CxxC-ZF Domain Is Needed for KDM2A to Demethylate Histone in rDNA Promoter in Response to Starvation
2014 Volume 39 Issue 1 Pages 79-92

Details
2014 Volume 39 Issue 1 Pages 79-92
The transcription of ribosomal RNA genes (rDNA) is a rate-limiting step in ribosome biogenesis and changes profoundly in response to environmental conditions. Recently we reported that JmjC demethylase KDM2A reduces rDNA transcription on starvation, with accompanying demethylation of dimethylated Lys 36 of histone H3 (H3K36me2) in rDNA promoter. Here, we characterized the functions of two domains of KDM2A, JmjC and CxxC-ZF domains. After knockdown of endogenous KDM2A, KDM2A was exogenously expressed. The exogenous wild-type KDM2A demethylated H3K36me2 in the rDNA promoter on starvation and reduced rDNA transcription as endogenous KDM2A. The exogenous KDM2A with a mutation in the JmjC domain lost the demethylase activity and did not reduce rDNA transcription on starvation, showing that the demethylase activity of KDM2A itself is required for the control of rDNA transcription. The exogenous KDM2A with a mutation in the CxxC-ZF domain retained the demethylase activity but did not reduce rDNA transcription on starvation. It was found that the CxxC-ZF domain of KDM2A bound to the rDNA promoter with unmethylated CpG dinucleotides in vitro and in vivo. The exogenous KDM2A with the mutation in the CxxC-ZF domain failed to reduce H3K36me2 in the rDNA promoter on starvation. Further, it was suggested that KDM2A that bound to the rDNA promoter was activated on starvation. Our results demonstrate that KDM2A binds to the rDNA promoter with unmethylated CpG sequences via the CxxC-ZF domain, demethylates H3K36me2 in the rDNA promoter in response to starvation in a JmjC domain-dependent manner, and reduces rDNA transcription.
Much has been discovered about the relationship between chromatin structures and transcription during the past two decades, and several chemical modifications of chromatin components have been reported (Berger, 2007). While DNA methylation at a cytosine residue located 5' to a guanosine in a CpG dinucleotide is an epigenetic mark associated with gene silencing in vertebrates (Deaton and Bird, 2011; Esteller, 2007; McStay and Grummt, 2008), there are regions, called CpG islands, which have high GC contents and contiguous unmethylated CpG dinucleotides, surrounding promoters and gene regulatory units in up to 70% of genes (Blackledge and Klose, 2011; Deaton and Bird, 2011; Gardiner-Garden and Frommer, 1987). Recent studies suggested that an unmethylated CpG dinucleotide was bound by a CxxC-zinc finger (CxxC-ZF) domain (Allen et al., 2006; Blackledge et al., 2010; Cierpicki et al., 2010; Lee et al., 2001; Song et al., 2011; Thomson et al., 2010; Voo et al., 2000) that was characterized by two CGXCXXC repeats and conserved cysteine residues that bound to two zinc ions (Long et al., 2013; Xu et al., 2011). It was reported that the CxxC-ZF domain was involved in targeting chromatin-modifying proteins to CpG islands, which may affect gene expression (Blackledge et al., 2010; Long et al., 2013; Smith and Shilatifard, 2010; Thomson et al., 2010). There are CxxC-ZF domain-containing enzymes, including KDM2B. Mouse embryo fibroblasts engineered to express KDM2B underwent immortalization in the absence of replicative senescence in a CxxC-ZF domain-dependent manner (Pfau et al., 2008). The KDM2B CxxC-ZF domain was also required for recruitment of polycomb repressive complex 1 to CpG islands and control of early differentiation of mouse embryonic stem cells by KDM2B (He et al., 2013; Wu et al., 2013). However, the CxxC-ZF domain was not required for KDM2B-mediated promotion of proliferation of iPS cells reprogrammed by Oct4, Sox2, and Klf4 (Liang et al., 2012). These results suggest that although the CxxC-ZF domain plays crucial roles in several cases, this domain is not always required for functions of KDM2B.
Another chemical modification of chromatin components is the methylation of lysine residues in histones. Highly specific enzymes that catalyse the addition of methyl groups, as well as proteins recognizing distinct methylated lysine residues, have been identified. While histone methylation determines the epigenetic state of a gene, recent discoveries of an increasing number of histone demethylases suggest that the regulation of histone methylation is involved in additional dynamic processes (Klose et al., 2006; Kustatscher and Ladurner, 2007; Stavropoulos and Hoelz, 2007). KDM2A, a paralogue of KDM2B mentioned above, is the first identified histone demethylase containing a JmjC domain as an absolutely required region (Tsukada et al., 2006). It demethylates di-methylated Lys 36 of histone H3 (H3K36me2) (Tsukada et al., 2006). The physiological roles of methylation of H3K36 are rather complex, especially in mammals. For example, H3K36me2 generated by Wolf-Hirschhorn syndrome candidate 1 (WHSC1/NSD2) along the euchromatin was reported to repress inappropriate transcription (Nimura et al., 2009). The demethylation of H3K36me2 by KDM2B was shown to promote the expression of NAD(P)H quinone oxidoreductase-1 and peroxiredoxin-4 (Polytarchou et al., 2008). These results suggest that H3K36me2 mediates the gene repression (Wagner and Carpenter, 2012). On the other hand, KDM2B targets the INK4b-ARF-INK4a locus and demethylates H3K36me2 to down-regulate the expression of INK4b and INK4a (He et al., 2008, 2011). Therefore, H3K36 methyl marks are gene-repression or -activation marks, depending on the target genes (Smith and Shilatifard, 2010). Another complexity is brought by the fact that the methyltransferases often create not only H3K36me2 but also H3K36me3 in cells (Nimura et al., 2009; Rahman et al., 2011). The production of H3K36me3 may involve another methyltransferase, SETD2, in mammalian cells (Wagner and Carpenter, 2012). The PWWP domain of DNMT3a binds to H3K36me3 far more strongly than to H3K36me2 (Dhayalan et al., 2010), suggesting that the production of H3K36me3 can result in different biological outputs from that of H3K36me2. However, the differences in physiological roles between H3K36me2 and H3K36me3 marks are not clear.
Regulation of cell growth ultimately depends on the control of ribosome synthesis (Grummt, 2003). Whereas the supply of ribosomal components needs the activities of three forms of nuclear RNA polymerase (pol I, pol II, and pol III) in eukaryotic cells, the control of pol I activity plays a central role in the regulation of ribosome biogenesis (Boulon et al., 2010; Chedin et al., 2007; Grewal et al., 2007). Pol I transcribes the eukaryotic ribosomal RNA genes (rDNA) to produce one precursor transcript, pre-ribosomal RNA (pre-rRNA), in nucleoli. To synthesize ribosomes on demand, the level of rDNA transcription is controlled (Boulon et al., 2010; Grummt and Ladurner, 2008). Recently, the relationship between structures of rDNA chromatin and rDNA transcription had been described (Olson, 2011). We previously found that KDM2A was localized in nucleoli, and reduced rDNA transcription in response to starvation (Tanaka et al., 2010). This reduction was accompanied by demethylation of H3K36me2 in the rDNA promoter. However, it remains to be demonstrated that the demethylase activity of KDM2A itself is required for the repression of rDNA transcription by KDM2A on starvation. Recently, it was suggested that KDM2A utilizes its CxxC-ZF domain to bind to CpG islands, resulting in removal of H3K36 methylation (Blackledge et al., 2010; Zhou et al., 2012). However, it remains to be clarified whether its CxxC-ZF domain is required for the activity of KDM2A in the rDNA promoter.
