Abstract
Background:
Endovascular treatment with balloon angioplasty plays a major role in revascularization of below-the-knee (BTK) arteries in patients with critical limb ischemia (CLI). However, with severely calcified lesions, achieving optimal revascularization with balloon angioplasty alone is difficult. Therefore, we are evaluating the safety and effectiveness of the Rotablator atherectomy system as an adjunctive device in the treatment of severely calcified lesions in BTK arteries in the RESCUE-BTK trial, a multicenter, single-arm, open-label, exploratory investigator-initiated clinical study of medical devices. In this paper we describe the design of the trial.
Methods and Results:
Seventeen patients with CLI in whom balloon angioplasty has failed are enrolled in the study. The primary endpoint is the procedural success rate of balloon angioplasty after rotational atherectomy. Success is defined as the fulfillment of 3 requirements upon assessment by the core laboratory: (1) final residual diameter stenosis <50%; (2) the absence of a delay in flow or vessel perforation in the target artery, or both; and (3) brisk antegrade flow to the foot. Key secondary endpoints are the number of complications associated with the trial procedures and the limb salvage rate. Participants are followed-up for 6 months after the trial procedures.
Conclusions:
The RESCUE-BTK trial will clarify the safety and effectiveness of the adjunctive use of the Rotablator system in severely calcified lesions of BTK arteries in patients with CLI.
With increases in the number of patients with diabetes mellitus who are dependent on dialysis, the prevalence of critical limb ischemia (CLI) is also increasing.1
CLI proceeds from ulcer to gangrene with progression of the disease state, and eventually necessitates lower extremity amputation. A meta-analysis of 6,118 patients with CLI who received conservative treatment reported a high rate (73–95%) of major amputation within 1 year.2
Therefore, it is important to improve the blood flow to the foot in order to suppress the development of these disease states and to avoid major amputation.
Below-the-knee (BTK) arterial lesions are the main complications of CLI. The current standard therapy for CLI that requires revascularization is endovascular treatment and bypass surgery. However, patients with CLI may be unlikely to undergo bypass surgery because of complex lesion structure, poor general condition, or limited access to dedicated vascular surgeons.3
In view of a wound healing rate of 74–80% and a limb salvage rate of 75–92% after infrapopliteal intervention, endovascular treatment can be reasonable for the treatment of CLI.4–6
However, when balloon angioplasty is used alone, the rate of procedural failure is 5.8% in patients not on dialysis and 12.3% in patients on dialysis.7
In such cases, severe calcification often hampers balloon passage or inflation. Furthermore, there are no additional effective therapeutic methods for these cases in Japan; as a result, major amputation is common, or patients undergo only continued observation.
The Rotablator atherectomy system (Boston Scientific, Natick, MA, USA) is widely used as an adjunctive device in the treatment of calcified lesions caused by coronary artery disease (CAD). The rate of technical success of rotational atherectomy for peripheral arterial disease (PAD) has been reported to be in the range 77–97%,8,9
but in Japan this procedure is not covered by insurance for the treatment of PAD. Furthermore, revascularization of the BTK arteries has been performed primarily with rotational atherectomy alone;1,2
thus, the adjunctive use of the Rotablator has not been fully evaluated.
Against this background, we thought that there is a strong need for the Rotablator system in the treatment of CLI. Thus, we planned this investigator-initiated clinical trial to examine the safety and effectiveness of the Rotablator as an adjunctive device in the treatment of severely calcified lesions in BTK arteries. The hypothesis of this study is that the adjunctive use of the Rotablator will lead to procedural success in cases of unsuccessful balloon angioplasty.
Herein we describe the design of the RESCUE-BTK trial.
Methods
Study Design
The RESCUE-BTK trial is an open-label, exploratory investigator-initiated clinical trial of clinical devices. The study is registered with the Japan Medical Association Center for Clinical Trials (JMACCT) Clinical Trial Registry (JMA-IIA00313).
Sample Size
We assumed that the proportion of successful balloon angioplasties after the trial procedure (P1) would be 85%. According to binomial probabilities, a sample size of 17 was required for attaining a power of 80% at a significance level of 2.5% for testing the null hypothesis (P1≤50%). Because it is expected that some patients will be excluded from the analysis population for assessing effectiveness on the basis of core laboratory results or withdrawal from the study, the number of patients included in this trial is expected to be approximately 25.
