Introduction
Membrane traffic between subcellular compartments is mediated by vesicular and tubular intermediates. Key regulators of these compartments’ formation are the ADP-ribosylation factor (ARF) family of small GTPases, which trigger budding of coated carrier vesicles by recruiting coat protein complexes and regulating phospholipid metabolism (D’Souza-Schorey and Chavrier, 2006). Like all other GTPases, ARFs cycle between a GDP-bound inactive state and a GTP-bound active state. Binding of GTP induces ARFs to undergo conformational changes that allow them to interact with a wide variety of effector proteins; these effectors include coat proteins, such as the COPI complex; the clathrin adaptor protein complexes and monomeric GGA proteins; and other proteins, such as arfaptins and arfophilins/Rab11-family interacting proteins.
ARF GTPases also interact with lipid modifying enzymes, including phospholipase D (PLD), phosphatidylinositol 4-kinase, and PtdIns(4)P 5-kinase. ARFs can stimulate PLD, an enzyme that hydrolyses phosphatidylcholine to form phosphatidic acid (PA) (Cockcroft, 2009), and thereby modifies membrane lipid composition (Brown et al., 1993; Cockcroft et al., 1994). This activity has been implicated in recycling of MHC class I molecules (Jovanovic et al., 2006), especially in the case of PLD2, which is involved in recycling of transferrin (Tfn) (Padron et al., 2006).
ARFs can be divided into three classes based on amino acid sequence similarity: class I contains ARF1–ARF3 (humans lack ARF2); class II comprises ARF4 and ARF5; and class III contains only a single protein, ARF6 (Kahn et al., 2006). Among these proteins, ARF1 and ARF6 have been the most thoroughly characterized (Gillingham and Munro, 2007). ARF1 mainly localizes to the Golgi apparatus, and is responsible for recruitment of COPI coat protein complexes from the cytosol to the Golgi membranes. ARF1 also regulates the recruitment of clathrin adaptor proteins, such as the AP-1 complex and GGA proteins, to the trans-Golgi network (TGN) and endosomes (D’Souza-Schorey and Chavrier, 2006). Furthermore, ARF1 is activated at the plasma membrane, where it is involved in dynamin-independent endocytosis (Beemiller et al., 2006; Cohen et al., 2007; Kumari and Mayor, 2008). On the other hand, ARF6 localizes mainly to the plasma membrane and the endosomal system, and regulates endocytic/recycling pathways and remodeling of the actin cytoskeleton (D’Souza-Schorey and Chavrier, 2006).
The exchange of GDP for GTP on ARFs is catalyzed by guanine nucleotide exchange factors (GEFs); the bound GTP is hydrolyzed to generate GDP with the help of GTPase-activating proteins (GAPs) (Gillingham and Munro, 2007; Shin and Nakayama, 2004). All ARF-GEFs identified to date contain a conserved Sec7 catalytic domain. The ARF-GEF family is divided into several subfamilies; these include the brefeldin A (BFA)–sensitive GBF/BIG subfamily, whose members localize to and function at the Golgi apparatus, and can activate class I and class II ARFs (Jackson and Casanova, 2000; Shin and Nakayama, 2004). We and others have shown that BIG2 activates class I ARFs (ARF1 and ARF3), localizes to recycling endosomes as well as the TGN, and is required for the integrity of recycling endosomes (Ishizaki et al., 2008; Shen et al., 2006; Shin et al., 2004). However, it has not yet been clarified whether ARF1 and ARF3 participate in endosome integrity downstream of BIG2, in part because it is not yet known whether these ARFs are associated with endosomal compartments. Here, we show that ARF1 and ARF3 localize to endosomal compartments, and that they are redundantly required for both integrity of recycling endosomes and efficient recycling of Tfn from endosomes to the plasma membrane, but not for retrograde transport from endosomes to the Golgi apparatus.
Materials and Methods
Plasmids
Construction of expression vectors for N-terminally EGFP-tagged Rab11a and Rab4 was described previously (Takahashi et al., 2011; Yamamoto et al., 2010). The EGFP-Rab5 construct was kindly provided by Marino Zerial (Max Planck Institute, Dresden, Germany). Construction of expression vectors for C-terminally HA- or EGFP-tagged ARFs was described previously (Makyio et al., 2012; Takatsu et al., 2002). A cDNA for the ARF1(N52R) mutant was generated by site-directed mutagenesis using a QuikChange Site-Directed Mutagenesis kit (Agilent Technologies) and cloned into pcDNA3-HAC, which includes sequence for a C-terminal HA tag (Hosaka et al., 1996).