In this study, we characterized the functions of two domains of KDM2A, JmjC and CxxC-ZF domains. We found that KDM2A binds to the rDNA promoter with unmethylated CpG dinucleotides via the CxxC-ZF domain, which is required for KDM2A to demethylate H3K36me2 in the rDNA promoter on starvation by the JmjC-dependent demethylase activity, and reduce rDNA transcription.
Point mutations were introduced into the cDNA for KDM2A used in this study without changes in the amino acid sequences by standard protocols. The mutations were introduced as ATGGAgCCgGAgGAgGAgcGcATcaGg, in which the ATG encodes the first Met and the mutated bases are shown by lowercase letters. The expression of protein from this KDM2A cDNA was resistant to the siRNA specific for KDM2A used in this study. The siRNA used here is described in the section on siRNA. The JmjC mutant KDM2A, which has a point mutation with His at 212 replaced with Ala, and does not have the histone demethylase activity, was described previously (Tanaka et al., 2010; Tsukada et al., 2006). The CXXC mutant KDM2A has point mutations in the CxxC-ZF domain, in which Cys at 571, 574, and 577 were replaced with Ala (Fig. 1A). The cDNAs encoding KDM2A, JmjC mutant, and CXXC mutant without any tag-sequences were cloned into a pUHC10-3 vector (Gossen et al., 1995), which harbors a tetracycline-responsive promoter, to produce ptetKDM2A, ptetmutantJmjCKDM2A, and ptetmutantCXXCKDM2A.

KDM2A proteins expressed under inducible promoter. (A) Human KDM2A protein with functional domains (upper panel) and mutations introduced in this work (lower panel) are shown. Functional domains in this figure were identified by the Uniprot database (Accession number: Q9Y2K7). His at 212 of the JmjC domain was changed to Ala in the JmjC mutant KDM2A, and cysteines at 571, 574, and 577 of the CxxC-ZF domain were changed to alanines in the CXXC mutant KDM2A. (B) Expression of wild-type and mutant KDM2A proteins. Two days after transfection of siRNA for endogenous KDM2A (KDM2A si) or control siRNA (contr si), cells were cultured in the presence or absence of doxycycline (Dox) for one day, and for two hours further with or without starvation. Cells were collected, and expression of KDM2A protein was detected by Western blotting using anti-KDM2A antibody (Tanaka et al., 2010). The results of the parental cells (MCF-7tet-on) and cells with the wild-type, JmjC mutant or CXXC mutant KDM2A under the doxycycline-induced promoter (MCF-7tet-KDM2A, MCF-7tet-mJmjC and MCF-7tet-mCXXC, respectively) are shown. β-actin was also detected as a loading control. (C) Subcellular localization of the exogenous KDM2A. Cells (MCF-7tet-on and MCF-7tet-KDM2A cells) were transfected with the siRNA for endogenous KDM2A (KDM2A si) or control siRNA (contr). Two days after transfection, cells were cultured in the presence or absence of Dox for one day. Cells were analyzed by indirect immunofluorescence technique using anti-KDM2A antibody (red) and nucleolin (green). The specimens were observed through a fluorescence and differential interference contrast (DIC) microscope, and representative images are shown. Anti-KDM2A antibody produced strong signals localized in the nucleoli with control siRNA (contr). Treatment with KDM2A siRNA reduced them. Treatment of MCF-7tet-KDM2A cells with Dox increased the signals. The subcellular pattern of the staining for the exogenous KDM2A was similar to that for endogenous KDM2A, because most of the signals for endogenous and exogenous KDM2A overlapped well with those for nucleolin (merge). Scale bar, 10 μm.
pUHD172-1neo encodes a tetracycline-sensitive transcription factor (rtTA) and neomycin-resistant protein (Gossen et al., 1995). pActHyg, which contains a hygromycin-resistance gene under the control of the actin promoter, was described previously (Tsuneoka et al., 1997). A pCAGGS vector that expresses the Flag-tagged wild-type or JmjC mutant KDM2A was reported previously (Tanaka et al., 2010). The cDNA for CXXC mutant KDM2A was recloned into a pCAGGS vector to express Flag-tagged KDM2A.
To express the GST fusion protein containing the CxxC-ZF domain of KDM2A protein, the cDNA encoding KDM2A from AA 548 to 648 was amplified by PCR with 5'-GGATCCGGTTAACACCTGTGAGGCCA-3' (adding a BamHI site) and 5'-GAATTCAATCTCATTGCAGATACAGC-3' (adding an EcoRI site) as primers using ptetKDM2A and ptetmutantCXXC plasmids as templates. The amplified 314 bp fragments were inserted into a pGEX-3X vector (GE Healthcare UK Ltd., Amersham Place, Buckinghamshire, England) to produce pGEX-CXXC(wild-type)KDM2A and pGEX-CXXC(mutant)KDM2A.
The human rDNA promoter sequence (1–155) was amplified by PCR using 5'-GCTGACACGCTGTCCTCTG-3' and 5'-TCGGACGCGCGAGAGAAC-3', and cloned into a pGEM-T-Easy vector (Promega, Madison, WI, USA, #A1360) by TA cloning to produce pGEMrDNApromoter. The DNA fragment with no CpG sequences (noCG fragment) was amplified from pBluesctipt II KS(–) by PCR using 5'-CTGGAATTCTTAAGGGATTTTGGT-3' and 5'-TCAGAATTCCTGAGATAGGTGCCT-3' (1859–2007 of pBluesctipt II KS(–), adding an EcoRI site to both primers), and cloned into pGEM-T-Easy vector (Promega) at an EcoRI/EcoRI site to produce pGEMnoCGsequense.
Cells and cell cultureHuman breast adenocarcinoma cell line MCF-7 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich Co., St. Louis, MO, USA, Cat# D5796) supplemented with 10% fetal calf serum (FCS). Cells were maintained at 37°C in an atmosphere containing 5% CO2 and 100% humidity. For starvation, cells were cultured in serum-free DMEM that did not contain glucose (Sigma-Aldrich, Cat# D5030).