Eligibility Criteria and Consent to Participate
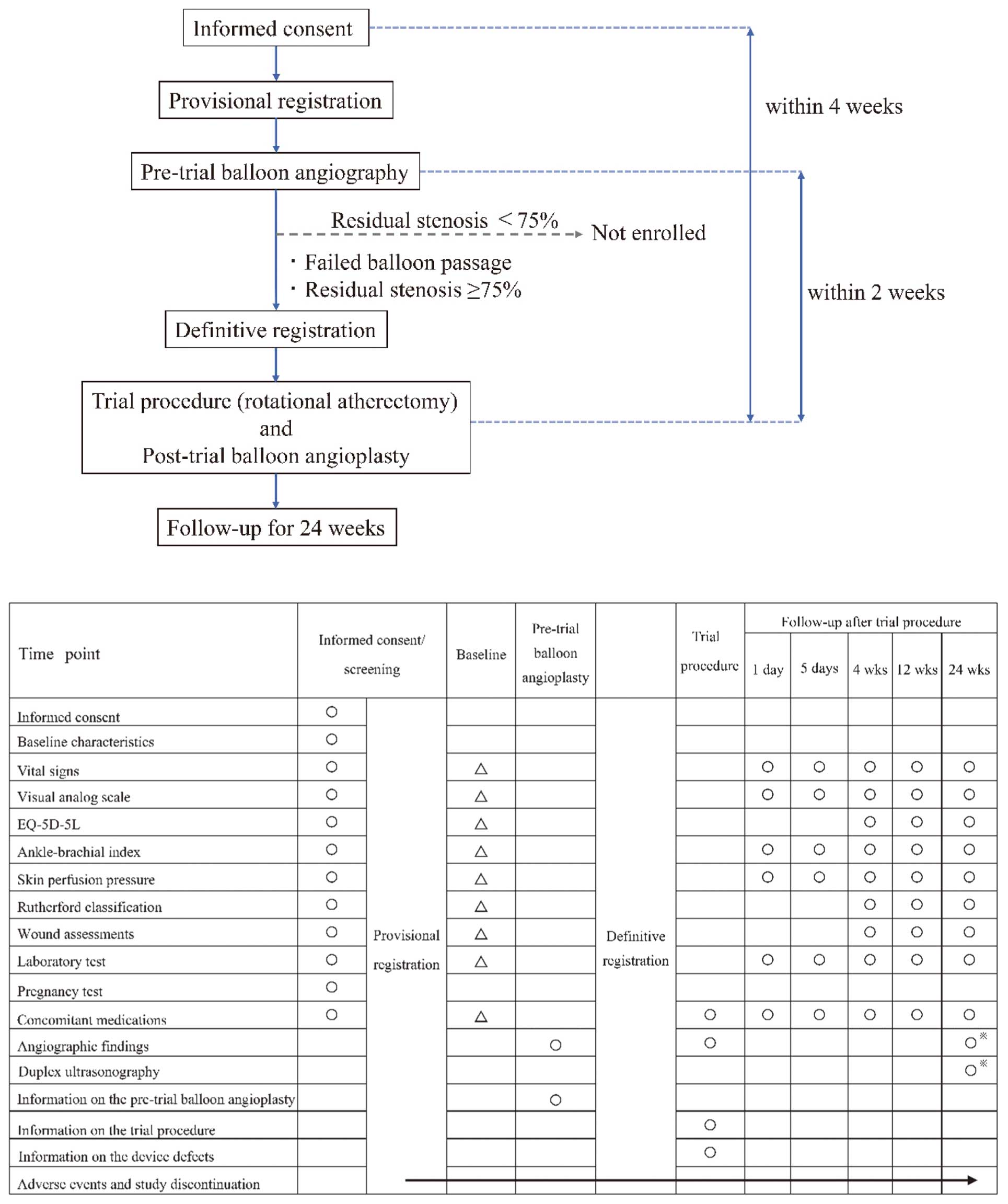
Subject eligibility is determined in 2 steps: provisional and definitive registration (Tables 1,2). Patients meeting the provisional criteria will be registered and then definitively registered on the basis of the results of pretrial balloon angioplasty. In both registration steps, patients will need to meet all the inclusion criteria and none of the exclusion criteria. All eligible participants are required to provide written informed consent.
