Abstract
Background:
Chronic angiotensin II (AngII) infusion promotes ascending aortic dilation in C57BL/6J mice. Meanwhile, vasohibin-2 (VASH2) is an angiogenesis promoter in neovascularization under various pathologic conditions. The aim of this study was to investigate whether exogenous VASH2 influences chronic AngII-induced ascending aortic dilation.
Methods and Results:
Eight–ten-week-old male C57BL/6J mice were injected with adenovirus (Ad) expressing either VASH2 or LacZ. One week after the injection, mice were infused with either AngII or saline s.c. for 3 weeks. Mice were divided into 4 groups: AngII+VASH2, AngII+LacZ, saline+VASH2, and saline+LacZ. Overexpression of VASH2 significantly increased AngII-induced intimal areas as well as the external diameter of the ascending aorta. In addition, VASH2 overexpression promoted ascending aortic medial elastin fragmentation in AngII-infused mice, which was associated with increased matrix metalloproteinase activity and medial smooth muscle cell (SMC) apoptosis. On western blot analysis, accumulation of apoptotic signaling proteins, p21 and p53 was increased in the AngII+VASH2 group. Furthermore, transfection of human aortic SMC with Ad VASH2 increased p21 and p53 protein abundance upon AngII stimulation. Positive TUNEL staining was also detected in the same group of the human aortic SMC.
Conclusions:
Exogenous VASH2 exacerbates AngII-induced ascending aortic dilation in vivo, which is associated with increased medial apoptosis and elastin fragmentation.
Thoracic aortic aneurysm (TAA) is a potentially fatal disease associated with enhanced rupture that frequently leads to sudden death. There is a poor prognosis for individuals with a ruptured aorta, even in a clinical environment.1–3
The number of surgical repairs of TAA is gradually increasing.4
Although thoracic endovascular aortic repair is increasingly common,5
surgical repair of ascending aorta and aortic arch remains challenging, with mortality rates >20% especially in rupture cases.4
Therefore, development of innovative, preventive therapy for TAA is a major unmet medical need.
Several human and experimental studies have described a role for angiotensin II (AngII) in the ascending aortic dilation.6–10
The first evidence came from an analysis of individuals with Marfan syndrome, in which an AngII type 1 (AT1) receptor antagonist mitigated ascending aorta dilation.8
This observation sparked multiple clinical trials to determine the efficacy of AT1
receptor antagonism in patients with TAA.11
Regarding TAA formation, 1 intensely studied mechanism is fibrillin-1 mutation12
and transforming growth factor (TGF)-β hyperactivity.13
Recently, in terms of AT1
receptor, AngII infusion into wild-type C57BL/6 mice has been established as a reproducible and indisputable model of ascending aortic dilation.14,15
Meanwhile, vasohibin (VASH) was first reported in 2004 as a negative regulatory system of angiogenesis, a molecule inhibiting angiogenesis and intimal thickening of microvessels.16
Furthermore, VASH includes at least 2 homologs, that is, VASH1 and VASH2. VASH1 has gathered attention mainly in the oncology field, as an endothelial cell-derived angiogenesis inhibitor induced by vascular endothelial growth factor (VEGF) under various pathologic conditions.17–20
In contrast, VASH2 is a mononuclear cell-derived protein mainly detected in cancer regions. Contrary to its name, VASH2 is an angiogenesis promoter.18,19
Downregulation of VASH2 or blocking of VASH2 suppresses tumor growth by inhibiting angiogenesis in endometrial cancer cells.21–23
Although VASH2 promotes epithelial-mesenchymal transition via TGF-β signaling,24
the role of VASH2 in the angiogenesis promotion pathway in large vessels remains unclear. In addition, the receptor of VASH2 has not been identified to date, and there is a paucity of evidence as to whether VASH2 is involved in aortic dilation.
Given these observations, we primarily hypothesized that exogenous VASH2 influences AngII-induced ascending aortic dilation through angiogenesis in the vascular wall of ascending aortas. The aim of this study was therefore to examine the effect of exogenous VASH2 on AngII-induced ascending aortic dilation in normal cholesterolemic mice.