The mammalian expression plasmids were introduced into cells using FuGENE 6 transfection reagent (Roche Applied Science, Indianapolis, IN, USA) according to the manufacturer’s instructions. To select cells carrying the doxycycline-dependent transcription factor (rtTA), MCF-7 cells were transfected with pUHD172-1neo (Gossen et al., 1995) and cultured in the presence of 800 μg/ml G418. After 3 wks, the colonies were picked up and a cell line that carried rtTA was selected and named MCF-7tet-on (the parental cells). MCF-7tet-on cells were further transfected with ptetKDM2A plus pActHyg, which confers hygromycin resistance, and cultured in the presence of 150 to 250 μg/ml hygromycin and 200 μ/ml G418. The selected colonies were picked up and cultured for 24 h in the presence of 1 μg/ml doxycycline (Dox), and the expression of KDM2A or its mutant proteins was detected by indirect immunofluorescence and Western blotting using an anti-KDM2A antibody (Tanaka et al., 2010).
siRNACells were transfected with stealth siRNA using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. The siRNA oligonucleotide sequence for KDM2A (5'-GAACCCGAAGAAGAAAGGAUUCGUU-3'), which is a stealth RNA-cognated partial nucleotide sequence for KDM2A mRNA (nn:4-28 in which nn indicates the nucleotide numbers from the A of the first Met of KDM2A mRNA), was previously described (Tanaka et al., 2010). Cells were also transfected with control stealth RNA (StealthTM RNAi Negative Control Medium GC Duplex, Invitrogen, Carlsbad, CA, USA).
AntibodiesGoat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, Santa Cruz CA, USA, sc-2054), goat anti-mouse IgG-HRP (Santa Cruz, sc-2005), Alexa 488-conjugated goat anti-mouse IgG (H+L) (A11029; Invitrogen), and Alexa 568-conjugated goat anti-rabbit IgG (H+L) (A11011; Invitrogen), mouse monoclonal anti-β-actin (AC-15) antibody (Sigma, St. Louis, MO, USA), mouse monoclonal anti-FLAG (M2) antibody (Sigma), mouse monoclonal anti-nucleolin antibody (C23 (MS-3): sc-8031; Santa Cruz), anti-trimethylated histone H3 lys36 antibody (Abcam, Cambridge, UK, ab9048), anti-histone H3 antibody (Abcam, ab1791), anti-dimethylated histone H3 lys36 antibody (Cell Signaling Technologies, Beverly, MA, USA, #9758), anti-GST antibody (Santa Cruz, #Z-5, sc-459), and control antibody (Cell Signaling, normal rabbit IgG #2729S) were purchased. The anti-KDM2A antibody was described previously (Tanaka et al., 2010).
Western blotting and immunofluorescence stainingCells were trypsinized and extracted in 3% SDS solution containing 100 mM Tris, pH 6.8, 50 mM DTT, and 20% glycerol. Cell extracts were separated on SDS-PAGE and transferred to a microporous PVDF membrane (Millipore, Bedford, MA, USA). After treatment with antibodies, bands were detected using an Immobilon Western system (Millipore, WBKLS0100) (Tanaka et al., 2010). For detection of histone H3, a BCIP-Solution Kit for Alkaline Phosphatase Staining (Nacalai Tesque, Kyoto, Japan, #03937-60) was used according to the manufacturer’s instructions.
For indirect immunofluorescence staining, cells grown on glass coverslips were fixed in methanol for 20 min at –20°C and incubated in 1% bovine serum albumin (BSA) in PBS. The rabbit antibody and/or mouse monoclonal antibody were added and incubated for 60 min at 37°C. After washing cells in 0.1% BSA in PBS three times, Alexa 488-conjugated anti-mouse IgG and Alexa 568-conjugated anti-rabbit IgG were added, incubated for 60 min at 37°C, and washed with 0.1% BSA three times. Finally, cells were embedded in Immunon (Thermo Shandon, Pittsburgh, PA, USA) and observed via confocal fluorescence microscopy.
To investigate the effect of KDM2A on H3K36me levels in nuclei, MCF-7 cells were transfected with the mammalian expression plasmids for KDM2As to transiently express the Flag-tagged KDM2A and KDM2A mutants. After being cultured for two days, H3K36me2, H3K36me3, and Flag-tag were detected by indirect fluorescence (Tanaka et al., 2010).
RNA preparation and quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)Total RNA was isolated from cells using a NucleoSpin RNA II kit (Takara Bio Inc., Otsu, Japan, #U0955C) according to the manufacturer’s instructions. Synthesis of single-strand cDNA was performed on total RNA (1 μg) by a Superscript III First-strand Synthesis system (Invitrogen) using random hexamers according to the manufacturer’s instructions. The products were diluted to 200 μl with distilled water, and 2.5 μl of the resultant single-strand cDNA was used as the template for qRT-PCR, using Thunderbird qPCR Mix (Toyobo Co., Ltd., Osaka, Japan, #QPS-201) with a Mx3000P QPCR system (Agilent Technologies, Inc., Santa Clara, CA, USA) according to the manufacturers’ instructions. The values were normalized using the amounts for a control mRNA, RNA polymerase II subunit a (Polr2a) mRNA (Dydensborg et al., 2006). The sets of PCR primers for amplification of the pre-rRNA, KDM2A mRNA, and Porl2a mRNAs used in this study were described previously (Tanaka et al., 2010).
StatisticsP values were calculated by two-tailed unpaired Student’s t-test.
Chromatin immunoprecipitations (ChIP)ChIP assays were performed using the Dynabeads system (Invitrogen) with buffer components from a ChIP assay kit (Millipore), according to the manufacturer’s instructions with some modifications. The immunoprecipitated DNA was purified by a Chelex-100-based DNA isolation procedure (Nelson et al., 2006), and the amount of the DNA fragment collected was measured by qRT-PCR. The primers used for H0 (rDNA from +1 to +155 from the transcriptional start site) were described previously (Tanaka et al., 2010). To detect specific binding, the values obtained by specific antibodies were subtracted from those performed simultaneously using a control antibody (normal rabbit IgG) and divided by total input. When detecting histone modifications, the values for the specific binding were normalized by the values for H3 (% of specific bound/input normalized by H3).
Sequential ChIP analyses were performed as described below. The first ChIP assay was performed as described above except that the immunoprecipitates were eluted twice by a ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris pH 8) supplemented with 10 mM DTT for 15 min at room temperature. The elutes were combined, and further analyzed for the second ChIP following the method described in the above paragraph. The eluate used for the second ChIP was diluted to ten times with ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, pH 8.1, and 167 mM NaCl), and used as the input of the second ChIP.
Electrophoretic mobility shift assay (EMSA)To obtain the rDNA promoter fragment, the plasmid pGEMrDNA promoter was cut with EcoRI, and the 181 bp-fragment was purified using a QIAquick Gel Extraction kit (Qiagen, Germantown, MD, USA, #28704). NoCG (161 bp) fragments were obtained from pGEMnoCGsequence, respectively, by the same procedure used for the rDNA fragment. The fragments were labelled with [32P]dATP using AmpliTaq DNA polymerase (Life Technologies, Carlsbad, CA, USA). To produce DNA fragments with methylated cytosine at the CpG site, the fragments were treated with the CpG methylase M.SssI enzyme (New England Biolabs, Ipswich, Massachusetts, USA, #0226S), according to the manufacturer’s instructions.