Antibodies and reagents
Sources of antibodies used in the present study were as follows. Polyclonal rabbit anti-TGN46 (Kain et al., 1998): a kind gift from Minoru Fukuda (Sanford-Burnham Medical Research Institute); polyclonal rabbit anti-golgin-97 (Yoshimura et al., 2004): a kind gift from Nobuhiro Nakamura (Kyoto Sangyo University); polyclonal rabbit anti-ARF4: Protein Tech; polyclonal rabbit anti-β-COP: ThermoScientific; monoclonal mouse anti-GM130, anti-EEA1, anti-CD4 (Leu3a), and anti-ARF3: BD Biosciences; monoclonal mouse antibodies against γ-adaptin (100.3), and FLAG (M2): Sigma; monoclonal mouse anti-Tfn receptor (TfnR): Zymed Laboratories; monoclonal mouse anti-ARF1: Enzo Life Sciences; monoclonal mouse anti-ARF5: Abnova; monoclonal mouse anti–β-actin and β-tubulin: Millipore; monoclonal rat anti-HA: Roche Applied Science; AlexaFluor-conjugated secondary antibodies: Molecular Probes; horseradish peroxidase-conjugated secondary antibodies: Jackson ImmunoResearch Laboratories. AlexaFluor-conjugated Tfn and epidermal growth factor (EGF) were purchased from Molecular Probes, and nocodazole was from Sigma–Aldrich.
Cell culture, RNAi suppression, immunofluorescence, and time-lapse imaging analysis
Culture of HeLa cells and transfection of expression plasmids were performed as described previously (Shin et al., 2005, 2004). HeLa cell lines stably expressing TfnR-EGFP were established as described previously (Takahashi et al., 2012). Knockdown of ARF1 or ARF3 were performed as described previously (Ishizaki et al., 2008, 2006; Man et al., 2011). Briefly, a pool of siRNAs targeted against the 3′-untranslated region of human ARF1 (PCR-amplified using primers 5′-CTCTCACTCCTCTTGCCCTC-3′ and 5′-GGCTGCCTGAAGGTGGGTAG-3′ for ARF1, or 5′-CGC-AACTCGCTTGTCCTTGG-3′ and 5′-CTAATAGCTATAATTA-CAGTGCTTG-3′ for ARF1′) or ARF3 (PCR-amplified using primers 5′-CAGACAGCCCTAACAAAGCAC-3′ and 5′-CAGA-GAGGAGGGTAACCAGTC-3′ for ARF3, or 5′-TCTGGATGA-TAGGACAGATGATG-3′ and 5′-ATCCATTTATGAGAAGTG-AGTGAG-3′ for ARF3′) was prepared using a BLOCK-iT RNAi TOPO transcription kit and a BLOCK-iT Dicer RNAi kit (Invitrogen). Cells were transfected with the siRNAs using Lipofectamine 2000 (Invitrogen) and incubated for 24 h. The transfected cells were then split and seeded onto culture dishes. After 24 h, the cells were transfected again with the siRNAs and incubated for 48 h. The transfected cells were then transferred to new culture dishes containing coverslips, incubated for another 24 h, and processed for immunofluorescence and immunoblot analyses as described previously (Ishizaki et al., 2008; Nishimoto-Morita et al., 2009; Shin et al., 1997). For time-lapse recording, the cells were incubated in HEPES-buffered modified Eagle’s medium, placed on a microscope stage that had been preincubated at 37°C, and observed using an FV1000D confocal microscope (Olympus). Images were acquired sequentially every 2–7 sec.
Tfn, EGF, and antibody uptake experiments
Assays for internalization and recycling Tfn and internalization of EGF were carried out according to previously described protocols (Shin et al., 2004) with some modifications. HeLa cells were transfected with a pool of siRNAs against LacZ as a control, or with siRNAs against ARF1 and/or ARF3 as described above. At 120 h post-transfection, cells were serum-starved for 3 h in MEM medium containing 0.2% BSA, and then incubated with AlexaFluor488- or AlexaFluor555-conjugated Tfn or AlexaFluor488-conjugated EGF on ice for 50 min. After washing out fluorescent Tfn or EGF with PBS on ice, cells were incubated in medium containing unlabelled holo-Tfn or in normal medium (for Tfn or EGF uptake, respectively) at 37°C for indicated times, and processed for immunofluorescence analysis. Immunofluorescence analysis was performed using an Axiovert 200 MAT microscope (Carl Zeiss) for epifluorescence images, or an LSM Pascal microscope (Carl Zeiss) for confocal images. The fluorescence intensity of Tfn was quantitated using MetaMorph imaging software (Molecular Devices).
