgfbr1M318R/+
and
Tgfbr2G357 W/+) identically developed vascular, craniofacial, and skeletal manifestations of LDS, but heterozygous knockout mice (Tgfbr1+/−
and
Tgfbr2+/−) did not develop any LDS features.10
This suggests that full-length variant TGFBR1 and TGFBR2 proteins improperly regulate the downstream signal pathways and cause LDS1 and LDS2.
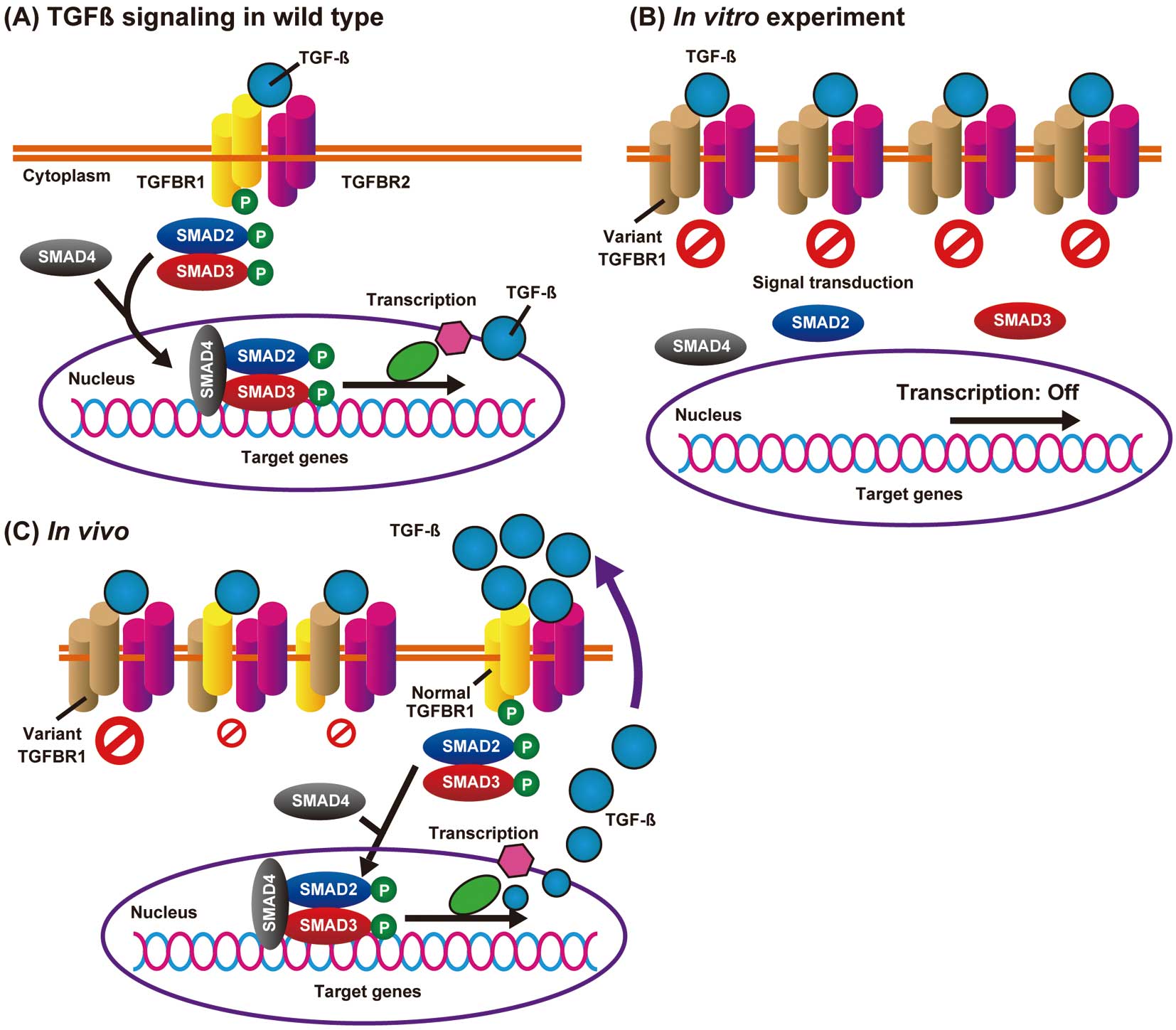
TGF-β Signaling Paradox in LDS
The mechanisms of how the missense variants cause LDS1 and LDS2 are still elusive. Although most variants have been verified or predicted to cause loss of function in the in vitro cultured cells (e.g., HEK293 cells;
Figure 1B),11,12
these variants enhanced TGF-β signaling in the aortic regions.10,13
The mechanism underlying the TGF-β signaling paradox remains elusive and is accepted as a scientific mystery.14
There has been speculation that increased TGF-β ligands in the LDS aortic wall activate intact TGFBR1/TGFBR2 complexes in vivo (Figure 1C), but the mechanisms of how TGF-β ligands are actively secreted remain to be determined.15
Very recently, MacFarlane et al reported a partial solution to this problem (Figure 2).16
The severely affected aortic root and ascending aorta are composed of 2 types of vascular smooth muscle cells (VSMCs): secondary heart field (SHF)- and cardiac neural crest (CNC)-derived VSMCs. In
Tgfbr1M318R/+
LDS mice these 2 types of VSMCs have distinct biological properties. SHF-derived VSMCs, but not CNC-derived VSMCs, showed impaired SMAD2/SMAD3 activation in response to TGF-β, increased expression of angiotensin II (AngII) type 1 receptor (Agtr1a), enhanced responsiveness to AngII, and higher expression of TGF-β ligands. In contrast, CNC-derived VSMCs had preserved TGF-β signaling potential; and CNC-specific, but not SHF-specific,
Smad2
deletion ameliorated aortic root aneurysm formation in
Tgfbr1M318R/+
mice. This suggests that TGF-β ligands secreted from SHF-derived VSMCs in an AngII type I receptor (AT1R)-dependent manner could activate CNC-derived VSMCs, thereby contributing to the in vivo TGF-β overactivity in LDS aortopathy. This might also explain the usefulness of an AT1R inhibitor, losartan, for preventing aortic root aneurysm formation in LDS (Figure 2).