To obtain the recombinant proteins, E. coli strain BL21(DE3)pLysS (Novagen, Madison, WI, USA, #69451) was transformed by pGEX-3X, pGEX-CXXC(wild-type)KDM2A, or pGEX-CXXC(mutant)KDM2A, and cultured in Luria-Bertani medium or 2xTB containing 0.25 mM ZnCl2 with 0.3 mM IPTG to produce GST and GST fusion proteins. The proteins were purified using Glutathione SepharoseTM4B beads (GE Healthcare) according to the manufacturer’s instructions. The production of the GST fusion proteins was checked by SDS-PAGE and CBB staining (Fig. 4A).
Electrophoretic mobility shift assays (EMSA) were performed by standard methods described previously (Blackledge et al., 2010; Tsuneoka and Mekada, 1998; Tsuneoka et al., 1997) with some modification. Recombinant GST-fusion proteins were dialyzed four times with BC100 buffer (50 mM Hepes, pH 7.9, 100 mM KCl, 10% glycerol, and 0.5 mM DTT) at 4°C. The dialyzed proteins were incubated in EMSA buffer (4% Ficoll 400, 20 mM Hepes 7.9, 150 mM KCl, 1 mM EDTA, 0.5 mM DTT, and 25 ng/μl poly-dAdT competitor DNA) for 10 min at room temperature, then radiolabeled rDNA promoter fragments were added, and incubated for 20 min at room temperature. The mixtures were electrophoresed in 7.5% polyacrylamide gel in 0.5 x TBE buffer at 100V at room temperature. Radioactivities were detected by a Fluorescence Image Analyzer FLA5100 (Fujifilm, Tokyo, Japan).
Measurement of methylated and unmethylated CpG in rDNA promoterDNA was isolated, and the level of DNA methylation was determined by the bisulphite method. using a MethylEasyTM Xceed rapid DNA bisulphite modification kit (Human Genetic Signatures Pty. Ltd., North Ryde, Australia) according to the manufacturer’s instructions with some modifications. In brief, input samples for ChIP were treated with protenase K, phenol-chloroform, de-fixation, and ethanol precipitation to obtain purified DNA. Cytosines in the DNA were converted to uracils. The modified DNA strands were amplified by PCR using primers that specifically amplify the fragment of the rDNA promoter from –184 to +48, cloned into pGEM-T-Easy vector, and sequenced. The PCR primers used were 5'-GTTTTTGGGTTGATTAGA-3' and 5'-AAAACCCAACCTCTCC-3'.
We previously reported that KDM2A bound to the rDNA promoter, and reduced H3K36me2 and rDNA transcription, in response to starvation (Tanaka et al., 2010). To investigate how KDM2A controls rDNA transcription, we established a cell line in which KDM2A was expressed under the doxycycline (Dox)-inducible (tet-on) promoter (Gossen et al., 1995). The cDNA for the exogenous KDM2A used in this study had a mutation to confer resistance to the specific siRNA for endogenous KDM2A. The cDNA under the tet-on promoter was introduced into parental cells, which were MCF-7 cells expressing the tet-on transcription factor (rtTA) (Gossen et al., 1995). The treatment of KDM2A siRNA reduced the expression of endogenous KDM2A (Fig. 1B). After a knockdown of endogenous KDM2A, the exogenous KDM2A was expressed by adding Dox (Fig. 1B). Western blotting (Fig. 1B) and immunofluorescence analysis (Fig. 1C) showed that the exogenous KDM2A had a molecular weight and subcellular localization pattern similar to those of endogenous KDM2A. The treatment of KDM2A siRNA decreased the reduction of rDNA transcription during starvation in both parental MCF-7tet-on and MCF-7tet-KDM2A cells in the absence of Dox (Fig. 2). The Dox treatment recovered the reduction of rDNA transcription in MCF-7tet-KDM2A but not in MCF-7tet-on cells (Fig. 2). The amount of KDM2A did not increase during starvation (Fig. 1B). In the experiments described in this report, KDM2A reduced rDNA transcription within two hours of starvation. Taken together, these results confirm our previous results (Tanaka et al., 2010) and indicate that the exogenous KDM2A mimics the function of endogenous KDM2A.

Effects of KDM2A on rDNA transcription during starvation. Two days after transfection with the siRNA for endogenous KDM2A (KDM2A si) or control siRNA (contr si), cells (MCF-7tet-on, MCF-7tet-KDM2A, MCF-7tet-mJmjC and MCF-7tet-mCXXC cells) were cultured in the presence or absence of Dox for one day. Cells were further cultured with or without starvation for two hours. RNA was isolated from cells, and the amount of pre-rRNA was measured by qRT-PCR. The values were normalized using the control RNA (Polr2a mRNA). The normalized value of pre-rRNA with starvation was divided by that without starvation to obtain the reduction due to starvation. To express the reductions of rDNA transcription by KDM2A, the values for the reduction due to starvation for each cell line were divided by that with the siRNA for KDM2A in the absence of Dox in the cell line. Error bars indicate standard deviations of each condition. *P<0.05.
Next, we established cell lines in which the JmjC mutant KDM2A or the CxxC mutant KDM2A was expressed under the tet-on system (MCF-7tet-mJmjC and MCF-7tet-mCXXC, respectively). In the JmjC mutant KDM2A, His 212 in the JmjC domain was replaced with Ala (Fig. 1A), and this mutation completely abolished the demethylase activity (Tanaka et al., 2010; Tsukada et al., 2006). In the CXXC mutant KDM2A, Cys 571, Cys 574, and Cys 577 in the CxxC-ZF domain were replaced with Ala (Fig. 1A). After a knockdown of endogenous KDM2A, expression of the JmjC mutant or the CXXC mutant KDM2A was induced. As shown in Fig. 1B, the JmjC mutant KDM2A and CXXC mutant KDM2A were expressed at a level comparable to the wild-type KDM2A in the presence of Dox. The treatment of KDM2A siRNA in these cells decreased the reduction of rDNA transcription during starvation and the reduction of rDNA transcription was not enhanced by induction of the JmjC or CXXC mutant KDM2A (Fig. 2). These results show that both JmjC and CxxC-ZF domains are required for the reduction of rDNA transcription on starvation.