Methods
Mice
All animal experiments conformed to the NIH guidelines (Guide for the Care and Use of Laboratory Animals). Eight–ten-week-old male C57BL/6J mice were obtained from Japan Charles River and maintained in the animal resource center of Okayama University. All mice were maintained in a barrier facility and fed a normal laboratory diet. Mice were injected with adenoviral vectors encoding for either VASH2 or LacZ. The mice were divided into 4 groups: AngII+VASH2 (n=22), AngII+LacZ (n=21), saline+VASH2 (n=10), and saline+LacZ (n=8). The experiment protocols were approved by the Ethics Review Committees for Animal Experimentation of Okayama University Graduate School of Medicine, Dentistry, and Pharmaceutical Sciences.
Adenovirus Injection
The mice were injected with adenoviral vectors (Ad LacZ or Ad hVASH2; 7.5×109 vp/100 μL) i.v. (via tail vein) under pentobarbital sodium (25 μg/g, i.p.) anesthesia as described previously.18
Injections started 1 week prior to AngII infusion, and were repeated at 2-week intervals as described previously.25
AngII Infusion
The mice were anesthetized using pentobarbital sodium (25 μg/g, i.p.). Saline or AngII (1,000 ng/kg/min; Bachem, Cat. no. H-1705) was infused into mice s.c. via Alzet mini-osmotic pumps (Model 2004; DURECT, Lot No. 10274-12) for 21 days as described previously.26,27
Blood Pressure
Systolic blood pressure (SBP) was measured in all mice in each group in the last week using a Softron tail cuff system (BP-98A), as described previously.28,29
Plasma Components
Plasma total cholesterol concentration was measured using a commercially available enzymatic kit (Waco Chemicals, Cat. no. 439-17501).30
VEGF concentration was measured using a commercially available enzymatic kit (R&D Systems, Cat. no. MMV00).
Polymerase Chain Reaction
To confirm the gene delivery after tail vein injection, we examined liver tissues on polymerase chain reaction (PCR). Liver tissues were collected with RNA later solution and stored at −80℃. RNA was purified using a commercially available assay kit (RNeasy Mini, QIAGEN, Cat. no. 74104). Reverse transcription was performed using a commercially available kit (iScript cDNA Synthesis Kit, Bio Rad, Cat. no.170-8890). VASH2 mRNA abundance in the liver tissues was analyzed using the following hVASH2 primers (Takara Bio): forward, 5'-TgCACACAgTCAAgAAggTC; reverse, 5'-TTCTCACTTgggTCggAgAg.31
The commercially available PCR Master-mix (GoTaq Green Master Mix, Promega, Cat. no. M7122) was used. cDNA (up to 200 μg) was mixed with 1 μL each primer, and 10 μL PCR buffer. PCR cycle parameters were as follows: hold 95℃ for 20 s, repeat 95℃ 3 s and 59.5℃ 30 s for 42 cycles, and hold 10℃.
Western Blot Analysis
Liver or aortic tissue lysates were extracted in RIPA lysis buffer and protein content was measured using the DC Protein Assay Kit (Bio Rad, Hercules, CA, USA). Protein extracts (10 μg) were resolved on SDS-PAGE (10% wt/vol) and transferred electrophoretically to PVDF membranes as described previously.27,32
After blocking with non-dry fat milk (5% wt/vol), membranes were probed with antibodies against VASH2 (clone 1760; provided by Tohoku University, Sendai, Japan),33,34
p53 (Abcam, Cat. no. ab131442), or p21 (Abcam, Cat. no. b109199), and β-actin (Sigma-Aldrich, Cat. no. A5441). Membranes were then incubated with appropriate secondary antibodies, and immune complexes were visualized on chemiluminescence (EMD Millipore, Cat. no. WBLUF0100, Cat. no. WBLUC0100) and quantified using a General Electric Imager (LAS 4000 mini).
Ascending Aortic Dilation Quantification
Intima areas of thoracic aortas were measured using an
en face
method as described previously.14
Ex vivo width of the middle of ascending aorta was measured toward the vertical direction of the tangential line of the ascending aortic greater curvature. Abdominal aortic dilation was measured using the ex vivo outer diameter of the abdominal aorta as described previously.28
Histology and Immunostaining
Histological staining and immunostaining were performed on formalin-fixed frozen sections, with appropriate negative controls.14,35
Histological stains included hematoxylin and eosin, Masson’s trichrome, and elastic-Van Gieson. Macrophages were detected in tissue sections of thoracic aorta using a rat anti-mouse CD68 (1:500, Serotec Raleigh, Cat. no. MCA1957). Angiogenetic endothelial cells were detected using a rabbit anti-mouse CD31 (1:500, Abcam, Cat. no. GR244952-5).