Table 1.
Inclusion and Exclusion Criteria for Provisional Registration
| Inclusion criteria |
| 1. Rutherford category 4 or 5 (because of atherosclerotic peripheral artery disease) |
| 2. Skin perfusion pressure <50 mmHg below the ankle joint of the affected limb |
3. Presence of ≥1 of the following risk factors: diabetes, maintenance dialysis (hemodialysis or peritoneal dialysis),
renal failure (eGFR <60 mL/min/1.73 m2), age ≥65 years, and presence of calcification in affected BTK arteries |
| 4. Age ≥20 years at the time of informed consent |
5. Agreement to participate in the study and provision of written informed consent by patient or legally acceptable
representative |
| Exclusion criteria |
| 1. Inability to receive contrast agents |
| 2. History of amputation above the knee joint or plans for major amputation surgery of the target lower limb |
| 3. Prior bypass surgery in the target lower limb |
| 4. Prior stent implantation in target vessel |
5. Prior endovascular intervention in the non-target vessel of the target lower limb within 1 week before provisional
registration |
| 6. Prior enrollment in this trial |
| 7. Presence of severe wound inflammation or any severe infectious disease |
8. History of myocardial infarction, ischemic stroke, or intracranial hemorrhage within 6 months before provisional
registration |
| 9. Serum creatinine level ≥2.0 mg/dL in a patient not on dialysis |
10. Any abnormal blood test result: platelet count <8.0×104/μL or ≥60.0×104/μL, white blood cell count <3,000/μL,
or hemoglobin <8.0 g/dL |
| 11. Inability to move using a wheelchair |
| 12. Life expectancy <1 year |
| 13. Pregnancy, breastfeeding, or possible pregnancy |
| 14. Contraindication to antiplatelet medications |
| 15. Participation or plans to participate in other clinical trials or interventional studies |
| 16. Any other reason, according to the investigator’s discretion |
Estimated glomerular filtration rate (eGFR) was calculated using the following formulas: 194 × serum creatinine−1.094×Age−0.287 in male patients; and 194 × serum creatinine−1.094×Age−0.287×0.739 in female patients. BTK, below the knee.
Table 2.
Inclusion and Exclusion Criteria for Definitive Registration
| Inclusion criteria |
1. Unsuccessful balloon angioplasty, defined as failed balloon passage or ≥75% residual stenotic diameter, for
target lesion within 2 weeks before the definitive registration |
| 2. Presence of calcification in the target lesion |
| 3. Target lesion between the distal popliteal artery and the distal crural artery |
| 4. Target lesion with a reference vessel with a visual diameter of 2.0–4.0 mm |
| 5. Target lesion length <100 mm |
| Exclusion criteria |
| 1. Failed passage of guide wire into target lesion |
| 2. Angiographic thrombus |
| 3. High risk for arterial perforation by trial procedures, according to the investigator’s discretion |
4. Prior endovascular intervention in non-target vessel of the target lower limb within 1 week before definitive
registration |
| 5. Prior enrollment in this trial |
| 6. Any other reason, according to the investigator’s discretion |
The study flow diagram and detailed time schedule are shown in the
Figure. Enrollment of study participants started in November 2017, and the follow-up period ended in April 2020.
Interventions
The following procedures were performed as part of the trial:
1. Selection of the target vessel: The target vessel was limited to one of the BTK vessels, with the operator identifying which vessel would achieve at least one straight-line flow to the foot.
2. Pretrial balloon angioplasty: The target lesion was limited to a lesion in which pretrial balloon angioplasty performed after provisional registration was unsuccessful. The investigators decided on balloon sizes and maximal pressures according to reference vessel diameter and lesion characteristics. Unsuccessful pretrial balloon angioplasty was defined as failure of balloon passage or ≥75% residual diameter stenosis. Visual assessment was used determine subject eligibility for the trial procedures; visual assessment by the core laboratory was used as the final assessment.
