Abstract
Gene editing methods were applied to the study of E-cadherin function in epithelial cells. The E-cadherin gene in epithelial DLD-1 cells was ablated using TALEN. The resultant cells showed round fibroblast-like morphology and had almost no Ca2+-dependent cell aggregation activity. E-cadherin re-expression in the knockout cells restored epithelial cell morphology and strong Ca2+-dependent cell-cell adhesion activity, indicating that the knockout cells retained the ability to support cadherin function. The knockout cells showed partial localization of desmoplakin and ZO-1 at intercellular contact sites. The transfectants expressing mutant E-cadherin lacking the cytoplasmic domain showed clear localization of desmoplakin and ZO-1 at cell-cell contact sites, although the cells had only weak Ca2+-dependent cell adhesion activity. Electron microscopy revealed the formation of intercellular junctions and apico-basal polarity in these cells. A portion of these cells occasionally formed an epithelial-like structure after prolonged culture. When the cells were treated with blebbistatin, the localization was enhanced. However, the localization was incomplete and contained defects. Double-knockout MDCK cells for the E-cadherin and cadherin-6 genes showed similar results, suggesting that the above properties were general. The present results showed that an epithelial-like structure could be formed without E-cadherin, but that the construction of mature epithelia requires E-cadherin.
Introduction
Epithelia are most basic tissue structure in multicellular organisms and show characteristic properties. One feature is that epithelial cells develop characteristic intercellular junctions such as tight junctions, adherens junctions, and desmosomes. Because of the apparent importance of epithelia, these junctions have been extensively studied and the basic properties such as overall structure, formation, and biological function of these structures have been well elucidated (Harmon and Green, 2013; Harris, 2012; Meng and Takeichi, 2009; Van Itallie and Anderson, 2014). As a result of these studies, many investigators think that the Ca2+-dependent cell adhesion protein E-cadherin plays a central role in the formation of intercellular junctions as well as in the formation of other epithelial features such as apico-basal polarity and cell structure.
Among different cadherins, E-cadherin has been extensively studied. The basic adhesion properties of cadherins have mainly been examined using a fibroblast cell line named L cells, despite the fact that E-cadherin is mostly expressed in epithelial cells (Meng and Takeichi, 2009), because a good E-cadherin-negative epithelial cell line has not been identified. This discrepancy raises a fundamental question whether the results obtained from the studies using fibroblasts can explain all the properties of E-cadherin in epithelial cells. Various properties can likely be elucidated using the fibroblasts, but to identify other properties, E-cadherin needs to be studied in epithelial cells. For example, the role of E-cadherin in apico-basal polarity-related processes cannot be studied in fibroblasts, because fibroblasts do not have this polarity. Hence, an E-cadherin-deficient cell line derived from epithelial cells that originally express E-cadherin needs to be used to characterize epithelia-related properties of E-cadherin. However, no such cell line is readily available, and the generation of an E-cadherin null cell line has been quite difficult, if not impossible. Indeed, E-cadherin-deficient epithelial cells such as A431D and MIA PaCa2 cells have been reported (Lewis et al., 1997; Ozaki et al., 2010), but they have not been used widely because of several difficulties such as the expression of other cadherins and no apico-basal polarity formation.
Recently, however, new gene editing methods, such as the transcription activator-like effector nuclease (TALEN) and clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 systems, have been developed (Cermak et al., 2011; Cong et al., 2013) and any given genes can be ablated in a short period of time. Currently, these methods have been primarily used for the studies of gene regulation in undifferentiated cells such as ES cells and fertilized eggs. The methods can also be used for the studies of gene function in differentiated cells, but few studies have been reported for these cases (Stroud et al., 2013; Incontro et al., 2014). One reason for the limited studies on differentiated cells may be that knockdown methods such as siRNA are widely available and can be easily applied to various studies (Lambeth and Smith, 2013). However, the knockdown method has several intrinsic problems. One problem is the inhibition of gene expression is variable and mostly incomplete and a significant level of gene expression remains in many cases, which may hinder some analyses. Gene knockout methods may be useful in these cases. In particular, re-constitution experiments are difficult for knockdown methods, especially for the application to complex multicomponent systems.