Gelatin Zymography
Protein of thoracic aortas were resolved on SDS-PAGE (7.5% wt/vol) polymerized in the presence of gelatin (2 mg/mL) to determine the abundance of matrix metalloproteinase (MMP)-2 and MMP-9. Gels were washed with Triton X-100 (2.5% vol/vol) and distilled water for 30 min. Gels were then incubated overnight at 37℃ in Tris buffer containing calcium chloride (5 mmol/L) and sodium azide (0.01% w/w), pH 8.0. After incubation, gels were stained with Coomassie Brilliant Blue, followed by de-staining with acetic acid (7% vol/vol) and methanol (40% vol/vol) as previously described.29,32,36
Gel images were captured using an imager. Unstained, translucent areas represented MMP activity.
TUNEL Staining
TUNEL staining was performed on formalin-fixed frozen sections of mouse aortas or incubated human aortic smooth muscle cells (SMC) as described in the fluorescein kit manual (Roche, Cat. no. 1 684 795).
Cell Culture
Human aortic SMC (6–8 passages, KURABO strain No. 01127: multi donor, the same batch) were seeded (1.0×105
cells) into each well of a 12-well dish. 1,800 MOI/100 μL Ad VASH2 or Ad LacZ was added to each well 24 h after the dissemination. Two hours later, cells were washed and substituted for appropriate volumes of FBS-free medium (HuMedia-SG-2 KURABO KS-2170S), and incubated for 48 h. AngII (1.0 μmol/L) were added to appropriate wells and cells were incubated for 24 h. Consequently, cells were divided into 4 groups: AngII+VASH2, AngII+LacZ, saline+VASH2, and saline+LacZ.
For TUNEL staining, human aortic SMC were plated at 1.0×104
cells into each well of an 8-well chamber slide (Iwaki, Cat. no. 5732-008) and stimulated by VASH2 and AngII as described herein.
Statistical Analysis
Data are presented as either mean±SD or mean±SEM. Student’s t-test was used to examine the difference between 2 groups. Two-way analysis of variance followed by the Holm–Sidak method was used to determine the difference in quantity of thoracic or abdominal aorta between the 4 groups. All datasets had variances and distributions that were consistent with the use of parametric tests. Analyses were performed using Sigma Plot for Windows (version 13.0, Systat Software). P<0.05 was deemed statistically significant.
Results
VASH2 Gene and Protein Expression
To confirm delivery of the VASH2 gene, PCR for VASH2 was conducted using mice liver tissues as described previously.25
The amplified bands of VASH2 were detected only in Ad VASH2-injected groups (Figure 1A). Western blotting for VASH2 protein abundance was also conducted. Similar to PCR results, western blot analysis using antibodies against VASH2 confirmed the presence of VASH2 protein bands only in Ad VASH2-injected groups (Figure 1B). These results confirmed gene delivery by adenovirus and protein expression of VASH2.
Mouse Characteristics
After 21 days of saline or AngII infusion, SBP was significantly higher in AngII-infused groups. No significant differences between the 4 groups were observed in body weight or plasma total cholesterol concentration. In addition, VASH2 injection had no influence on circulating VEGF concentration (Supplementary Table).
Exogenous VASH2 Increased AngII-Induced Ascending Aortic Dilation
Ascending and abdominal aortic dilation was observed in several aortas of AngII-infused groups (Supplementary Figure 1A). Intima area of ascending aortas was measured with an en face method (Supplementary Figure 1B). The area of ascending aorta was significantly increased in AngII-infused groups compared with saline-infused groups (P<0.0001). Furthermore, the area in the AngII+VASH2 group was significantly larger than in the AngII+LacZ group (19.78±0.40 mm3
vs. 17.74±0.44 mm3, P=0.0004;
Figure 2A).