Uptake of anti-CD4 or anti-FLAG antibody by cells stably expressing CD4-furin or FLAG-TGN38 was assayed as described previously (Ishizaki et al., 2008; Shin et al., 2005, 2004; Takahashi et al., 1995) with some modifications. Briefly, HeLa cells stably expressing CD4-furin or FLAG-TGN38 were mock-treated with a pool of siRNAs against LacZ or treated with a pool of siRNAs against ARF1 and/or ARF3 for 120 h; prior to antibody uptake, cells were incubated with 15 mM sodium butyrate for 16 h. The cells were then incubated for 60 min with AlexaFluor488-conjugated Tfn and monoclonal anti-CD4 (Leu3a) or anti-FLAG (M2) antibody at 4°C or 19.5°C, respectively; subjected to an acid wash (0.5% acetic acid, pH 3.0, 50 mM NaCl) in the anti-FLAG uptake assay; and finally incubated at 37°C for indicated periods of time.
Results
Depletion of ARF1 and ARF3 does not affect Golgi structure
We previously showed that BIG2 localizes to recycling endosomes as well as the Golgi apparatus, and that its GEF activity is required for the integrity of these compartments (Shin et al., 2004). Because BIG2 has GEF activity toward class I ARFs (ARF1 and ARF3), we asked whether ARF1 and/or ARF3 are implicated in the integrity and function of recycling endosomes. In order to investigate specific functions of ARF1 and ARF3, we knocked down these genes using RNAi. In order to avoid overexpression artifacts, we chose not to overexpress ARF mutants: because not only class I ARFs but also other ARFs share some ARF-GEFs, GAPs, and effector proteins, overexpression of ARF mutants, either in a GDP- or GTP-bound form, may sequester these proteins.
We designed two different pools of siRNAs targeted against non-coding regions of ARF1 and ARF3 mRNAs (see Materials and Methods). HeLa cells were transfected with the siRNA pools against ARF1 or ARF3 alone, or both pools in combination; we confirmed specific and efficient depletion of the ARF1 and ARF3 proteins by immunoblotting (Supplemental Fig. S1A). siRNA treatment did not decrease the amount of ARF4, ARF5, or actin, although ARF5 expression was slightly increased in ARF1-depleted cells (Supplemental Fig. S1A).
We were unable to directly examine depletion of ARF1 and ARF3 by immunofluorescence analysis, because the available antibodies did not work well in immunofluorescence. We therefore identified individual siRNA-transfected cells by co-transfection of an EGFP-encoding plasmid along with siRNAs against ARF1 and/or ARF3 (Fig. 1A insets). Transfection of siRNAs against ARF1 or ARF3 alone, or both genes in combination, did not significantly alter the distribution of the cis-Golgi marker GM130 (Fig. 1Ab–b‴) or the TGN marker TGN46 (Fig. 1Aa–a‴), indicating that depletion of class I ARFs does not significantly affect Golgi structure. Although depletion of ARF1 and ARF3 causes a slight increase of β-COP (a subunit of the COPI complex)-positive puncta in cytoplasm (Volpicelli-Daley et al., 2005), it does not affect the Golgi localization of β-COP (Fig. 1Ac–c‴). Therefore, depletion of class I ARFs does not disrupt the recruitment of β-COP to the Golgi complex.

We next examined whether endosomal compartments are morphologically affected by knockdown of ARF1 and/or ARF3, using several endosomal markers. Simultaneous knockdown of ARF1 and ARF3 induces extensive tubulation of TfnR-positive early/recycling endosomes (Fig. 1Af‴); although TfnR-positive endosomes were tubulated to some extent in cells depleted of ARF1 alone (f′), the tubular structures were much more prominent in cells double knockdowns (f‴). In contrast, distribution of neither early endosomal EEA1 (d–d‴) nor late endosomal Lamp-1 (e–e‴) was significantly altered in single or double-knockdown cells. Treatment of cells with independently derived pools of siRNAs against ARF1 and ARF3 resulted in the same phenotypes (Supplementary Fig. S1B and C), confirming that tubulation of TfnR-positive endosomes is a specific phenotype induced by depletion of ARF1 and ARF3. These observations together indicate that ARF1 and ARF3 play redundant roles in the integrity of TfnR-positive endosomes, but not early or late endosomes.