However, the gene knockout methods contain one potential drawback in the application of the methods to differentiated cells in addition to possible off-target effects. Most proteins interact with various other proteins, which may affect directly or indirectly the expression and/or function of other genes. Hence, the knockout of a gene may result in the alteration of properties in the target cells. Alternatively, a gene knockout may not significantly alter the properties in the target cells at least in some cases. An α-catenin-deficient DLD-1 cell line was isolated, and the re-expression of α-catenin in the cells rescued the cells to the parent-like cells (Lewis et al., 1997). Hence, the α-catenin-deficient cells were successfully used for the structure-function analysis of α-catenin by expressing various α-catenin mutants in the cells (Watabe-Uchida et al., 1998). The results suggest the possibility that the deletion of structural genes in differentiated cells, such as epithelial cells, does not drastically alter the properties of the cells and the re-expression of the gene in deficient cells may rescue the original properties of the cells. If this assumption were held in many other cases, then the approach using the TALEN and CRISPR/Cas9 systems would become a powerful tool to analyze the function of various proteins even in differentiated cells.
In this report, we generated E-cadherin knockout cells from epithelial cell lines using newly developed gene editing methods, the TALEN and CRISPR/Cas9 systems and the resultant cells were used for the characterization of E-cadherin properties in epithelial cells. The results confirmed various features of E-cadherin that had been characterized using the L cells fibroblast cell line. In addition, the present study clarified some unsettled questions and suggested a new role for E-cadherin in the formation of epithelial structure. This approach appears to be very promising. We describe the results and discuss the feasibility of this approach.
Materials and Methods
Reagents
The following antibodies were used: mouse anti-E-cadherin (Takara, Kyoto, Japan), anti-P-cadherin (Takara, Kyoto, Japan), mouse anti-desmoplakin (Progen, Heidelberg, Germany), mouse anti-desmocollin-2 (Invitrogen, Carlsbad, CA, USA), mouse anti-desmoglein-2 (Progen), mouse anti-occludin (Invitrogen), mouse anti-pan-cadherin (Sigma), rabbit anti-claudin-1 (Invitrogen), rabbit anti-ZO-1 (Invitrogen), mouse anti-N-cadherin (Invitrogen), rabbit anti-β-catenin (Sigma-Aldrich, St. Louis, MO, USA), mouse anti-γ-catenin (BD Transduction Laboratories, Lexington. KY, USA), mouse anti-E-cadherin (BD Transduction Laboratories), and mouse anti-HA (Nacalai, Kyoto, Japan). The polyclonal anti-β-actin antibody was a gift from Dr. S. Imaoka. Lipofectamine LTX and Dynabeads were obtained from Invitrogen. Blebbistatin and 12-O-Tetradecanoylphorbol 13-acetate (TPA) were obtained from Calbiochem (San Diego, CA, USA).
Cell culture
DLD-1 and MDCK cells were obtained from ATCC. An alpha-catenin-deficient DLD-1 clone (DLD-1(Δα)) was isolated as an adhesion-deficient clone by subcloning the original DLD-1 cells in our laboratory by using a procedure similar to the method described by van Hengel et al. (1997). Cells were cultured in Dulbecco’s modified Eagle’s medium-F12 (1:1) mixture containing 10% fetal bovine serum, streptomycin, and penicillin at 37°C in a 5% CO2 atmosphere. In some cases, cells were incubated with blebbistatin (50 nM) or TPA (100 nM) for 2–6 hr and then subjected to further analysis.
DNA constructs
cDNAs for human E-cadherin and N-cadherin were isolated from a human brain cDNA library. The resultant cDNAs were cloned into the pEF1 expression vector using appropriate restriction sites. Expression constructs of human E-cadherin and N-cadherin and mutant constructs of human E-cadherin were described previously (Ozaki et al., 2010). The transfection of culture cells was performed using Lipofectamine LTX (Invitrogen).