3. Trial procedure: The trial procedures were performed within 4 weeks after informed consent was obtained and within 2 weeks after pretrial balloon angioplasty. An appropriate-sized burr was selected, and the Rotablator procedures were performed according to the standard operating procedures of this trial. In principle, a single lesion per vessel was subjected to the trial procedures. If, as a result of the trial procedures and post-trial balloon angioplasty, a lesion in the peripheral region of the target vessel was identified as requiring the use of the trial device, the application of trial procedures to a single additional lesion was permitted. If a lesion was found in the target vessel that did not require the trial device but was likely to need revascularization, balloon angioplasty was allowed.
4. Post-trial balloon angioplasty: After the trial procedures, a balloon with a size comparable to that of the reference vessel diameter was selected, and balloon angiography was performed. Final angiography was performed to confirm blood flow into the target lesion and the peripheral lesion, and treatment was completed. No restrictions were imposed for the treatment of no-flow/slow-flow phenomenon.
Endpoints
The primary endpoint of the study was the procedural success rate of balloon angioplasty. Success was defined as the fulfillment of the following 3 requirements upon assessment by the core laboratory: (1) final residual diameter stenosis <50%; (2) no complications, such as delayed flow and vessel perforation in the target artery; and (3) brisk antegrade flow to the foot. Secondary endpoints are summarized in
Table 3. A cardiovascular event was defined as a composite of cardiovascular-related death, non-fatal myocardial infarction, non-fatal stroke, or transient ischemic attack. A major adverse limb event was defined as a composite of target vessel revascularization or major amputation surgery of the target lower limb.
Table 3.
Secondary Endpoints
| Effectiveness |
1. Change in residual diameter stenosis by quantitative vessel analysis (core laboratory; pre- and post-trial
procedures, after balloon angioplasty) |
| 2. Limb salvage rate (4, 12, and 24 weeks after trial procedures) |
| 3. Amputation-free survival (24 weeks after trial procedures) |
4. Change in visual analog scale score (at baseline, and 1 and 5 days and 4, 12, and 24 weeks after trial
procedures) |
| 5. Change in ankle-brachial index (at baseline, and 1 and 5 days and 4, 12, and 24 weeks after trial procedures) |
6. Change in skin perfusion pressure (at baseline, and 1 and 5 days and 4, 12, and 24 weeks after trial
procedures) |
| 7. Change in Rutherford classification (at baseline and 4, 12, and 24 weeks after trial procedures) |
| 8. Wound assessment (at baseline and 4, 12, and 24 weeks after trial procedures) |
9. Change in residual diameter stenosis by visual estimation (on site; final angiography after endovascular
intervention) |
| 10. Balloon catheter passing rate (in cases of failed balloon passage; pre- and post-trial procedures) |
| Safety |
| 1. Number of patients with distal embolization (up to 5 days after trial procedures) |
| 2. Number of re-occlusions (up to 24 weeks after trial procedures) |
| 3. Number of serious adverse events associated with the trial procedures (up to 4 weeks after trial procedures) |
| 4. Number of deaths (up to 24 weeks after trial procedures) |
| 5. Number of cardiovascular events (up to 24 weeks after trial procedures) |
| 6. Number of major adverse limb events (up to 24 weeks after trial procedures) |
| 7. Number of target vessel revascularizations (up to 24 weeks after trial procedures) |
The system for electronic data capture and data management has been validated to meet the regulatory requirements. In addition, on-site monitoring including source document verification and audit were conducted.
Study Organization
The study recruited 10 hospitals in Japan specializing in the treatment of PAD and with extensive experience in using the Rotablator in the treatment of CAD. The independent safety monitoring board is comprised of 3 individual experts in vascular surgery, radiology, and cardiology. Furthermore, angiographic data were independently evaluated at a core laboratory. Details regarding study organization are provided in the
Supplementary File.
Statistical Methods
Two analysis populations were defined: Population 1, consisting of the whole population of patients who underwent the trial procedure and were followed-up for at least one of the endpoints of effectiveness; and Population 2, a subpopulation consisting only of patients in whom balloon angioplasty was unsuccessful before the trial procedure, as rated by the core laboratory. The endpoints of effectiveness will be assessed in Population 1, whereas secondary analyses will be performed with Population 2 to support the conclusions based on the primary analyses. Safety endpoints will be assessed for all patients undergoing the trial procedure.