In addition, the ex vivo width of the middle ascending aorta was significantly increased in AngII-infused groups compared with saline-infused groups (P<0.0001). The expansion of ascending aortic width was significantly larger in the AngII+VASH2 group compared with the AngII+LacZ group (1.35±0.12 mm vs. 1.47±0.11 mm, P=0.0009;
Figure 2B).
Cellular Changes of Ascending Aorta
Histological staining of ascending aortas identified erythrocyte accumulation on the aortic media in the AngII+VASH2 group. The medial thickness was also increased in the AngII-infused groups. Elastin fragmentation in media was observed only in the AngII+VASH2 group (Figure 3). We also performed CD68 and CD31 staining of ascending aortas. Neither prominent macrophage accumulation nor neovascularization in tunica media was observed (Supplementary Figure 2).
MMP Expression in Thoracic Aorta
Based on the observed increased elastin fragmentation only in the AngII+VASH2 group, next we examined the activity of MMP in ascending aortas on gelatin zymography. Upregulation of the latent form of MMP-2 was detected in the AngII-infused groups. This is consistent with a previous report.37
Furthermore, the cleaved form of the MMP-2 band was highly prominent in aortic extracts from the AngII+VASH2 group. MMP-9 was faintly detected in aortic tissue from AngII-infused groups (Supplementary Figure 3).
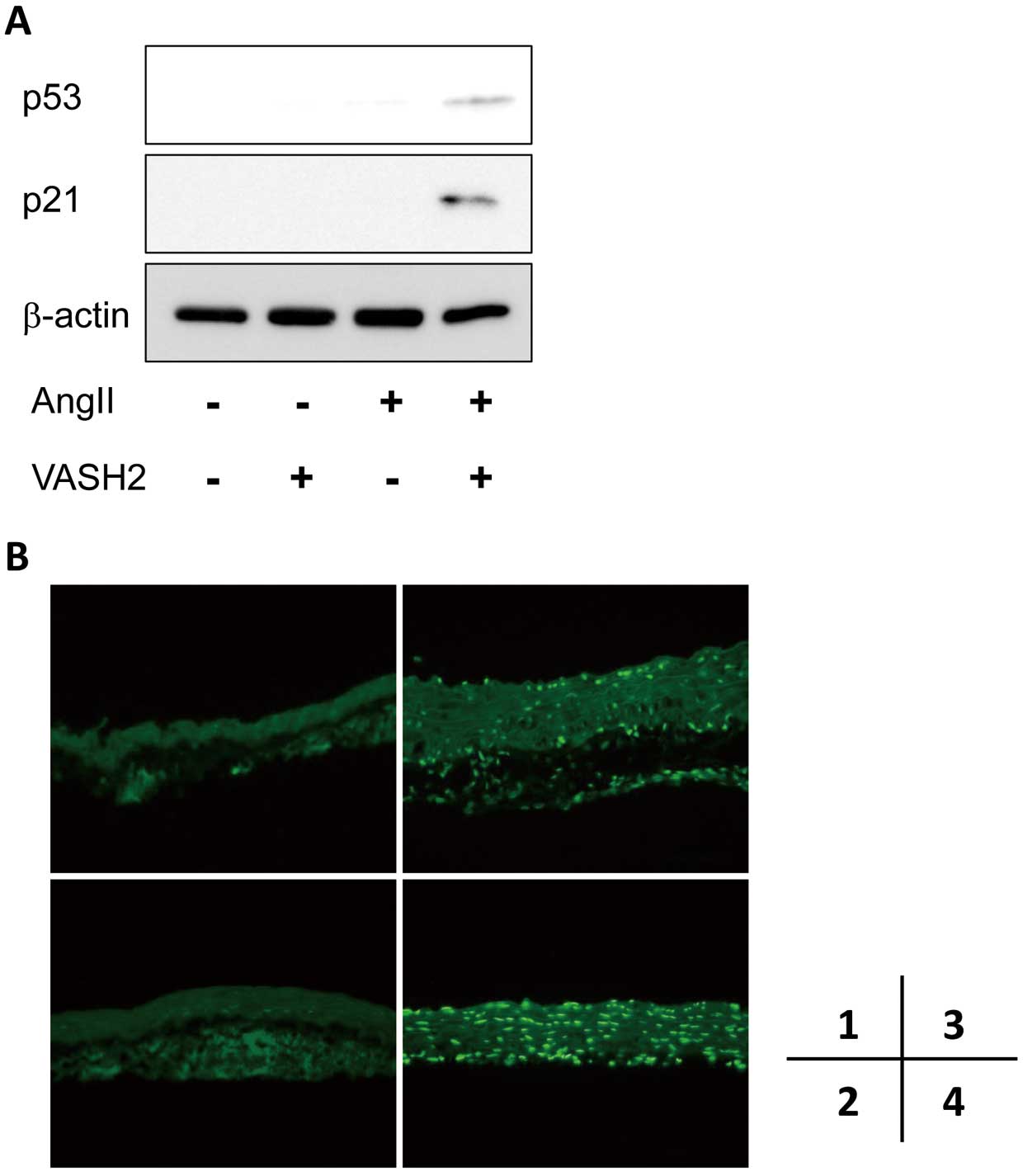
Protein Expression in Thoracic Aorta
AngII has been reported to induce apoptosis or premature senescence of vascular SMC (VSMC) via a p53- or p21-related pathway.38,39
Interestingly, on western blotting using ascending aortic lysates, an increased protein abundance of p53 and p21 was seen in the AngII+VASH2 group (Figure 4A). Positive TUNEL staining was also observed in the ascending aortic wall in the AngII+VASH2 group (Figure 4B). This suggests that the observed increased AngII-induced ascending aortic dilation upon exogenous VASH2 was associated with accelerated aortic medial SMC apoptosis.
p53 and p21 Protein Abundance and Apoptosis in Aortic SMC In Vitro
Based on the observed increased p53 and p21 protein abundance and TUNEL-positive staining in ascending aortas in vivo, we further hypothesized that VSMC are compromised by VASH2. To test and confirm this hypothesis, human aortic SMC were transfected with Ad VASH2 or Ad LacZ and incubated with or without AngII. In consistent with in vivo data, the protein abundance of p53 and p21 were detected only in the AngII+VASH2 group (Figure 5A). Positive TUNEL staining was greatly increased in the AngII+VASH2 group (Figure 5B). These results further confirm that the VASH2 overexpression promotes AngII-induced aortic SMC apoptosis.