To confirm that these tubular structures represent endocytic compartments, we monitored changes in the distribution of endocytosed Tfn. To this end, control cells or cells doubly depleted of ARF1 and ARF3 were incubated at 37°C for various time periods in the continuous presence of AlexaFluor488-conjugated Tfn (Fig. 1B). After 5 min incubation, when most Tfn had reached the early endosomes (Maxfield and McGraw, 2004; Sonnichsen et al., 2000), Tfn was observed in punctate structures in both control and double-knockdown cells, but tubulation of Tfn-positive structures was not evident in the double-knockdown cells. However, after longer incubation periods (30 and 60 min), when Tfn had reached the recycling endosomes (Maxfield and McGraw, 2004; Sonnichsen et al., 2000), endocytosed Tfn was observed in tubulated structures in the double-knockdown cells, indicating that the tubular structures are generated from recycling endosomes rather from early endosomes. These observations are in agreement with a previous report showing that in cells depleted of ARF1 and ARF3 by shRNA treatment, Tfn internalized for 60 min is present in tubular structures; those authors did not investigate earlier time points of internalization (Volpicelli-Daley et al., 2005).
We next examined dynamics of the Tfn/TfnR-positive tubular structures by time-lapse imaging. When TfnR-EGFP–expressing cells were incubated with AlexaFluor555-conjugated Tfn at 37°C for 30 min, endocytosed Tfn was extensively colocalized with TfnR-EGFP in both control cells and the cells doubly depleted of ARF1 and ARF3 (Fig. 1C), indicating that endocytosed Tfn is accessible to the TfnR-positive tubular structures induced by depletion of ARF1 and ARF3. In control cells, a number of vesicular and short tubular structures positive for Tfn and TfnR could be observed moving around the cytoplasm (Fig. 1C, upper panels; and supplemental Video S1). In striking contrast, in the double-knockdown cells, endocytosed Tfn and TfnR were observed in prominent tubular structures that extended and retracted intermittently (Fig. 1C, lower panels; and supplemental Video S2). Thus, the Tfn-TfnR–positive tubular compartments are dynamic rather than static structures.
To confirm that the endosome tubulation specifically resulted from depletion of ARF1 and ARF3 rather than an off-target effect, we performed rescue experiments. When C-terminally EGFP-tagged ARF1 or ARF3 was exogenously expressed in double-knockdown cells, the tubular compartments containing endocytosed Tfn were almost completely restored to punctate structures (Fig. 2Ab and c, asterisks, and B). In contrast, the tubulation phenotype could not be restored by exogenous expression of EGFP or ARF6-EGFP (Fig. 2Aa and d, asterisks, and B). These observations confirm that the siRNA-mediated depletion of class I ARFs leads to endosome tubulation, as well as showing that ARF1 and ARF3 play redundant roles in the integrity of recycling endosomes.
TfnR-containing tubular compartments are positive for Rab4 and Rab11 but not Rab5
We next asked which endosomal Rab proteins localize to the tubular structures induced by depletion of ARF1 and ARF3. As shown in Fig. 3, Rab4 and Rab11, which are localized respectively to early/recycling endosomes and recycling endosomes, were observed on the tubular structures induced by the ARF1/ARF3 double knockdown. In contrast, early endosomal Rab5 was not associated with the tubules. Taken together, these observations indicate that simultaneous depletion of ARF1 and ARF3 induces tubules from Rab4- and/or Rab11-positive endosomes, but not from the Rab5-positive early endosomal compartments. These results are consistent with those of our previous study showing that overexpression of a BIG2 dominant negative mutant induces tubulation of Rab4- and Rab11-positive compartments (Shin et al., 2004).
TfnR-positive tubular structures are formed in a microtubule-dependent manner
Because the tubular structures induced by depletion of ARF1 and ARF3 are dynamically extended and retracted, as shown in supplemental Video S2, we next asked whether formation of the tubular structures is dependent on microtubules. To this end, we examined the morphological changes of endosomes after treatment of control and double-knockdown cells with nocodazole for various intervals. In the absence of nocodazole, TfnR-positive tubular structures induced by depletion of ARF1 and ARF3 were extended along the microtubules (Fig. 4B (–), insets). After 5 min nocodazole treatment, both the microtubule network and TfnR-positive tubular structures could be observed, but some were shortened (Fig. 4A and B, 5 min panels); some TfnR-positive tubules remained aligned along the microtubules (Fig. 4B, 5 min, insets). After longer treatments, microtubules were gradually disrupted, and no TfnR-positive tubules were observed (Fig. 4A and B, 10–60 min). After 60 min or more treatment, almost all the TfnR-positive endosomes were evenly distributed as cytoplasmic puncta in both control and double-knockdown cells (Fig. 4A and B, 60 min). These observations suggest that the tubular endosomal structures induced by depletion of ARF1 and ARF3 are extended in a microtubule-dependent manner, and that neither ARF1 nor ARF3 is required for association of endosomes with microtubules.