Patient demographic data and endpoints will be analyzed descriptively: continuous variables will be reported as the mean±SD or as the median and interquartile range; categorical variables will be reported as frequencies and percentages with corresponding exact (Clopper-Pearson) 95% confidence intervals. The first occurrence of events will be estimated using the Kaplan-Meier method.
Details regarding the analyses will be prespecified in the statistical analysis plan. All analyses will be performed using SAS/STAT version 9.2 or later (SAS Institute, Cary, NC, USA).
Discussion
For this trial, we considered it appropriate to adopt a single-arm design for 2 reasons. First, a meta-analysis showed that 95% of patients with CLI who do not undergo angioplasty or surgical revascularization required amputation within 1 year;2
therefore, it is ethically unacceptable assign patients to a control group. Second, the primary endpoint was defined as the procedural success rate of balloon angioplasty after rotational atherectomy. For patients in whom endovascular treatment for calcified lesions of the BTK arteries was unsuccessful, it would be generally impossible to select a further treatment option. Thus, the RESCUE-BTK trial was designed as a single-arm study.
In previous studies of Rotablator atherectomy, the procedure was performed primarily as a stand-alone procedure for the treatment of PAD, and 25–50% of patients had complications, such as arterial spasm, thrombosis, dissection, vessel perforation, distal embolism, and no reflow.8,9
Shortening of the duration of use and target lesion length, as well as restricting burr size and the number of target lesions in the RESCUE-BTK trial are expected to reduce the risk of complications. Therefore, we considered it desirable to use the Rotablator as an adjunctive device for balloon angioplasty in cases of unsuccessful balloon angioplasty. Based on the results of a previous clinical study,8
we set the upper limit of lesion length at <100 mm. The lower limit of reference vessel diameter (2 mm) was also set on the basis of results regarding the safe use of the Rotablator. In principle, the target lesion is a single lesion in which pretrial balloon angioplasty has been unsuccessful. Furthermore, for thromboprophylaxis, standard antiplatelet therapy with aspirin and a thienopyridine will be initiated ≥2 days before the trial procedures and continued over the first 2 weeks postoperatively.
One major problem after interventions in lower extremity arteries is the low rate of vessel patency. Loss of vessel patency results in no healing, worsening of the ulcer, or the appearance of new lesions in the foot. Previous studies showed that restenosis occurs early after balloon angioplasty,10
with a 1-year rate of restenosis of 75%.11
Thus, target lesion revascularization rates may be high after the trial procedures, but it is clinically important to be able to dilate target lesions with balloon angioplasty after Rotablator atherectomy, even if restenosis occurs repeatedly.
Other methods and devices have recently been developed for atherectomy in BTK arteries, such as directional atherectomy, laser atheroablation, orbital atherectomy and chronic total occlusion devices. However, there have been few reports comparing the success rate and complications of atherectomy devices. A recent retrospective study showed that the adjunctive use of the Rotablator resulted in a low complication rate of 1.8% and a high technical success rate of 94.5%;12
therefore, the RESCUE-BTK trial will provide further insights into the safety, effectiveness, and applicability of the Rotablator.
Acknowledgments
The authors gratefully acknowledge S. Kimura for secretarial work and M. Uotani and K. Hirase as clinical research coordinators at the National Cerebral and Cardiovascular Center (Osaka, Japan). The authors also thank M. Yamamoto at the Japan Medical Association Center for Clinical Trials (Tokyo, Japan) for advice on study implementation.
Sources of Funding
This work was supported by a clinical trial promotion project of the Japan Medical Association, which is funded by a Health Labour Sciences Research Grant from the Ministry of Health, Labour and Welfare of Japan.
Disclosures
The Rotablator atherectomy systems are being provided by Boston Scientific based on the policy of Japanese Good Clinical Practice.
S.I. reports receiving a grant from the Japan Society for the Promotion of Science outside the submitted work.
O.K. reports receiving honoraria for lectures and advisory board fees from Boston Scientific outside the submitted work.
M.A. reports receiving personal fees from Daiichi Sankyo and Otsuka outside the submitted work.