Knockout using TALEN and CRISPR/Cas9 systems
Gene disruption was conducted using the TALEN and CRISPR/Cas9 systems. TALEN #1 and #2 plasmids were constructed by the protocol described by Cermak et al. (2011) with modification (Sakuma et al., 2013a), and TALEN #3 plasmid was constructed as described by Sakuma et al. (2013b) using the following kits obtained from Addgene (Cambridge, MA, USA): Golden Gate TALEN and TAL Effector Kit (#1000000024) with TALEN Construction Accessory Pack (#1000000030) for TALEN #1 and #2; Platinum Gate TALEN Kit (#1000000043) for TALEN #3. CRISPR/Cas9 plasmid was constructed using the pX330 vector (Addgene, #42230) according to the method described by Cong et al. (2013) with some modifications. The SSA assay and Cel 1 assay were performed according to the method of Kim et al. (2009) and Kulinski et al (2000), respectively. The expression of the target protein in the resultant cells was analyzed by immunoblot analysis. To confirm the deletion or addition at the target site region in the genomic DNA, a DNA fragment containing the region was amplified by PCR from the genomic DNA purified from the cells using a DNeasy Blood & Tissue Kit according to the manufacturer’s protocol (Qiagen, Valencia, CA, USA). Alternatively, genomic PCR was directly performed from the cell suspension using KOD FX Neo (Toyobo, Tokyo, Japan). PCR products were subcloned into a vector and sequenced using an ABI PRISM 3100 Genetic Analyzer.
Immunofluorescence staining
Cells cultured on cover slips were fixed with cold methanol or paraformaldehyde. After washing the cells, the cells were permeated with 0.1% Triton X-100 for 5 min and incubated with 1% BSA in Tris-buffered saline (TBS). The resultant cells were reacted with a specific antibody for 3 hr at room temperature. Then, the cells were washed with TBS three times and reacted with secondary antibody conjugated with Alexa Fluor 488 or 518 for 1.5 hr. The resultant samples were briefly stained with DAPI and examined using a Nikon Eclips TE2000-U fluorescence microscope. In some cases, photographs were taken with a Nikon A1 laser confocal microscope (Nikon, Tokyo, Japan).
Immunoprecipitation
Immunoprecipitation was performed according to the method described by Ozaki et al. (2010). Briefly, cells were solubilized in 0.5% NP-40 in HBS for 30 min. After centrifugation, the supernatant was reacted with antibody-bound Dynabeads (Invitrogen) for 3 hr. The resultant samples were washed with HBS; and bound proteins were extracted with SDS-sample buffer and examined by immunoblot analysis or directly stained using the silver staining method.
Electron microscopy
Electron microscopy was performed as described previously (Obata and Usukura, 1992). Briefly, cells cultured on cover slips were fixed with paraformaldehyde and glutaraldehyde, and then osmium acid. The resultant cells were embedded in Epon after dehydration and sectioned. The sections were stained with uranium and lead and were subjected to electron microscopy using a Hitachi H7500 (Hitachi High-Technologies, Tokyo, Japan).
Other methods
The cell aggregation assay, SDS-PAGE, and imunoblot analysis were performed as described previously by Tai et al. (2010). The cell dissociation assay was performed according the method described by Nagafuchi et al. (1994). Silver staining was conducted using a kit from Daiichikagaku Co. following the manufacturer’s instructions.
Results
Generation of E-cadherin knockout DLD-1 cells
We generated E-cadherin knockout cells of an epithelial adenocarcinoma cell line, DLD-1 cells, using TALEN to clarify the function of E-cadherin in epithelia. Three TALEN constructs for the human E-cadherin gene were made using the Addgene Golden Gate TALEN and TAL Effector Kit or Platinum Gate TALEN Kit (Fig. 1). Fig. 1A shows the TALEN target sites for the E-cadherin gene knockout. The activities of the E-cadherin TALEN constructs were examined by SSA assay (Fig. 1B). Construct #3 showed the highest activity among the constructs in the assay and cut the target sequence as demonstrated by the Cel 1 assay (Fig. 1C). Then, construct #3 was introduced into DLD-1 cells by lipofection. After 14 days of culture, we obtained 5 clones that did not express E-cadherin by immunoblot analysis and determined the genomic sequences around the target site of the clones. The results showed that two clones contained in-frame deletions around the target site and 3 clones had out-of-frame deletions each of which generated a stop codon in the sequence downstream of the deletions (Fig. 1D). In subsequent experiments, we primarily used clone #4, but the other clones also showed similar results.