We next asked whether ARF1 and ARF3 are localized to endosomes, since there has been no report unequivocally demonstrating endosomal localization of class I ARFs. This lack of information regarding their endosomal localization is due, in part, to lack of specific antibodies capable of detecting endogenous ARF1 and ARF3 as well as to the predominantly cytosolic distributions of exogenously expressed ARF1 and ARF3 as detected by immunofluorescence analysis. We first attempted to overcome this problem by extracting cytosolic proteins prior to fixation of cells expressing ARF1-EGFP or ARF3-EGFP, but failed to detect any distinct endosomal signals (data not shown). We then observed ARF1-EGFP or ARF3-EGFP in living cells. As shown in Fig. 5A and supplemental Videos S3 and S4, we could observe distinct Golgi localization of ARF1-EGFP and ARF3-EGFP. On the other hand, we could only very faintly detect punctate structures positive for ARF1-EGFP and ARF3-EGFP, which were superimposed on punctate signals of endocytosed AlexaFluor555-Tfn. During the course of the aforementioned rescue experiments, however, we noticed that in ARF-knockdown cells, punctate signals of ARF1-EGFP and ARF3-EGFP were more evident than in control cells. When ARF1-EGFP was expressed in cells depleted of ARF1, we detected both perinuclear signals representing the Golgi apparatus and punctate signals of ARF1-EGFP distributed throughout the cytoplasm, many of which overlapped with endocytosed Tfn (compare Fig. 5B, top panels, and supplemental Video S5, with Fig. 5A, upper panels, and supplemental Video S3). This observation can be explained by assuming that endogenous ARF1 competes with exogenously expressed ARF1-EGFP for a limited number of binding sites on endo-somal membranes. On the other hand, endosomal localization of ARF3-EGFP could be visualized in cells depleted of both ARF1 and ARF3 (Fig. 5B, middle panels, and supplemental Video S6), but not those depleted of ARF3 alone (data not shown). Moreover, ARF3(FF)-EGFP, in which Leu9 and Ile13 within the N-terminal amphipathic helix are replaced with Phe residues that occupy the corresponding positions in ARF1, could be detected on Tfn-positive structures in ARF3-knockdown cells (Fig. 5B, bottom panels, and supplemental Video S7). In light of a recent report showing that ARF3(FF)-EGFP is recruited onto membranes more efficiently than ARF3-EGFP (Manolea et al., 2010), it is likely that ARF1 binds more effectively than ARF3 to endosomal membranes, due to the bulky aromatic amino acids in the N-terminal amphipathic helix; however, the precise molecular details remain unknown. Overall, these observations indicate that some populations of ARF1 and ARF3 are associated with membranes of Tfn-positive endosomes as well as with Golgi membranes (see Discussion).

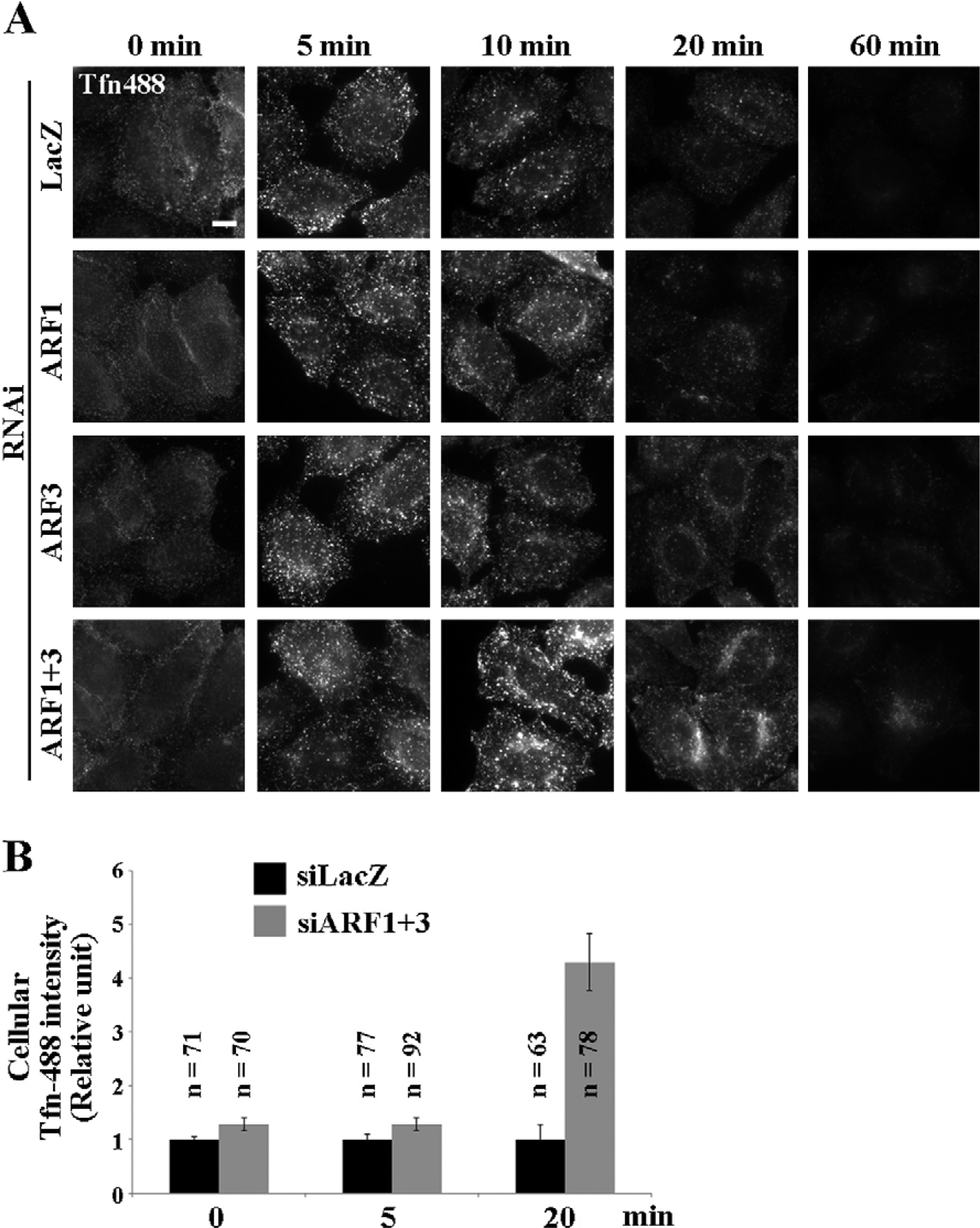