KDM2A with a mutation in CxxC-ZF domain retains the demethylase activity in overall nucleiTo check the demethylase activity of these mutant KDM2As in vivo, the expression plasmids for Flag-tagged KDM2As were transiently transfected, and H3K36me2 in nuclei was observed by indirect immunofluorescence. When Flag-tagged wild-type KDM2A was overexpressed, the signals for H3K36me2 but not H3K36me3 were reduced (Fig. 3A and Fig. 3B). Overexpression of Flag-tagged JmjC mutant KDM2A or GFP reduced the signals for neither H3K36me2 nor H3K36me3, showing that the demethylase activity of KDM2A reduced H3K36me2. When Flag-tagged CXXC mutant KDM2A was overexpressed, the signals for H3K36me2 but not H3K36me3 were reduced (Fig. 3A and Fig. 3B). The reduction of H3K36me2 by Flag-tagged CXXC mutant KDM2A was even severer than that by wild type KDM2A (Fig. 3B). These results suggest that the CXXC mutant KDM2A has the demethylase activity. Further, in MCF-7tet-mCXXC as well as MCF-7tet-KDM2A cells, Dox-treatment reduced H3K36me2 but not H3K36me3 (Fig. 3C). In MCF-7tet-mJmjC cells, Dox-treatment did not reduce H3K36me2 (Fig. 3C), showing that the reduction of H3K36me2 was due to the enzyme activity of KDM2A itself. These results confirm the results described above (Fig. 3A and Fig. 3B) that the CXXC mutant KDM2A retains the demethylase activity.

Reduction of H3K36me2 by KDM2A in overall nuclei. (A) MCF-7 cells were transiently transfected with expression vectors encoding the Flag-tagged wild-type, JmjC mutant, CXXC mutant KDM2A, or GFP. Cells were analysed by indirect immunofluorescence with anti-Flag (green), anti-H3K36me2 (red), and anti-H3K36me3 (red) antibodies. GFP (green) was detected using anti-GFP antibody (in the row “GFP”). The specimens were observed through a fluorescence and differential interference contrast (DIC) microscope, and representative images are shown. Transfected cells were detected by their green colour. One cell with a positive signal for the exogenous proteins in one field is indicated by an arrowhead. Scale bar, 10 μm. (B) The numbers of cells in which the KDM2As or GFP overexpression had not reduced H3K36me2 or H3K36me3 were counted. Error bars indicate standard deviations. (C) Two days after transfection of siRNA for endogenous KDM2A, cells (MCF-tet-on, MCF-7tet-KDM2A, MCF-7tet-mJmjC and MCF-7tet-mCXXC cells) were cultured in the presence or absence of doxycycline (Dox) for one day and lysed. Cell lysates were analysed by Western blotting using anti-KDM2A, anti-H3K36me2, and anti-H3K36me3 antibodies. The membrane on which H3K36me2 was detected was re-probed with anti-histone H3 antibody.
Since the rDNA promoter contains CpG dinucleotides (Gagnon-Kugler et al., 2009; Ghoshal et al., 2004) (also refer to Fig. 5B), we examined whether the CxxC-ZF domain was involved in binding of KDM2A to the rDNA promoter. First, we tested whether KDM2A CxxC-ZF domain could bind to rDNA promoter in vitro. We produced a recombinant GST-fusion protein containing the wild type or mutant CxxC-ZF domain of KDM2A (Fig. 4A) and performed an electrophoretic mobility shift assay (EMSA) with the rDNA promoter fragment, which contains 21 CpG dinucleotides. As shown in Fig. 4B and Fig. 4C, when the unmethylated rDNA promoter fragment was incubated with the recombinant protein of the wild type CxxC-ZF domain, a band shift was observed. This shifted band was abolished by the specific antibody to GST. The disappearance of the shifted band by the same antibody was reported before (Lutzner et al., 2012). These results confirm that the shifted band consists of the GST fused CxxC-ZF domain molecule. The control GST protein and the recombinant mutant CxxC-ZF domain did not produce the band shift, even when the amounts of proteins used were increased up to three fold (Fig. 4B). The band shift was not observed when the rDNA promoter fragment was methylated (Fig. 4B), nor was it observed when a DNA fragment with no CpG sequences was used as the probe (Fig. 4C). Further, the shifted band was decreased by addition of the cold unmethylated rDNA promoter fragment, but neither the cold methylated rDNA promoter fragment nor the cold DNA fragment with no CpG sequences (Fig. 4D). These results indicate that binding of the CxxC-ZF domain to the rDNA promoter fragment is dependent on unmethylated CpG dinucleotides.

Binding of CxxC-ZF domain of KDM2A to unmethylated rDNA promoter in vitro. (A) The recombinant GST proteins, GST protein fused with the CxxC-ZF domain (GST-WTCXXC), and the mutant CxxC-ZF domain (GST-mutantCXXC) of KDM2A were produced and analyzed by SDS-PAGE. The proteins were stained by CBB. (B) The radiolabeled unmethylated rDNA promoter fragment was incubated with GST fusion recombinant protein containing the CxxC-ZF domain of KDM2A (WT) or the mutant CxxC-ZF domain (C571A, C574A, C577A as shown in Fig. 1A) (m), or the control GST protein (v) and electrophoresed. The specific antibody to GST was added to the reaction mixture in the indicated lane. In lanes labelled “3x prot.”, three-fold amounts of the recombinant proteins were added to the reaction mixtures. In the lanes labelled “methylated probe”, the rDNA promoter fragment was methylated using M.SssI enzyme before radiolabeling. (C) EMSA was performed for the radiolabeled unmethylated rDNA promoter fragment (rDNA promoter) and DNA fragment that had no CpG sequences (no CpG) as described in B. (D) EMSA was performed with excess amounts of cold competitor fragments, including the unmethylated rDNA promoter (unmethylated), methylated rDNA promoter (methylated) and the DNA fragment with no CpG dinucleotides (no CpG). The competitor fragments used in these experiments were three-fold (×3) or ten-fold (×10) more than the radiolabeled probe.