Discussion
The initial purpose of the present study was to determine the influence of exogenous VASH2 on AngII-induced ascending aortic dilation. The study showed that overexpression of VASH2 exacerbated AngII-induced ascending aortic dilation. In contrast to the primary hypothesis, the underlying mechanism consisted not of VASH2-induced angiogenesis but instead, VASH2-enhanced vascular SMC apoptosis and elastin degradation.
VASH2 has been shown to promote angiogenesis during tumor development.21,31,33
In the present study, however, there was no evidence of neovascularization with VASH2 under AngII infusion in the aortas. Interestingly, in the present study VASH2 overexpression was strongly associated with aortic medial SMC apoptosis.
Apoptosis or premature senescence of vascular SMC has been shown via p53- or p21-related pathways stimulated by AngII.38,39
And in the present study, increased protein abundance of p53 and p21 was observed in the vascular wall of the ascending aorta in the AngII+VASH2 group. A similar result was also observed in cultured human aortic SMC transfected with VASH2 and AngII. SMC apoptosis occurs in many arterial diseases, including atherosclerosis, angioplasty restenosis, and abdominal aneurysm formation.40,41
Pan caspase inhibitor has also been shown to prevent the initiation of AngII-induced aortic aneurysm formation in mice.42
In addition, chronic apoptosis of SMC accelerates atherosclerosis and promotes calcification and medial degeneration.43
Furthermore, p21-dependent premature senescence of SMC accelerates development of atherosclerosis.39
Thus, it is likely that SMC apoptosis is closely associated with several vascular pathologies. In the current study, VASH2-enhanced cell apoptosis was observed in ascending aorta expansion in AngII-induced mice, which might lead to loss of SMC and inter-lamellar hemorrhage. Indeed, medial erythrocytes in ascending aortic aneurysm were identified in the AngII-induced ascending aortic dilation in a mouse study.14,15
Consistent with the present findings, loss of SMC of ascending aortic aneurysm in humans has been observed.14
Given these observations, it is suggested that overexpression of VASH2 enhanced AngII-induced ascending aortic dilation via SMC apoptosis.