TGFBR1
Truncating STK Domain Variants
As described previously, LDS1 and LDS2 are induced by loss-of-function missense variants in or near the STK domain, and full-length variant proteins seem to function improperly. Interestingly,
TGFBR1
has been reported to be a causative gene for MSSE,6
and most MSSE variants are reported to be located in the extracellular ligand-binding domain, and truncating variants (nonsense, frameshift) in the STK domain. We recently encountered, however, a Japanese familial case of LDS involving a novel splice donor site variant in intron 5 in
TGFBR1
(c.973+1G>A), and noted that the in silico-predicted effect of in-frame exon 5 skipping (168 bp) had the same effect as a splice acceptor site variant in intron 4 causing MSSE (c.806-2A>C;
Figure 3A).6,7
Exon 5 encodes part of the STK domain, and LDS and MSSE family members do not present with overlap features; thus, we performed ex vivo minigene splicing assays of 2 variants (Figure 3B,C) to elucidate the mechanism by which these 2 apparently similar variants produce different system diseases.7

On analysis of the resulting transcripts, the LDS variant was found to produce 2 types of in-frame products as a result of exon 5 skipping (r.806_973del, p.Asp269_Gln324del), and the activation of a cryptic donor splice at a site 9 bp upstream of the 5’ natural splice donor site (r.965_973del, p.Thr323_Gly325del;
Figure 3B). The results were verified on reverse transcription-polymerase chain reaction using RNA from blood samples, and these 2 LDS-inducing variant proteins exerted dominant-negative effects at least in vitro. In contrast, the MSSE variant activated a cryptic acceptor site at 76 bp downstream of the 3’ natural splice acceptor site, which produced an out-of-frame transcript (r.807_882del, p. Asn270Thrfs*8), and was expected to cause haploinsufficiency because of nonsense-mediated mRNA decay (Figure 3C). Our results support the previously proposed but not yet approved mechanism6
that loss-of-function and haploinsufficiency-causing variants in the STK domain induce LDS and MSSE, respectively.