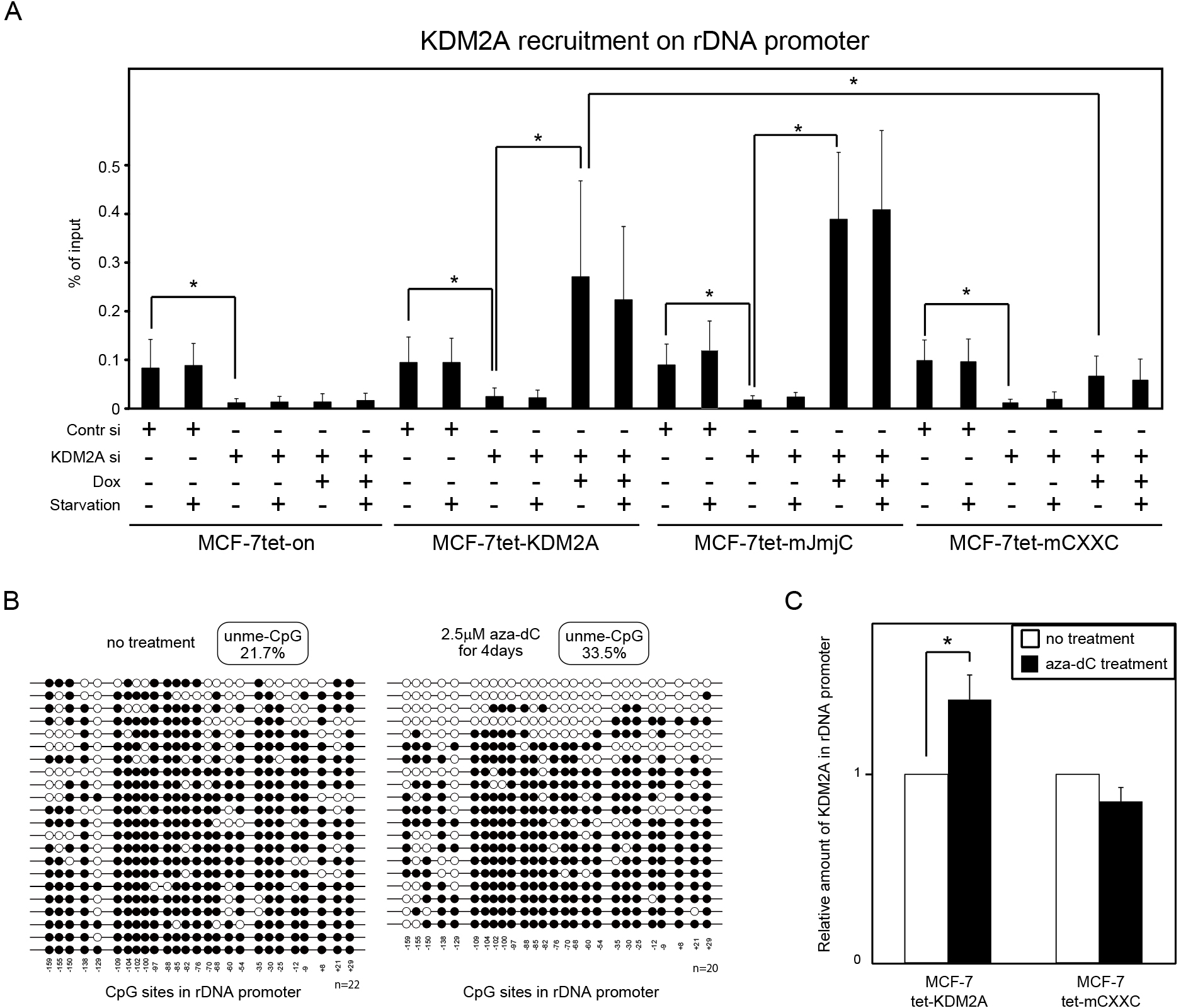
Binding of KDM2A to rDNA promoter in vivo. (A) Cells were cultured in various conditions as described in Fig. 2. ChIP assays were performed to detect KDM2A in the rDNA promoter. The results are shown as the percentage of input. Error bars indicate standard deviations. *P<0.05. (B) Dox-treated MCF-7tet-KDM2A cells were cultured in the absence (left panel) or presence (right panel) of 2.5 μM 5-aza-2'-deoxycytidine (aza-dC) for four days. DNA was isolated, and the level of DNA methylation was determined by the bisulphite method. Open and closed circles represent unmethylated and methylated cytosine in CpG sequences, respectively. The amount of unmethylated CpG in the rDNA promoter was increased from 21.7% to 33.5%. (C) MCF-7tet-KDM2A and MCF-7tet-mCXXC cells were transfected with siRNAs for KDM2A and cultured in the presence of Dox, with or without 2.5 μM aza-dC for four days. KDM2A on the rDNA promoter was detected by ChIP using anti-KDM2A antibody. The values with the aza-dC treatment were divided by the values without the aza-dC treatment. Error bars indicate standard deviations. *P<0.05.
The binding of KDM2A to rDNA promoter in cells was tested by ChIP analysis. Without the KDM2A siRNA and Dox treatments, the rDNA promoter fragments were collected by anti-KDM2A antibody at similar levels in all four cell lines produced here (Fig. 5A). KDM2A siRNA treatment severely reduced the amounts of the rDNA promoter fragments collected by anti-KDM2A antibody (Fig. 5A), showing that anti-KDM2A antibody actually collected the rDNA promoter fragments bound by KDM2A. Dox-treatments of MCF-7tet-KDM2A and MCF-7tet-mJmjC cells but not of MCF-7tet-on cells increased binding of KDM2A to the rDNA promoter (Fig. 5A). Dox-treatment of MCF-7tet-mCXXC cells increased binding of KDM2A to the rDNA promoter but the increase was smaller than that of MCF-7tet-KDM2A cells (Fig. 5A). Starvation did not increase binding of KDM2A to the rDNA promoter (Fig. 5A). Together, these results indicate that KDM2A is recruited to the rDNA promoter before starvation and the CxxC-ZF domain but not JmjC dependent demethylase activity is involved in recruitment of KDM2A to rDNA promoter in vivo.
To confirm that KDM2A binds to the rDNA promoter with unmethylated CpG dinucleotides in vivo, we investigated binding of the wild type and the CXXC mutant KDM2A to rDNA promoter in 5-aza-2'-deoxycytidine (aza-dC)-treated cells. The increase in the DNA unmethylation level due to aza-dC treatment was confirmed (Fig. 5B). MCF-7tet-KDM2A and MCF-7tet-mCXXC cells were transfected with KDM2A siRNA and cultured in the presence of Dox and 2.5 μM aza-dC for four days. Fig. 5C shows that aza-dC treatment elevated the amount of wild-type KDM2A on the rDNA promoter 1.4 fold. In contrast, the aza-dC treatment did not increase the amount of the CXXC mutant KDM2A on the rDNA promoter (Fig. 5C). These results suggest that the binding activity of the KDM2A CxxC-ZF domain to unmethylated CpG dinucleotides is involved in binding of KDM2A to the rDNA promoter in vivo.
CxxC-ZF domain is required for KDM2A to demethylate H3K36me2 in rDNA promoter in response to starvationNext, we investigated the effects of KDM2A on the levels of H3K36me2 in the rDNA promoter. Starvation reduced H3K36me2 in the rDNA promoter, and the KDM2A siRNA treatment abolished the reduction (Fig. 6A). Dox-treatment of MCF-7tet-KDM2A cells recovered the reduction of H3K36me2 on starvation, while it did so in neither MCF-7tet-on nor MCF-7tet-mJmjC cells (Fig. 6A). Dox-treatment of MCF-7tet-mCXXC cells also did not reduce H3K36me2 in the rDNA promoter on starvation (Fig. 6A). H3K36me3 in the rDNA promoter was not reduced by starvation (Fig. 6B). These results demonstrate that the CxxC-ZF domain is required for KDM2A to demethylate H3K36me2 in the rDNA promoter in response to starvation.