The resultant E-cadherin knockout cells (DLD1(ΔE)) showed no E-cadherin expression by immunoblot analysis, whereas other adhesion proteins were expressed at levels similar to those in wild type DLD-1 cells (Fig. 2A). DLD-1(ΔE) cells showed round fibroblast-like cell morphology (Fig. 2B) and had nearly no Ca2+-dependent cell aggregation activity (Fig. 2C). Then, we performed immunoprecipitation experiments using an anti-β-catenin antibody to examine whether the knockout cells expressed other classical cadherins, because classical cadherins tightly associate with β-catenin (Ozawa et al., 1990). The results indicated that β-catenin co-precipitated with E-cadherin in DLD-1 cells and DLD-1(Δα) cells. However, β-catenin did not co-precipitate with any clear bands in the range of 100–130 kDa in DLD-1(ΔE) cells, although smear was detected in the range (Fig. 2D). E-cadherin was not detected in the knockout cells by immunostaining using antibody against the cytoplasmic domain of E-cadherin (Fig. 2E) and β-catenin was not localized at the intercellular contact sites (data not shown). However, the tight junction marker ZO-1 showed dot-like clusters that were not localized well at intercellular contact sites and the desmosome marker desmoplakin (DSP) also revealed patch-like accumulation that was localized diffusely on the cell surface (Fig. 2E) (Demlehner et al., 1995).

Another E-cadherin-defective cell line lacking α-catenin expression has been isolated from DLD-1 cells (van Hengel et al., 1997). We independently obtained similar α-catenin-deficient cells from DLD-1 cells (DLD-1(Δα)) and compared the properties with those of DLD-1(ΔE) cells. DLD-1(Δα) cells showed a round fibroblastic cell shape and revealed no or very weak Ca2+-dependent cell adhesion activity by a cell aggregation assay (van Hengel et al., 1997). By immunofluorescence staining, E-cadherin was hardly detected at intercellular contact region, whereas DSP formed plaques and was diffusely localized on the cell surface (Fig. 2E). ZO-1 formed dot-like clusters, but they were not localized at the cell-cell adhesion sites. These properties were very similar to those of the DLD-1(ΔE) cells.