Genetic Variance and Disease Phenotypes
Genetic tests for suspected genetic diseases are not only for diagnosis. Variant types and sites in a single gene are known to influence the age of onset, severity, and pattern of affected organs, for example
FBN1
(MFS),
LMNA
(laminopathy),
DMD
(muscular dystrophy), and
TTR
(hereditary amyloidogenic transthyretin amyloidosis) in the cardiovascular field, and understanding the genotype-phenotype relationship can aid in genetic counseling and in determining the treatment strategy. In contrast, to the best of our knowledge, completely distinct system and/or organ diseases induced by 1 gene mutation have been rarely reported.17
We recently reported the relationship between
FBN1
genotype and severe aortic events (aortic root replacement, type A aortic dissection, and related death), and the main conclusion was that MFS patients with
FBN1
truncating variants had a higher risk of aortic events compared with patients with missense and in-frame variants, and male patients had an increased risk.18
In addition, we encountered a patient with Malan syndrome with a single base insertion in exon 2 of
NFIX, complicated with type A aortic dissection.19
Variants in
NFIX
induce 2 major types of syndrome affecting skeletal and neural development: Malan syndrome, which is characterized by Sotos-like overgrowth and macrocephaly and is mainly induced by variants affecting the evolutionarily conserved N-terminal DNA-binding/dimerization domain (exons 2 and 3); and the more severe Marshall-Smith syndrome, which is caused by frameshift and splice variants in exon 6–8 to escape nonsense-mediated RNA decay.20
Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) are neuromuscular diseases characterized by progressive muscle degeneration and weakness, and are caused by variants in the dystrophin gene (DMD) on the X chromosome. Truncating
DMD
variants induce the severe type of DMD, and missense or in-frame variants induce the relatively mild type of BMD.21
Variants that maintain the translational reading frame (in-frame) generally result in an abnormal but partially functional dystrophin; thus, genetic treatment approaches to restore the normal reading frame by CRISPR-Cas9 genome editing or after modifying the pre-messenger RNA splicing by “exon skipping”, especially for the more severely affected DMD male patients, have been vigorously investigated.22,23
Many patients with DMD have variable-sized deletions spanning exons 47–50; thus, an antisense oligonucleotide (eteplirsen) directed against the exon 51 splicing enhancer region of pre-mRNA, leading to its exclusion from mRNA, is approved for the treatment of DMD patients who have a confirmed variant that is amenable to exon 51 skipping (Figure 4).22

Genetic tests for hereditary aortic aneurysm and dissection (HTAAD) to guide precision medicine have been covered by health insurance in Japan since 2016, and gene therapy for HTAAD has also been expected.5
There is no evidence to indicate that haploinsufficiency due to nonsense or out-of-frame variants predisposes to LDS caused by
TGFBR1/TGFBR2
or to non-syndromic HTAAD caused by
ATCA2
and
MYH11
; thus, gene therapy approaches to correct or destroy the affected allele could be theoretically applied to such variants. The biggest obstacle to success, however, might be a lack of established systems for the gene delivery to aortic VSMC, and various delivery methods are being developed.24,25
Conclusions
We briefly reviewed the recent understanding of the molecular mechanism of LDS and the unresolved problems, including the TGF-β signaling paradox and the mechanism by which
TGFBR1
variants cause 2 distinct system diseases, LDS and MSSE. Recent basic research using the LDS mice model provides crucial insights into the pathogenic mechanisms of LDS, and the establishment of an integrated clinical and genomic information system for genetic diseases would also contribute to the discovery of promising clues to the mechanism behind gene mutation.9
Further analysis is also warranted to analyze genetic variants of unknown clinical and biological significance, as in the present study, to deepen the understanding of the disease mechanism.
Disclosures
I.K. is a member of
Circulation Reports
’ Editorial Team. T.F. is affiliated with an endowed department sponsored by Actelion Pharmaceuticals Japan, Otsuka Pharmaceutical, Nipro Corporation, Terumo Corporation, Senko Medical Instrument, Century Medical, KCI Licensing, Abbott Medical Japan, Mochida Pharmaceutical, Nippon Shinyaku and Teijin Pharma Limited. The other authors declare no conflicts of interest.
References
- 1.
Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 2005; 37: 275–281.
- 2.
Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med 2006; 355: 788–798.
- 3.
MacCarrick G, Black JH 3rd, Bowdin S, El-Hamamsy I, Frischmeyer-Guerrerio PA, Guerrerio AL, et al. Loeys-Dietz syndrome: A primer for diagnosis and management. Genet Med 2014; 16: 576–587.
- 4.
Meester JAN, Verstraeten A, Schepers D, Alaerts M, Van Laer L, Loeys BL. Differences in manifestations of Marfan syndrome, Ehlers-Danlos syndrome, and Loeys-Dietz syndrome. Ann Cardiothorac Surg 2017; 6: 582–594.
- 5.
Takeda N, Komuro I. Genetic basis of hereditary thoracic aortic aneurysms and dissections. J Cardiol 2019; 74: 136–143.
- 6.
Goudie DR, D’Alessandro M, Merriman B, Lee H, Szeverenyi I, Avery S, et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet 2011; 43: 365–369.
- 7.