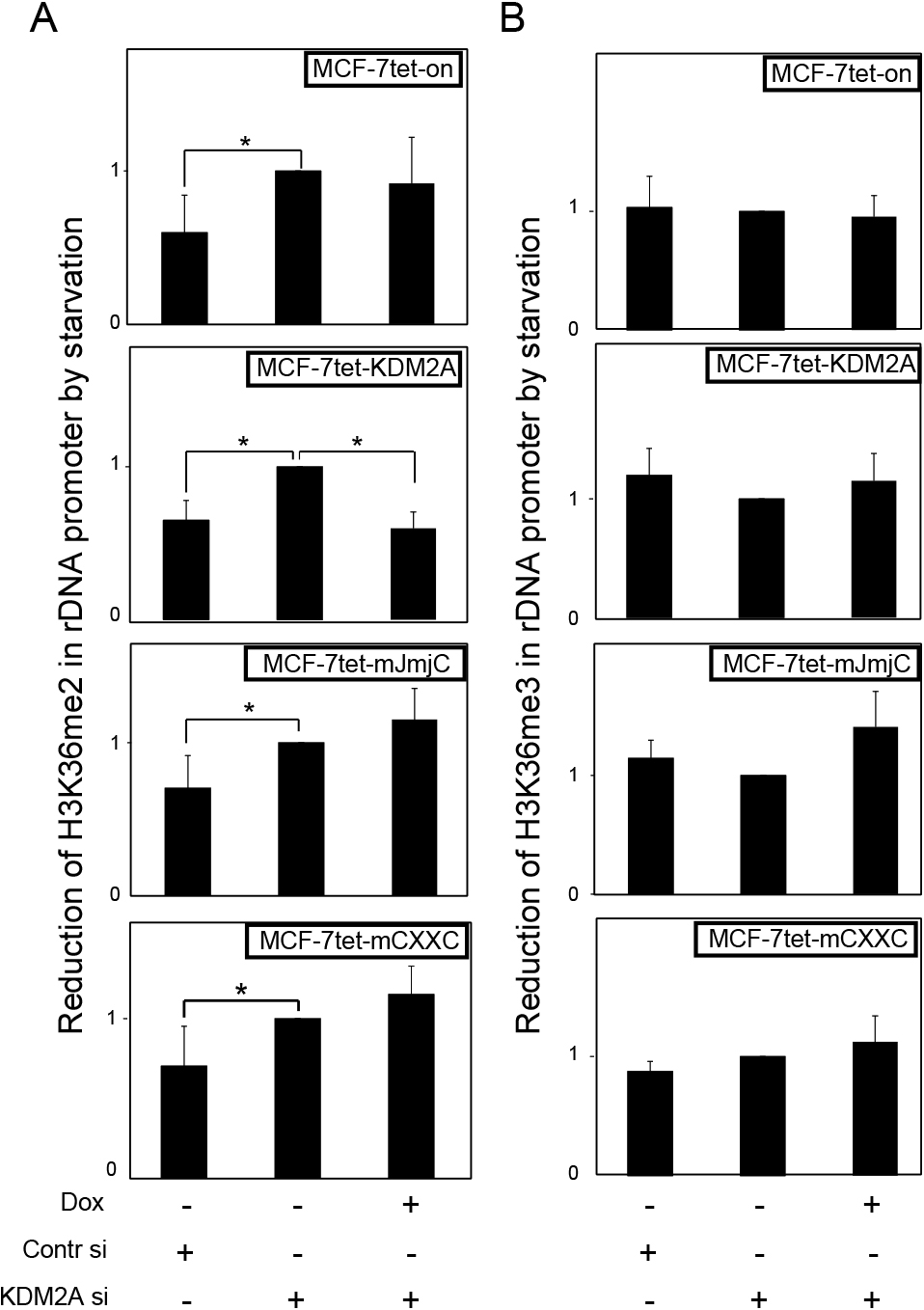
Effects of KDM2A on H3K36 methylation in rDNA promoter. Cells were cultured in various conditions as described in Fig. 2. The levels of methylation at H3K36 in the rDNA promoter were measured by ChIP, using specific antibodies to H3K36me2 (A) and H3K36me3 (B). The values for each cell line in various conditions with starvation were divided by those without starvation to express the reduction of H3K36 methylation in the rDNA promoter during starvation. Error bars indicate standard deviations of each condition. *P<0.05.
When H3K36me2 in the rDNA promoter was reduced by KDM2A on starvation (Fig. 6A), binding of KDM2A to the rDNA promoter did not increase (Fig. 5A). These results suggest that the H3K36me2 demethylation by KDM2A bound to the rDNA promoter is activated by starvation. If so, the amounts of H3K36me2 in the rDNA promoter bound by KDM2A should be reduced by starvation. To test this possibility, sequential ChIP analyses were performed. First, DNA fragments were collected using anti-KDM2A antibody from MCF-7tet-KDM2A and MCF-7tet-mJmjC cells with the treatments of KDM2A siRNA and Dox, and then the fragments were re-collected using anti-H3K36me2 or anti-H3K36me3 antibody. The results showed that H3K36me2 in the KDM2A-binding rDNA promoter was reduced during a two hour starvation (Fig. 7A), but that H3K36me2 in the JmjC mutant KDM2A-binding rDNA promoter was not (Fig. 7A). The level of H3K36me3 in the rDNA promoter was hardly changed (Fig. 7B).

The levels of H3K36me2 in rDNA promoter bound by KDM2A were affected by cell-culturing conditions. (A and B) MCF-7tetKDM2A and MCF-7tetmJmjC cells were cultured in various conditions as described in Fig. 2, and cell lysates were analysed by sequential ChIP assay. First, DNA fragments were collected using anti-KDM2A antibody, and then re-collected using anti-H3K36me2 (A) or anti-H3K36me3 antibody (B). The results are shown as values relative to those without starvation. (C) Quick recovery of H3K36me2 levels in the rDNA promoter to initial levels. Cells cultured for two hours in starvation conditions were cultured for another two hours in growth medium (+2h&–2h), or continuously cultured under starvation conditions for four hours (+4 h). DNA fragments were collected. Error bars indicate standard deviations. *P<0.05.
Next, cells cultured for two hours under starvation conditions were further cultured for another two hours in growth medium or continuously cultured under starvation conditions for another two hours. H3K36me2 in the KDM2A-binding rDNA promoter was recovered to the initial level after two hour culture in growth medium, while it was continuously low in cells under starvation conditions for four hours (Fig. 7C). H3K36me2 in the mutant JmjC KDM2A-binding rDNA promoter did not change after re-culturing in the growth medium (Fig. 7C). Together, these results suggest that the H3K36me2 demethylation by KDM2A bound to the rDNA promoter is controlled by cell culturing conditions.
To investigate the mechanisms by which KDM2A reduces rDNA transcription on starvation, we established cell lines in which exogenous KDM2A could be induced (Fig. 1). After a knockdown of endogenous KDM2A, exogenous KDM2A was expressed. In cells with exogenous wild-type but not the JmjC mutant KDM2A, the levels of H3K36me2 but not H3K36me3 in the rDNA promoter were reduced on starvation (Fig. 6), and the reduction of rDNA transcription due to starvation was enhanced (Fig. 2). These results demonstrate that the demethylase activity of KDM2A itself is required for the reduction of rDNA transcription by KDM2A. In these experiments, the levels of H3K36me2 in the rDNA promoter and rDNA transcription were reduced within two hours under starvation conditions. Together, these results show that KDM2A and H3K36me2 marks in the rDNA promoter function as components of a signaling pathway to control rDNA transcription in response to cell-culturing conditions (Fig. 8), which is consistent with the suggestion that the post-translational modifications of histones are part of the signaling pathways (Smith and Shilatifard, 2010). These results also showed one example of the difference between H3K36me2 and K3K36me3. H3K36me2 but not H3K36me3 may function as a signal transferring molecule in the rDNA promoter.