Several studies have reported that TGF-β cascade and MMP are associated with ascending aortic dilation.44,45
In the current study, overexpression of VASH2 enhanced both the latent form of MMP-2 and the active form of MMP-2. In support of this, activation of MMP-2 and MMP-9 has been shown to promote elastin degradation in aortic aneurysm formation.46
Thus, the current observations suggest that VASH2 may be a potential trigger of matrix degradation in the ascending aorta via MMP activation.
We also examined ex vivo outer diameter of abdominal aortic aneurysm (AAA;
Supplementary Figure 4). No significant differences were detected between the AngII-infused groups. The incidence of AAA, however, was extremely low in both groups, as reported in previous studies using normocholesterolemic mice.29
In the current study, we used normocholesterolemic mice in order to examine the effect of VASH2 on AngII-induced ascending aortic dilation. AngII infusion reproducibly generated a higher incidence of ascending aortic dilation in normocholesterolemic mice, whereas AngII generated a higher incidence of AAA under hypercholesterolemic condition.26,30,47
Further studies are warranted to elucidate the effect of VASH2 on AngII-induced AAA using hypercholesterolemic LDLr−/− or apoE−/− mice.
Conclusions
Exogenous VASH2 significantly exacerbated AngII-induced ascending aortic dilation. VASH2-induced SMC apoptosis may be associated with the acceleration of AngII-induced ascending aortic dilation. Therefore, VASH2 antagonism could be a potential therapeutic target to attenuate AngII-induced ascending aortic dilation. Further studies are warranted, however, to determine the molecular mechanisms by which VASH2 promotes AngII-induced SMC apoptosis and contributes to the development of AngII-induced vascular pathologies.
Acknowledgments
The authors thank Dr. Hisashi Sawada for his useful comments in the measurement of ascending aorta width.
Funding
This work was supported by Grant-in-Aid for Scientific Research C from The Ministry of Education, Culture, Sports, Science and Technology, Japan and by the National Institutes of Health (Grant R01HL130086).
Disclosures
The authors declare no conflicts of interest.
Supplementary Files
Please find supplementary file(s);
http://dx.doi.org/10.1253/circrep.CR-19-0008
References
- 1.
Guidelines for diagnosis and treatment of aortic aneurysm and aortic dissection (JCS 2006). J Cardiol 2007; 50: 547–577.
- 2.
Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE Jr, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation 2010; 121: e266–e369.
- 3.
JCS Joint Working Group. Guidelines for diagnosis and treatment of aortic aneurysm and aortic dissection (JCS 2011): Digest version. Circ J 2013; 77: 789–828.
- 4.
Masuda M, Okumura M, Doki Y, Endo S, Hirata Y, Kobayashi J, et al. Thoracic and cardiovascular surgery in Japan during 2014: Annual report by The Japanese Association for Thoracic Surgery. Gen Thorac Cardiovasc Surg 2016; 64: 665–697.
- 5.
Kanaoka Y, Ohki T, Toya N, Ishida A, Tachihara H, Hirayama S, et al. Technical challenges in endovascular repair of complex thoracic aortic aneurysms. Ann Vasc Dis 2012; 5: 21–29.
- 6.
Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312: 117–121.
- 7.
Pannu H, Tran-Fadulu V, Papke CL, Scherer S, Liu Y, Presley C, et al. MYH11 mutations result in a distinct vascular pathology driven by insulin-like growth factor 1 and angiotensin II. Hum Mol Genet 2007; 16: 2453–2462.
- 8.
Brooke BS, Habashi JP, Judge DP, Patel N, Loeys B, Dietz HC 3rd. Angiotensin II blockade and aortic-root dilation in Marfan’s syndrome. N Engl J Med 2008; 358: 2787–2795.
- 9.
Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: Attenuation by CCR2 deficiency in apoE−/− mice. Clin Sci (Lond) 2010; 118: 681–689.
- 10.
Rateri DL, Moorleghen JJ, Balakrishnan A, Owens AP 3rd, Howatt DA, Subramanian V, et al. Endothelial cell-specific deficiency of Ang II type 1a receptors attenuates Ang II-induced ascending aortic aneurysms in LDL receptor−/− mice. Circ Res 2011; 108: 574–581.