Volpicelli-Daley et al. previously reported that treatment of cells with shRNAs for ARF1 and ARF3 inhibited Tfn recycling by monitoring Tfn internalization over a 60 min time course (Volpicelli-Daley et al., 2005). In our double-knockdown cells, however, Tfn signals in the perinuclear region after 60 min of internalization were stronger than in control cells (Fig. 1B, 60 min), suggesting that at this time-point Tfn had already accumulated within the cell at higher levels, possibly due to the inhibition of recycling. To examine the recycling of Tfn more quantitatively, control and knockdown cells were incubated with AlexaFluor488-conjugated Tfn at 4°C for 50 min, washed to remove unbound Tfn, and then allowed to internalize Tfn at 37°C for various time periods. Internalization of Tfn was not significantly affected by knockdown of ARF1, ARF3, or both genes simultaneously (Fig. 6A and B, 5 min, and Fig. 1B). In control cells, Tfn signals on punctate intra-cellular structures were markedly decreased after 20 min incubation at 37°C, due to release of internalized Tfn to the extracellular medium. In considerable contrast, a significant level of Tfn remained inside cells doubly depleted of ARF1 and ARF3 (Fig. 6A and B, 20 min). After 60 min, Tfn disappeared almost completely from control cells and single-knockdown cells, whereas some Tfn was still retained within double-knockdown cells. Thus, recycling of Tfn is considerably delayed, although not completely blocked, by the simultaneous depletion of ARF1 and ARF3.
The phenotypes of cells doubly depleted of ARF1 and ARF3, namely, tubulation of endosomal compartments and slowing of Tfn recycling, are reminiscent of previously reported phenotypes in PLD2-knockdown cells (Padron et al., 2006). ARF1 and ARF3 were purified as cytosolic factors that stimulate PLD activity (Cockcroft et al., 1994; Sung et al., 1999); PA, a cone-shaped lipid produced through hydrolysis of phosphatidylcholine catalyzed by PLD, favors membranes with negative curvature and thus may affect vesicle budding and membrane fission (Donaldson, 2009). We therefore were interested in whether PLD is implicated in the phenomena observed in cells doubly depleted of ARF1 and ARF3. To examine this, we exploited an ARF1(N52R) mutant. ARF1(N52R) and ARF6(N48I) lack the ability to stimulate PLD, but retain the ability to stimulate PtdIns4P 5-kinase (Jones et al., 1999; Skippen et al., 2002). Given that ARF1(N52R) binds GTPγS as efficiently as ARF1(WT) (Jones et al., 1999), and that ARF6(N48I) can be activated by ARNO (an ARF-GEF) and inactivated by GIT1 (an ARF-GAP) (Vitale et al., 2002), the ARF mutants seem to function normally, except for their defect in PLD activation. Unlike ARF1(WT), ARF1(N52R) did not rescue the tubulation of Tfn-positive endosomes induced by the knockdown of ARF1 and ARF3 (Fig. 2Ae′ and B), suggesting that PLD activity participates in the integrity of recycling endosomes regulated by class I ARFs.