Re-expression of E-cadherin in E-cadherin knockout DLD-1(ΔE) cells
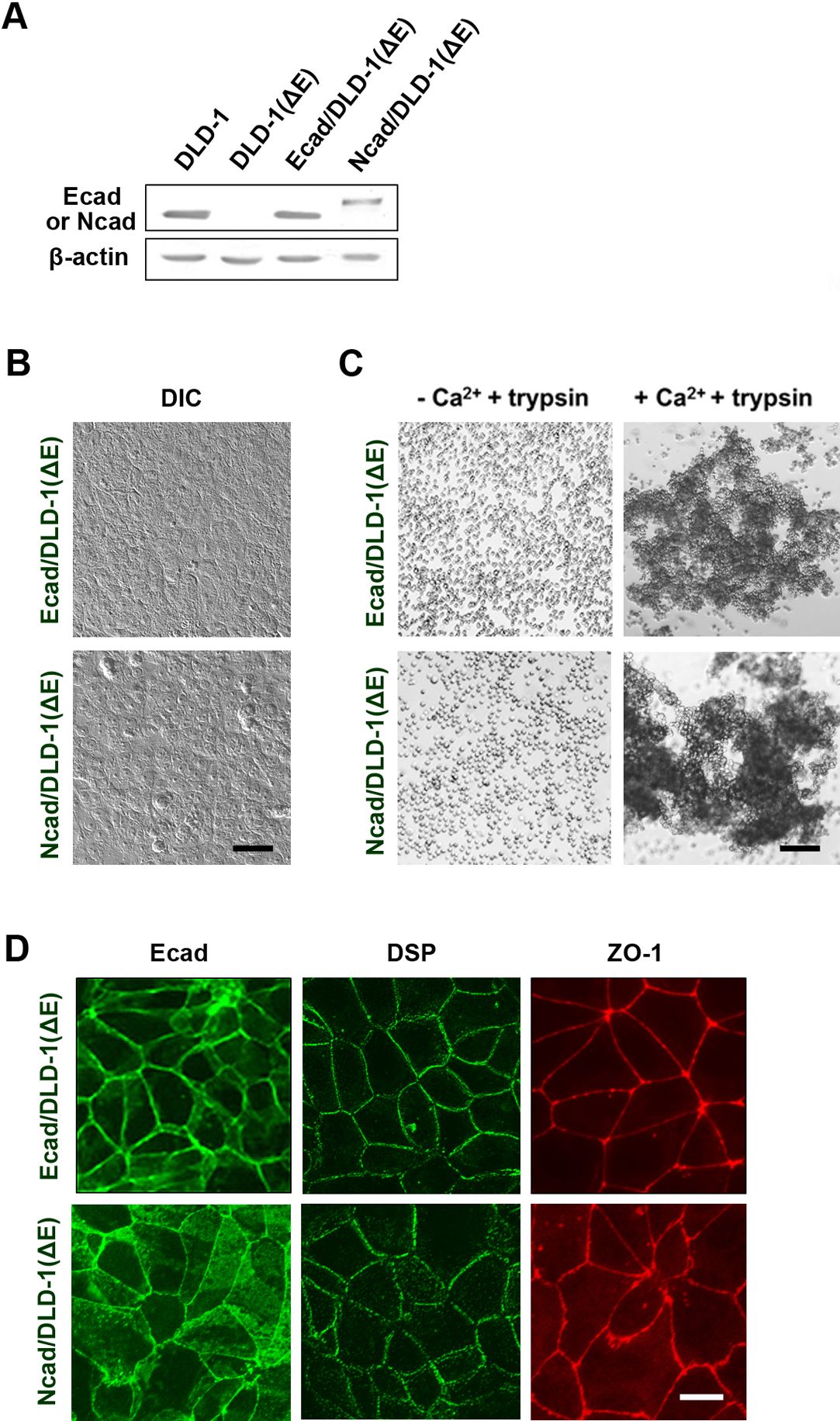
E-cadherin was re-expressed from cDNA in DLD-1(ΔE) cells using the calcium phosphate precipitation method (Fig. 3). After screening with G418, multiple clones were obtained. The transfectants expressed E-cadherin (Fig. 3A) and showed typical epithelial morphology (Fig. 3B). A cell aggregation assay clearly indicated that the resultant cells had strong Ca2+-dependent cell adhesion activity (Fig. 3C). Moreover, immunofluorescence staining demonstrated that E-cadherin, DSP, and ZO-1 were localized at intercellular contact sites as in the parental DLD-1 cells (Fig. 3D) and showed possible apico-basal polarity.
N-cadherin was expressed in the knockout cells to examine whether other cadherin could form epithelial structure. The transfectants revealed properties similar to those of the E-cadherin transfectants (Fig. 3). The transfectants showed epithelial morphology and N-cadherin, DSP, and ZO-1 were localized at intercellular contact sites (Fig. 3D).
Together, these results indicate that DLD-1(ΔE) cells retained basic epithelial properties, thereby the re-expression of E-cadherin recovered epithelial structure and strong cell adhesion activity. Therefore, the knockout cells can be used for the study of E-cadherin function in epithelial cells.
Expression of deletion constructs of E-cadherin in DLD-1(ΔE) cells
Next, we made three deletion constructs of the E-cadherin cytoplasmic domain, expressed the constructs in the DLD-1(ΔE) cells, and examined the properties of the resultant transfectants. First, we examined the transfectants of an E-cadherin mutant lacking the entire cytoplasmic domain, Ecad(ΔCP) (Fig. 4A and B). The transfectants showed fibroblast-like morphology (Fig. 4C). By immunofluorescence staining, Ecad(ΔCP) was weakly localized at cell-cell contact sites (Fig. 4E) and DSP clusters and ZO-1 were localized at intercellular contact sites, which were clearer than those in DLD-1(ΔE) cells (Fig. 2E). However, a significant number of the clusters were localized in other parts of the cells.