- 11.
Moltzer E, Essers J, van Esch JH, Roos-Hesselink JW, Danser AH. The role of the renin-angiotensin system in thoracic aortic aneurysms: Clinical implications. Pharmacol Ther 2011; 131: 50–60.
- 12.
Bellini C, Korneva A, Zilberberg L, Ramirez F, Rifkin DB, Humphrey JD. Differential ascending and descending aortic mechanics parallel aneurysmal propensity in a mouse model of Marfan syndrome. J Biomech 2016; 49: 2383–2389.
- 13.
Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: Insights from predisposing genes and mouse models. Annu Rev Med 2017; 68: 51–67.
- 14.
Rateri DL, Davis FM, Balakrishnan A, Howatt DA, Moorleghen JJ, O’Connor WN, et al. Angiotensin II induces region-specific medial disruption during evolution of ascending aortic aneurysms. Am J Pathol 2014; 184: 2586–2595.
- 15.
Trachet B, Piersigilli A, Fraga-Silva RA, Aslanidou L, Sordet-Dessimoz J, Astolfo A, et al. Ascending aortic aneurysm in angiotensin II-infused mice: Formation, progression, and the role of focal dissections. Arterioscler Thromb Vasc Biol 2016; 36: 673–681.
- 16.
Watanabe K, Hasegawa Y, Yamashita H, Shimizu K, Ding Y, Abe M, et al. Vasohibin as an endothelium-derived negative feedback regulator of angiogenesis. J Clin Invest 2004; 114: 898–907.
- 17.
Sato Y, Sonoda H. The vasohibin family: A negative regulatory system of angiogenesis genetically programmed in endothelial cells. Arterioscler Thromb Vasc Biol 2007; 27: 37–41.
- 18.
Kimura H, Miyashita H, Suzuki Y, Kobayashi M, Watanabe K, Sonoda H, et al. Distinctive localization and opposed roles of vasohibin-1 and vasohibin-2 in the regulation of angiogenesis. Blood 2009; 113: 4810–4818.
- 19.
Sato Y. The vasohibin family: Novel regulators of angiogenesis. Vascul Pharmacol 2012; 56: 262–266.
- 20.
Sato Y. The vasohibin family: A novel family for angiogenesis regulation. J Biochem 2013; 153: 5–11.
- 21.
Koyanagi T, Saga Y, Takahashi Y, Suzuki Y, Suzuki M, Sato Y. Downregulation of vasohibin-2, a novel angiogenesis regulator, suppresses tumor growth by inhibiting angiogenesis in endometrial cancer cells. Oncol Lett 2013; 5: 1058–1062.
- 22.
Koyanagi T, Suzuki Y, Saga Y, Machida S, Takei Y, Fujiwara H, et al. In vivo delivery of siRNA targeting vasohibin-2 decreases tumor angiogenesis and suppresses tumor growth in ovarian cancer. Cancer Sci 2013; 104: 1705–1710.
- 23.
Koyanagi T, Suzuki Y, Komori K, Saga Y, Matsubara S, Fujiwara H, et al. Targeting human vasohibin-2 by a neutralizing monoclonal antibody for anti-cancer treatment. Cancer Sci 2016; 108: 512–519.
- 24.
Norita R, Suzuki Y, Furutani Y, Takahashi K, Yoshimatsu Y, Podyma-Inoue KA, et al. Vasohibin-2 is required for epithelial-mesenchymal transition of ovarian cancer cells by modulating TGF-beta signaling. Cancer Sci 2017; 108: 419–426.
- 25.
Nasu T, Maeshima Y, Kinomura M, Hirokoshi-Kawahara K, Tanabe K, Sugiyama H, et al. Vasohibin-1, a negative feedback regulator of angiogenesis, ameliorates renal alterations in a mouse model of diabetic nephropathy. Diabetes 2009; 58: 2365–2375.
- 26.
Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000; 105: 1605–1612.
- 27.
Umebayashi R, Uchida HA, Kakio Y, Subramanian V, Daugherty A, Wada J. Cilostazol attenuates angiotensin II-induced abdominal aortic aneurysms but not atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2018; 38: 903–912.
- 28.
Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol 2007; 27: 380–386.
- 29.