Depletion of ARF1 and ARF3 does not affect retrograde transport from endosomes to the Golgi complex or lysosomal transport
Rat TGN38 and its human orthologue, TGN46, cycle between the plasma membrane and the TGN at a very low rate (Ladinsky and Howell, 1993; Reaves et al., 1993), although at steady state these proteins localize mainly to the TGN. During retrograde transport from the plasma membrane to the TGN, TGN38/TGN46 passes through early/recycling endosomes (Ghosh et al., 1998; Mallet and Maxfield, 1999; Maxfield and McGraw, 2004). We therefore asked whether depletion of ARF1 and ARF3 affects retrograde transport of TGN38 from early/recycling endosomes to the TGN. To this end, we performed an antibody uptake experiment using a HeLa-derived cell line stably expressing exoplasmically FLAG-tagged TGN38. At steady state, FLAG-TGN38 colocalized with golgin-97; its Golgi localization was not significantly changed by the simultaneous knockdown of ARF1 and ARF3 (Fig. 7A). This observation is consistent with the Fig. 1A, which shows no significant difference in Golgi structure (as visualized by staining for GM130 and TGN46) between control and double-knockdown cells. We next analyzed the retrograde transport of FLAG-TGN38 by following extracellularly applied anti-FLAG antibody (Fig. 7B and C). FLAG-TGN38–expressing HeLa cells treated with control siRNAs (LacZ) or siRNAs against ARF1 and ARF3 were incubated with anti-FLAG antibody for 50 min at 19.5°C to allow accumulation of internalized FLAG-TGN38, mainly in early endosomes; after an acid wash to remove cell surface–bound antibodies, internalization was allowed to proceed at 37°C for various time periods. In both control and double-knockdown cells, anti-FLAG antibody was observed mainly on peripheral punctate structures at 0 min (Fig. 7B). After 15 min at 37°C, we could detect partial colocalization of internalized FLAG-TGN38 and golgin-97 (Fig. 7B, 15 min); after 60 min, the majority of FLAG-TGN38 had been transported to the TGN, and colocalized extensively with golgin-97 (B, 60 min) in both control and double-knockdown cells; semi-quantitative analysis of retrograde transport of FLAG-TGN38 to the Golgi revealed no significant difference between the control and double-knockdown cells (Fig. 7C). Therefore, ARF1 and ARF3 are dispensable for retrograde transport of TGN38 from early/recycling endosomes to the TGN.

Next we asked whether ARF1 and ARF3 are involved in another retrograde pathway from late endosomes to the TGN. To this end, we made use of a cell line stably expressing a CD4-furin chimera (Mallet and Maxfield, 1999; Maxfield and McGraw, 2004: Ishizaki et al., 2008 #2991; Schapiro et al., 2004). At steady state, CD4-furin colocalized with golgin-97 in both control and double-knockdown cells (Fig. 7D). We analyzed retrograde transport of CD4-furin by following extracellularly applied anti-CD4 antibody (Fig. 7E); CD4-furin-expressing cells treated with LacZ siRNAs (control) or siRNAs against ARF1 and ARF3 were incubated with anti-CD4 antibody for 50 min at 4°C; after washing with PBS, internalization was allowed to proceed at 37°C for various time periods. Immunofluorescence and subsequent semi-quantitative analysis did not reveal any significant difference over the course of incubation between the control and double-knockdown cells (Fig. 7E and F). After 60 min incubation, most CD4-furin had reached the golgin-97–positive Golgi structures (Fig. 7F, black bars).
We also examined the effects of the ARF1+ARF3 knockdown on the lysosomal transport of endocytosed EGF, but did not detect any significant difference between the control and double-knockdown cells (Fig. S2A). These observations together suggest that ARF1 and ARF3 are not required for retrograde transport from early/recycling endosomes or late endosomes to the TGN or lysosomal transport.
Discussion
We have shown that ARF1 and ARF3 localize to TfnR-positive endosomes, and play redundant roles in the integrity of these endosomal compartments, which are positive for Rab4 and Rab11 but not for Rab5. Moreover, we have demonstrated that ARF1 and ARF3 are required for efficient recycling of Tfn through endosomes, but not retrograde transport from endosomes to the Golgi apparatus.