p120-catenin is generally thought to play critical roles in cadherin function (Pieters et al., 2012), and p120-catenin has been suggested to be involved in polarity formation (Nelson et al., 2013). However, only the regulatory role in E-cadherin recycling has been established (Miyashita and Ozawa, 2007). Hence, we expressed mutant E-cadherins Ecad(ΔDM/p120A) and Ecad(Δ18) that had a mutated or deleted p120-catenin binding site in DLD-1(ΔE) cells (Ohkubo and Ozawa, 1999; Ozaki et al., 2010) (Fig. 4A and B). The transfectants showed an epithelial cell shape very similar to that of the parental cells (Fig. 4C). Immunoprecipitation of the resultant transfectants revealed reduced p120-catenin binding activity (Fig. 4D). Immunofluorescence staining demonstrated that ZO-1 localized at cell-cell contact sites of the transfectants but was irregularly localized in some regions (Fig. 4E). The Localization patterns of E-cadherin and DSP were minimally changed.
When the Ecad(ΔCP) transfectants of DLD-1(ΔE) cells were cultured for 3–5 days, a portion of the cells showed epithelial-like localization of DSP and ZO-1 and cell arrangement (Fig. 4F). DLD-1(ΔE) cells occasionally showed similar properties.
The cell adhesion activities of the above transfectants were examined and quantified. First, we quantified the results of the cell aggregation assay (Fig. 5A). DLD-1(ΔE) cells showed nearly no cell adhesion activity, whereas the E-cadherin transfectants of DLD-1(ΔE) cells showed strong cell aggregation activity. The Ecad(ΔCP) transfectants of DLD-1(ΔE) cells showed weak but significant Ca2+-dependent cell adhesion activity, and the Ecad(ΔDM/p120A) transfectants of DLD-1(ΔE) cells showed strong but slightly weaker activity than that of wild type E-cadherin transfectants. The Ecad(Δ18) transfectants of DLD-1(ΔE) cells showed moderate cell aggregation activity. We also performed a cell dissociation assay and quantified the results (Fig. 5B). The results were mostly comparable to those of the cell aggregation assay. These results suggest that the localization of ZO-1 and DSP are roughly correlated with the cell adhesion activity of the cells used.

DLD-1(ΔE) cells showed occasional weak localization of DSP and ZO-1 at intercellular contact sites (Fig. 2E), and the Ecad(ΔCP) transfectants of DLD-1(ΔE) cells showed the localization of DSP and ZO-1 at cell-cell contact sites (Fig. 4E), which led us to speculate that immature desmosomes and tight junctions might be formed in E-cadherin-deficient or E-cadherin-compromised cells under certain conditions. First, we cultured DLD-1(ΔE) cells for 4 days and examined the localization of DSP and ZO-1 (Fig. 6). The results demonstrated that ZO-1 clearly formed linear but discontinuous localization at intercellular contact sites in DLD-1(ΔE) cells and the DSP plaques became localized to sites similar to those of ZO-1 (Fig. 6A). Desmocollin (DSC), desmoglein (DSG), and occludin (OCLN) also showed a similar localization (data not shown). These results suggest the formation of some type of cell-cell junctions. Indeed, a cell dissociation assay indicated that DLD-1(ΔE) cells had very weak but clear Ca2+-dependent cell adhesion activity (Fig. 6B). However, they never showed strong cell adhesion activity. In contrast, careful examination revealed the ring-like localization of DSP and ZO-1 in some of the cells that had nearly no cell-cell contact with other cells (Fig. 6A), suggesting the partial formation of polarity, possibly apico-basal polarity, in these cells: Similar localization suggesting apico-basal polarity formation was already reported by Baas et al. (2004). However, the polarity seems to be incomplete. DLD-1(Δα) cells also showed ring-like localization of ZO-1 and DSP (Fig. 6A).