M.K. reports receiving grants and personal fees from Takeda, Astellas, Sanofi, Pfizer, Ono, Novartis, Mitsubishi Tanabe, Kyowa Hakko Kirin, Abbott, and Otsuka, grants from the Japanese government, Japan Heart Foundation, Japan Cardiovascular Research Foundation, Calpis, and Nihon Kohden, and personal fees from Daiichi-Sankyo, Bayer, Boehringer, Kowa, Sumitomo Dainippon, Sawai, MSD, Shionogi, AstraZeneca, Asahi Kasei Medical, Novo Nordisk, Fujifilm RI, and Japan Medical Data outside the submitted work.
S.Y. reports receiving grant support from Takeda and Abbott and lecture fees from Daiichi Sankyo and Bristol-Myers Squibb outside the submitted work.
S.I., S.Y. are members of
Circulation Reports’ Editorial Team.
The remaining authors report no conflicts of interest.
IRB Information
This study was conducted in compliance with the Declaration of Helsinki and Japanese Good Clinical Practice. The study protocol was approved by the institutional review boards (IRBs) of all participating institutes, as well as by the National Cerebral and Cardiovascular Center IRB (No. 1010).
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circrep.CR-20-0024
References
- 1.
Patel VI, Mukhopadhyay S, Guest JM, Conrad MF, Watkins MT, Kwolek CJ, et al. Impact of severe chronic kidney disease on outcomes of infrainguinal peripheral arterial intervention. J Vasc Surg 2014; 59: 368–375.
- 2.
Wolfe JH, Wyatt MG. Critical and subcritical ischaemia. Eur J Vasc Endovasc Surg 1997; 13: 578–582.
- 3.
Kawarada O, Zen K, Hozawa K, Ayabe S, Huang HL, Choi D, et al. Contemporary critical limb ischemia: Asian multidisciplinary consensus statement on the collaboration between endovascular therapy and wound care. Cardiovasc Interv Ther 2018; 33: 297–312.
- 4.
Lo RC, Darling J, Bensley RP, Giles KA, Dahlberg SE, Hamdan AD, et al. Outcomes following infrapopliteal angioplasty for critical limb ischemia. J Vasc Surg 2013; 57: 1455–1463.
- 5.
Kawarada O, Fujihara M, Higashimori A, Yokoi Y, Honda Y, Fitzgerald PJ. Predictors of adverse clinical outcomes after successful infrapopliteal intervention. Catheter Cardiovasc Interv 2012; 80: 861–871.
- 6.
Fernandez N, McEnaney R, Marone LK, Rhee RY, Leers S, Makaroun M, et al. Predictors of failure and success of tibial interventions for critical limb ischemia. J Vasc Surg 2010; 52: 834–842.
- 7.
Kawarada O, Yokoi Y, Higashimori A, Fujihara M, Sakamoto S, Ishihara M, et al. Impact of end-stage renal disease in patients with critical limb ischaemia undergoing infrapopliteal intervention. EuroIntervention 2014; 10: 753–760.
- 8.
The Collaborative Rotablator Atherectomy Group (CRAG). Peripheral atherectomy with the rotablator: A multicenter report. J Vasc Surg 1994; 19: 509–515.
- 9.
Henry M, Amor M, Ethevenot G, Henry I, Allaoui M. Percutaneous peripheral atherectomy using the rotablator: A single-center experience. J Endovasc Surg 1995; 2: 51–66.
- 10.
Schmidt A, Ulrich M, Winkler B, Klaeffling C, Bausback Y, Braunlich S, et al. Angiographic patency and clinical outcome after balloon-angioplasty for extensive infrapopliteal arterial disease. Catheter Cardiovasc Interv 2010; 76: 1047–1054.
- 11.
Liistro F, Porto I, Angioli P, Grotti S, Ricci L, Ducci K, et al. Drug-eluting balloon in peripheral intervention for below the knee angioplasty evaluation (DEBATE-BTK): A randomized trial in diabetic patients with critical limb ischemia. Circulation 2013; 128: 615–621.
- 12.
Yamamoto Y, Kawarada O, Ando H, Anzai H, Zen K, Tamura K, et al. Effects of high-speed rotational atherectomy in peripheral artery disease patients with calcified lesions: A retrospective multicenter registry. Cardiovasc Interv Ther, doi:10.1007/s12928-020-00643-9.