Although earlier studies have reported that exogenously expressed ARF1 and ARF3 are mainly localized to the Golgi as well as distributed throughout the cytoplasm (Fig. 5A) (Cavenagh et al., 1996; Kawamoto et al., 2002; Manolea et al., 2010), here we have used time-lapse imaging to demonstrate the endosomal localization of ARF1-EGFP and ARF3-EGFP exogenously expressed in cells depleted of endogenous ARF1 and/or ARF3. We believe that some populations of ARF1 and ARF3 localize to TfnR-positive endosomes as well as to the Golgi; that their membrane association is saturable; and that membrane association of ARF1 is more stable than that of ARF3, on the basis of the following observations: (i) Endosomal localization of ARF1-EGFP is detectable in ARF1-depleted cells; (ii) endosomal localization of ARF3-EGFP is observed in cells depleted of both ARF1 and ARF3, but not in cells depleted of ARF3 alone; and (iii) endosomal localization of the ARF3(FF)-EGFP mutant, which has a larger hydrophobic surface and is less temperature-sensitive than ARF3-WT (Manolea et al., 2010), is detectable in ARF3-depleted cells.
ARF1 is required for the COPI coat recruitment to the budding vesicles in early secretory pathway (D’Souza-Schorey and Chavrier, 2006). Our study shows that depletion of class I ARFs does not abolish the recruitment of β-COP and affect Golgi structure (Fig. 1A). We think that other ARFs as well as class I ARFs may be redundantly implicated in the recruitment of β-COP and Golgi organization, though incomplete knockdown of ARF1 and ARF3 cannot be excluded.
Although internalized TGN38 and Tfn were colocalized to the tubular endosomes (Supplemental Fig. S2B), TGN38 was normally transported from these compartments to the Golgi complex despite the perturbation of Tfn recycling. Thus, morphological changes of endosomes induced by depletion of ARF1 and ARF3 do not appear to cause general endosome dysfunction; we conclude that ARF1 and ARF3 are specifically involved in the Tfn recycling pathway.
We previously showed that loss of function of BIG2, induced via RNAi or overexpression of the BIG2 dominant-negative mutant BIG2(E738K), induces tubulation of recycling endosomal compartments; however, neither BIG2 depletion nor BIG2(E738K) overexpression inhibits Tfn recycling (Ishizaki et al., 2008; Shin et al., 2004). Furthermore, treatment of cells with BFA, which inhibits the GBF/BIG subfamily of ARF-GEFs, induces tubulation of Rab4- and Rab11-positive recycling endosomal compartments, but does not inhibit Tfn recycling (Damke et al., 1991; Lippincott-Schwartz et al., 1991; Sonnichsen et al., 2000; Wood and Brown, 1992). Given that BIG2 activates ARF1 and ARF3, these data appear to be inconsistent with our observations that depletion of ARF1 and ARF3 induces tubulation of Rab4- and Rab11-positive endosomes (Fig. 2) and inhibits recycling of Tfn from recycling endosomes (Fig. 6). A simple explanation for this apparent contradiction is that BIG2 depletion, BIG2(E738K) overexpression, and BFA treatment do not completely inhibit activation of ARF1 and ARF3, whose activity required for Tfn recycling, although any of these treatments is sufficient to impair the integrity of recycling endosomes. BFA-insensitive ARF-GEFs do exist, e.g. ARNO, which activates ARF1 as well as ARF6 (Cohen et al., 2007); therefore, some populations of ARF1 may be activated by such BFA-insensitive ARF-GEFs on endosomal membranes, and participate in Tfn recycling even under conditions that perturb BFA-sensitive ARF-GEFs. PLD hydrolyzes phosphatidylcholine to generate PA, which is a cone-shaped lipid that favors membranes with negative curvature and can affect vesicle budding and membrane fission (Donaldson, 2009). The enzymatic activity of PLD is stimulated by ARFs (Cockcroft et al., 1994; Sung et al., 1999); PLD also interacts with dynamin and stimulates its GTPase activity (Lee et al., 2006). Thus, PLD can participate in vesicle budding and membrane fission by producing PA, as well as by stimulating the GTPase activity of dynamin. Because tubulation of recycling endosomes upon depletion of ARF1 and ARF3 cannot be rescued by exogenous expression of the ARF1(N52R) mutant, which lacks the ability to stimulate PLD (Fig. 4), ARF1 and ARF3 may influence vesicle budding or membrane fission from recycling endosomes by regulating PLD activity; therefore, depletion of ARF1 and ARF3 results in membrane tubulation. Furthermore, depletion of PLD2 inhibits recycling of TfnR from recycling endosomes, but does not appear to affect endocytosis (Padron et al., 2006). Taken together, these observations indicate that ARF1 and ARF3 are engaged in endosomal integrity and Tfn recycling, in part by regulating PLD activity. The molecular mechanisms by which ARF1 and ARF3 selectively regulate the Tfn recycling pathway is an issue to be addressed in future studies.