Uchida HA, Poduri A, Subramanian V, Cassis LA, Daugherty A. Urokinase-type plasminogen activator deficiency in bone marrow-derived cells augments rupture of angiotensin II-induced abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol 2011; 31: 2845–2852.
- 30.
Uchida HA, Kristo F, Rateri DL, Lu H, Charnigo R, Cassis LA, et al. Total lymphocyte deficiency attenuates AngII-induced atherosclerosis in males but not abdominal aortic aneurysms in apoE deficient mice. Atherosclerosis 2010; 211: 399–403.
- 31.
Takahashi Y, Koyanagi T, Suzuki Y, Saga Y, Kanomata N, Moriya T, et al. Vasohibin-2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol Cancer Res 2012; 10: 1135–1146.
- 32.
Subramanian V, Uchida HA, Ijaz T, Moorleghen JJ, Howatt DA, Balakrishnan A. Calpain inhibition attenuates angiotensin II-induced abdominal aortic aneurysms and atherosclerosis in low-density lipoprotein receptor-deficient mice. J Cardiovasc Pharmacol 2012; 59: 66–76.
- 33.
Kitahara S, Suzuki Y, Morishima M, Yoshii A, Kikuta S, Shimizu K, et al. Vasohibin-2 modulates tumor onset in the gastrointestinal tract by normalizing tumor angiogenesis. Mol Cancer 2014; 13: 99.
- 34.
Masuda K, Tanabe K, Ujike H, Hinamoto N, Miyake H, Tanimura S, et al. Deletion of pro-angiogenic factor vasohibin-2 ameliorates glomerular alterations in a mouse diabetic nephropathy model. PLoS One 2018; 13: e0195779.
- 35.
Daugherty A, Rateri DL, Lu H, Inagami T, Cassis LA. Hypercholesterolemia stimulates angiotensin peptide synthesis and contributes to atherosclerosis through the AT1A receptor. Circulation 2004; 110: 3849–3857.
- 36.
Haas TL, Davis SJ, Madri JA. Three-dimensional type I collagen lattices induce coordinate expression of matrix metalloproteinases MT1-MMP and MMP-2 in microvascular endothelial cells. J Biol Chem 1998; 273: 3604–3610.
- 37.
Jimenez E, Perez de la Blanca E, Urso L, Gonzalez I, Salas J, Montiel M. Angiotensin II induces MMP 2 activity via FAK/JNK pathway in human endothelial cells. Biochem Biophys Res Commun 2009; 380: 769–774.
- 38.
Dimmeler S, Zeiher AM. Reactive oxygen species and vascular cell apoptosis in response to angiotensin II and pro-atherosclerotic factors. Regul Pept 2000; 90: 19–25.
- 39.
Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, et al. Angiotensin II induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a p21-dependent pathway. Circulation 2006; 114: 953–960.
- 40.
Lopez-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol 1997; 150: 993–1007.
- 41.
Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis 1997; 8: 623–631.
- 42.
Yamanouchi D, Morgan S, Kato K, Lengfeld J, Zhang F, Liu B. Effects of caspase inhibitor on angiotensin II-induced abdominal aortic aneurysm in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2010; 30: 702–707.
- 43.
Clarke MC, Littlewood TD, Figg N, Maguire JJ, Davenport AP, Goddard M, et al. Chronic apoptosis of vascular smooth muscle cells accelerates atherosclerosis and promotes calcification and medial degeneration. Circ Res 2008; 102: 1529–1538.
- 44.
Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: At the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ Res 2013; 113: 327–340.
- 45.
Nagasawa A, Yoshimura K, Suzuki R, Mikamo A, Yamashita O, Ikeda Y, et al. Important role of the angiotensin II pathway in producing matrix metalloproteinase-9 in human thoracic aortic aneurysms. J Surg Res 2013; 183: 472–477.
- 46.
Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest 2002; 110: 625–632.
- 47.
Lu H, Howatt DA, Balakrishnan A, Graham MJ, Mullick AE, Daugherty A. Hypercholesterolemia induced by a PCSK9 gain-of-function mutation augments angiotensin II-induced abdominal aortic aneurysms in C57BL/6 mice: Brief report. Arterioscler Thromb Vasc Biol 2016; 36: 1753–1757.