These results indicated that DLD-1(ΔE) cells as well as DLD-1(Δα) cells had intrinsic localization activities of desmosome and tight junction proteins at intercellular contact sites after a prolonged culture even in the absence of E-cadherin. In addition to these results, TPA was reported to enhance the localization of DSP in DLD-1(Δα) cells (van Hengel et al., 1997). Therefore, we postulated that the treatment of cells with some types of reagents might enhance the accumulation and/or localization of these proteins. When the cells were incubated with various reagents, blebbistatin a non-muscle myosin II inhibitor showed a profound effect on the localization of DSP and ZO-1 (Fig. 6C). After incubation, the DLD-1(ΔE) cells and DLD-1(Δα) cells formed good contacts with each other. Furthermore, DSP and ZO-1 localized to the cell-cell contact sites, although cell compaction was not detected. TPA enhanced DSP localization in the DLD-1(Δα) cells as reported previously (van Hengel et al., 1997), but TPA did not enhance ZO-1 localization. TPA also increased the localization of DSP, but not ZO-1, in DLD-1(ΔE) cells.
Examination of cell junction formation by electron microscopy
Next, the ultrastructure of these cells was examined by electron microscopy (Fig. 7) to confirm the formation of desmosomes and tight junctions. DLD-1(ΔE) cells cultured for 1 day formed half desmosomal structures (Demlehner et al., 1995) and only occasionally formed desmosomes (Fig. 7A), whereas DLD-1(ΔE) cells cultured for 3 days formed tight junction-like structures at the subapical region of the lateral membrane and desmosomes at the lower region (Fig. 7B). Ecad(ΔCP) transfectants of DLD-1(ΔE) cells clearly formed desmosomes and tight junctions or tight junction-like structures at regions similar to those in DLD-1 cells (Fig. 7C). The above results also suggest the formation of apico-basal polarity in these cells. Ecad(ΔDM/p120A) transfectants showed the formation of desmosomes and tight junction-like structures similar to those of wild type DLD-1 cells (Fig. 7D). DLD-1(ΔE) cells treated with blebbistatin clearly showed the formation of desmosomes (Fig. 7E). These results are roughly comparable to those of immunofluorescence staining (Fig. 2, Fig. 3, Fig. 4).

The above results indicated that DLD-1(ΔE) cells showed partial localization of DSP and ZO-1 at intercellular contact sites, the formation of immature apico-basal polarity, and even epithelial cell arrangement. However, these properties may be exceptional, because DLD-1 is an adenocarcinoma cell line. We ablated the E-cadherin and cadherin-6 genes in MDCK II cells and characterize the properties of the knockout cells to examine whether the above properties were specific to DLD-1 cells (Fig. 8). We obtained 2 clones that had deletions or an addition at the target sites that disrupted the reading frames and formed new stop codons (Fig. 8B). In this study, clone #2 was primarily used. An immunoblot analysis showed no expression of E-cadherin and cadherin-6 in the cells (Fig. 8C). Immunoprecipitation using an anti-β-catenin antibody also indicated that no significant band in the range of 100–130 kDa co-precipitated with β-catenin, indicating the expression of no other classical cadherins (Fig. 8D). The knockout cells showed round fibroblast-like cell morphology and had no significant Ca2+-dependent cell aggregation activity (Fig. 8E and F). However, the re-expression of E-cadherin in the knockout cells restored the epithelial cell shape and strong Ca2+-dependent cell aggregation activity (Fig. 8F). Moreover, the re-expressed E-cadherin was localized at cell-cell contact sites (Fig. 8G).

When the double-knockout cells were cultured for 3–5 days, DSP plaques were linearly but partially localized at cell-cell contact sites and ZO-1 was linearly localized at cell-cell contact sites (Fig. 9A). The localization of DSP and ZO-1 was clearer than that in DLD(ΔE) cells. Interestingly, the cells that had nearly no contact with other cells formed a ring-like accumulation of DSP and ZO-1 (Fig. 9B), suggesting that the knockout cells formed polarity, possibly apico-basal polarity. Moreover, a portion of the knockout cells formed epithelial-like structure.
When the double-knockout cells were incubated with blebbistatin, the cells closely contacted each other (Fig. 9C) and DSP and ZO-1 were localized at intercellular contact sites. The cell arrangement was very similar to that of epithelia, but compaction was hardly observed. TPA slightly enhanced the localization of DSP but not ZO-1.