Fujiwara T, Takeda N, Hara H, Morita H, Kishihara J, Inuzuka R, et al. Distinct variants affecting differential splicing of TGFBR1 exon 5 cause either Loeys-Dietz syndrome or multiple self-healing squamous epithelioma. Eur J Hum Genet 2018; 26: 1151–1158.
- 8.
Hata A, Chen YG. TGF-beta signaling from receptors to Smads. Cold Spring Harb Perspect Biol 2016; 8: a022061.
- 9.
Jondeau G, Ropers J, Regalado E, Braverman A, Evangelista A, Teixedo G, et al. International registry of patients carrying TGFBR1 or TGFBR2 mutations: Results of the MAC (Montalcino Aortic Consortium). Circ Cardiovasc Genet 2016; 9: 548–558.
- 10.
Gallo EM, Loch DC, Habashi JP, Calderon JF, Chen Y, Bedja D, et al. Angiotensin II-dependent TGF-beta signaling contributes to Loeys-Dietz syndrome vascular pathogenesis. J Clin Invest 2014; 124: 448–460.
- 11.
Cardoso S, Robertson SP, Daniel PB. TGFBR1 mutations associated with Loeys-Dietz syndrome are inactivating. J Recept Signal Transduct Res 2012; 32: 150–155.
- 12.
Horbelt D, Guo G, Robinson PN, Knaus P. Quantitative analysis of TGFBR2 mutations in Marfan-syndrome-related disorders suggests a correlation between phenotypic severity and Smad signaling activity. J Cell Sci 2010; 123: 4340–4350.
- 13.
Hara H, Takeda N, Fujiwara T, Yagi H, Maemura S, Kanaya T, et al. Activation of TGF-beta signaling in an aortic aneurysm in a patient with Loeys-Dietz syndrome caused by a novel loss-of-function variant of TGFBR1. Hum Genome Var 2019; 6: 6.
- 14.
Akhurst RJ. The paradoxical TGF-beta vasculopathies. Nat Genet 2012; 44: 838–839.
- 15.
Takeda N, Hara H, Fujiwara T, Kanaya T, Maemura S, Komuro I. TGF-beta signaling-related genes and thoracic aortic aneurysms and dissections. Int J Mol Sci 2018; 19: E2125.
- 16.
MacFarlane EG, Parker SJ, Shin JY, Kang BE, Ziegler SG, Creamer TJ, et al. Lineage-specific events underlie aortic root aneurysm pathogenesis in Loeys-Dietz syndrome. J Clin Invest 2019; 129: 659–675.
- 17.
Kohn A, Chang C. The relationship between hypermobile Ehlers-Danlos syndrome (hEDS), postural orthostatic tachycardia syndrome (POTS), and mast cell activation syndrome (MCAS). Clin Rev Allergy Immunol, doi:10.1007/s12016-019-08755-8.
- 18.
Takeda N, Inuzuka R, Maemura S, Morita H, Nawata K, Fujita D, et al. Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ Genom Precis Med 2018; 11: e002058.
- 19.
Oshima T, Hara H, Takeda N, Hasumi E, Kuroda Y, Taniguchi G, et al. A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection. Hum Genome Var 2017; 4: 17022.
- 20.
Schanze D, Neubauer D, Cormier-Daire V, Delrue MA, Dieux-Coeslier A, Hasegawa T, et al. Deletions in the 3‘ part of the NFIX gene including a recurrent Alu-mediated deletion of exon 6 and 7 account for previously unexplained cases of Marshall-Smith syndrome. Hum Mutat 2014; 35: 1092–1100.
- 21.
Takeshima Y, Yagi M, Okizuka Y, Awano H, Zhang Z, Yamauchi Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet 2010; 55: 379–388.
- 22.
McNally EM, Wyatt EJ. Mutation-based therapy for Duchenne muscular dystrophy: Antisense treatment arrives in the clinic. Circulation 2017; 136: 979–981.
- 23.
Long C, Li H, Tiburcy M, Rodriguez-Caycedo C, Kyrychenko V, Zhou H, et al. Correction of diverse muscular dystrophy mutations in human engineered heart muscle by single-site genome editing. Sci Adv 2018; 4: eaap9004.
- 24.
Akimoto S, Suzuki JI, Aoyama N, Ikeuchi R, Watanabe H, Tsujimoto H, et al. A novel bioabsorbable sheet that delivers NF-kappaB decoy oligonucleotide restrains abdominal aortic aneurysm development in rats. Int Heart J 2018; 59: 1134–1141.
- 25.
Hosaka K, Manfredsson FP, Hoh BL. Localized intra-arterial gene delivery using AAV. Methods Mol Biol 2019; 1937: 259–265.