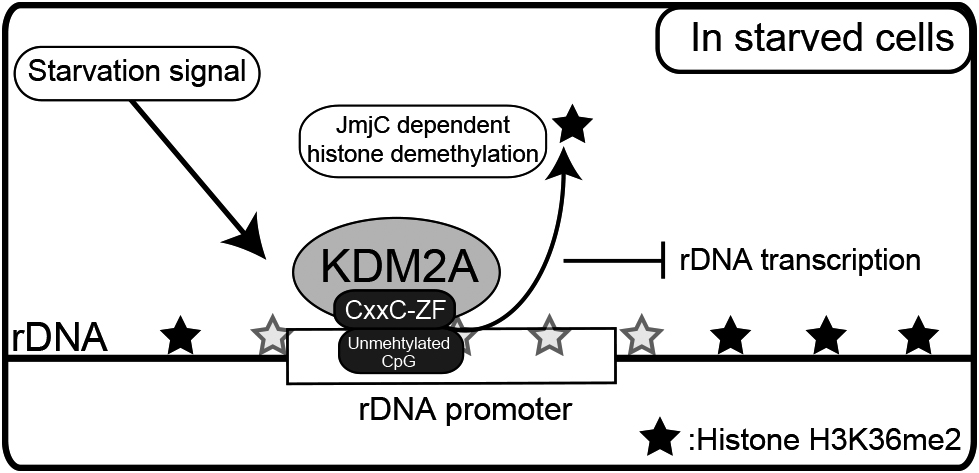
Model for control of rDNA transcription by KDM2A. KDM2A binds to the unmethylated rDNA promoter through its CxxC-ZF domain before starvation. KDM2A binding to the rDNA promoter is activated by starvation to demethylate H3K36me2 in its JmjC domain-dependent manner, and then reduces rDNA transcription.
We investigated whether the CxxC-ZF domain was involved in the binding of KDM2A to the rDNA promoter. The EMSA with the rDNA promoter fragment showed that the recombinant GST fusion protein containing KDM2A CxxC-ZF domain bound to the rDNA promoter fragment with unmethylated CpG, but it did not so to both the methylated rDNA promoter fragment and the DNA fragment with no CpG sequences in vitro (Fig. 4). The shifted band was decreased by addition of antibody against GST or the cold unmethylated rDNA promoter fragment, but neither the cold methylated rDNA promoter fragment nor the cold DNA fragment with no CpG sequences (Fig. 4). Mutation of the CxxC-ZF domain abolished the binding (Fig. 4). KDM2A bound to the rDNA promoter dependent on the CxxC-ZF domain in vivo (Fig. 5); further, the amount of KDM2A binding to the rDNA promoter increased when cells were cultured in the presence of aza-dC to increase unmethylated CpG sequences (Fig. 5), and this elevation of the binding was also dependent on the CxxC-ZF domain (Fig. 5). These results demonstrate that KDM2A uses the activity of the CxxC-ZF domain binding to unmethylated CpG sequences to bind to the rDNA promoter. The JmjC mutant KDM2A bound to the rDNA promoter with an efficiency similar to the wild-type KDM2A (Fig. 5A), showing that the demethylase activity is not required for binding of KDM2A to the rDNA promoter.
We detected that the CXXC mutant KDM2A weakly but reproducibly bound to rDNA promoter (Fig. 5), suggesting that KDM2A possesses other binding activity or activities to rDNA promoter chromatin in addition to the CxxC-ZF domain. The JmjC domain recognizes H3K36me2 marks, which are substrates for KDM2A. KDM2A also has plant homeodomain (PHD) finger. PHD fingers are present in a variety of eukaryotic proteins and were shown to recognize the modified and unmodified histone H3 tail (Musselman and Kutateladze, 2011). These regions of KDM2A may contribute to the binding of KDM2A to the rDNA promoter chromatin.
Fig. 3A showed that the CXXC mutant KDM2A was localized in nucleoli as wild type KDM2A. Recently we found that some KDM2A deletion mutant, that contained the CxxC-ZF domain but lacked amino acid sequence at the C-terminal side of the CxxC-ZF domain, was not localized in nucleoli (unpublished data). These results suggest that there is a nucleolar localization signal in KDM2A, which does not exist within the CxxC-ZF domain. The previous report showed that KDM2B, a paralogue of KDM2A, and the CXXC mutant KDM2B were localized to nucleoli, and had a nucleolar localization signal outside of the CxxC-ZF domain (Frescas et al., 2007). These results suggest that the CxxC-ZF domains of KDM2A and KDM2B do not localize proteins to nucleoli. Because KDM2A functions on the rDNA promoter, the mechanism of the nucleolar localization of KDM2A is an important issue, which remains as an opened question.
It was reported that KDM2B reduced the rDNA transcription (Frescas et al., 2007). In their report, the amount of chromatin-bound KDM2B increased by culturing in starved conditions, and serum re-addition transiently abolished this binding (Frescas et al., 2007). These results suggest that the binding of KDM2B to genomic DNA may be controlled to regulate the activity of KDM2B. On the other hand, KDM2A continuously bound to the rDNA promoter before and after starvation, and the level of H3K36me2 in the rDNA promoter bound by KDM2A was reduced on starvation (Fig. 7). After re-culturing in growth medium following starvation, the level of H3K36me2 in the rDNA promoter bound by KDM2A was recovered (Fig. 7). These results suggest that the demethylase activity of KDM2A in the rDNA promoter is controlled in response to cell culturing conditions. While KDM2A had been shown to bind to the P2RX4 gene promoter and demethylate H3K36me2 in the promoter (Blackledge et al., 2010), H3K36me2 in the P2RX4 promoter was not reduced on starvation (Supplementary Fig. 1B). There results suggest that starvation controls the KDM2A demethylase activity locally in the rDNA promoter. Recently, it was reported that in the rDNA promoter of NIH3T3 cells, serum-deprivation increased the amounts of chromodomain helicase DNA-binding protein 4 (CHD4), histone H3K4me3 and H3K27me3, and changed the position of promoter-associated nucleosome (Xie et al., 2012). These results with our results suggest that the chromatin structure of the rDNA promoter is a target for the signalling pathway from starvation to regulate rDNA transcription. Further studies of these molecular mechanisms would contribute to understand how rDNA transcription is controlled in response to environmental conditions.
We thank Ms. Saori Takahashi, Ms. Kazumi Tachikawa, Mr. Eiji Hoshino, and Ms. Kana Miyamoto (Takasaki University of Health and Welfare) for technical assistance. This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (Grant-in-Aid for Scientific Research (C): No. 21570204, No. 23770209, No. 25840011, and No. 25440090; Grant-in-Aid for Scientific Research on Innovative Areas: No. 23114721).