Discussion
The E-cadherin gene in epithelial DLD-1 cells was successfully ablated by TALEN in this study (Fig. 1). However, the immunoprecipitation of DLD-1(ΔE) cells with anti-β-catenin antibody showed smear in the rage of 100–130 kDa (Fig. 2D). We think that the smear does not indicate the expression of abnormal E-cadherin or other classical cadherins in the cells because of the following reasons. (a) Antibody against the cytoplasmic domain of E-cadherin did not detect the smear in immunoblot analysis nor show E-cadherin staining in immunofluorescence staining (Fig. 2A and E). (b) Antibody against β-catenin did not reveal the localization of β-catenin at cell-cell contact sites. Furthermore, pan-cadherin antibody that recognized cadherin-6 and antibodies against P-cadherin, cadherin-4, and cadherin-5 we had did not detect any band in immunoblot analysis of DLD-1(ΔE) cells (data not shown). Hence, we concluded that DLD-1(ΔE) cells did not express significant amount of classical cadherins, although the cells expressed a high level of β-catenin. The significant expression of β-catenin in the knockout cells (Fig. 2A) may be due to the expression of truncated APC protein in DLD-1 cells (Yang et al., 2006).
The efficiency of E-cadherin knockout was not high and the knockout clones were less than 1% of the total clones isolated after screening. This frequency is apparently much lower than that reported for fertilized eggs (Peng et al., 2014). When the CRISPR/Cas9 system was used for the E-cadherin knockout, the efficiency of the knockout increased, but it was still less than 1%. We examined several other CRISPR/Cas9 constructs, but the results did not drastically change. In contrast, the knockout of desmosomal cadherin genes, which are closely related to the classical cadherin genes, yielded a much higher success rate (Fujiwara et al., in press). A good tool to design efficient targets may be necessary in the cases of the low efficiency.
One concern regarding the application of these methods is the off-target effects. However, the re-expression of E-cadherin as well as N-cadherin in the knockout cells restored epithelial morphology and strong cell adhesion activity, indicating that the knockout cells can support the basic function of classical cadherins. The results do not necessarily indicate that the knockout cells have no off-target mutations. However, we did not try to directly detect the possible off-target effects that concern many investigators, because we do not have a simple general method to detect these effects. Instead, we used multiple target sites and employed both the TALEN and CRISPR/Cas9 systems to confirm the results, and the resultant E-cadherin knockout cells showed essentially the same results. Taken together, these results indicated that the knockout cells retained the basic epithelial properties to support E-cadherin function. We think that the knockout cells are applicable to the study of classical cadherin function in epithelial cells, even though the cells may have some off-target effects and/or secondary effects due to the knockout. The results also suggest that this type of approach can be applied to the studies of various other genes in differentiated cells in culture.
Epithelial cells show characteristic features. One feature is the well-developed intercellular junctions. The present study clarifies a couple of the functions of E-cadherin in junction formation in epithelial cells using a new gene-editing method. E-cadherin knockout DLD-1(ΔE) cells became round or fibroblastic and did not form appreciable desmosomes or tight junctions, although the junction proteins were expressed in the cells (Fig. 2A). However, the junction proteins such as DSP and ZO-1 accumulated and were localized at the intercellular contact sites of DLD-1(ΔE) cells, especially when the cells were cultured for a prolonged time (3–5 days). Furthermore, the cells showed very weak but clear Ca2+-dependent cell adhesion activity in a cell dissociation assay (Fig. 6B). The partial formation of desmosomes or desmosome-like structures may be responsible for this activity. Indeed, electron microscopy demonstrated the formation of desmosomes or desmosome-like structures in the transfectants (Fig. 7). The results indicate that the localization of junctional proteins and possibly junction formation do not necessarily require E-cadherin in DLD-1 cells. However, the localization of these proteins was weak and incomplete in DLD-1(ΔE) cells compared with parental DLD-1 cells.
These features seem to be general among vertebrate epithelial cells, because E-cadherin and cadherin-6 double-knockout MDCK II cells showed similar properties (Fig. 8 and Fig. 9). Capaldo and Macara (2007) reported that the knockdown of E-cadherin in MDCK cells disrupted the formation but not maintenance of cell junctions, which were partially different from our results. We have no good explanation for them, but it may be due to the difference in the methods used.
Examination of transfectants of different E-cadherin mutants showed that the localization of the junction proteins was roughly correlated with the strength of cell adhesion activity (Fig. 4 and Fig. 5). The results suggest that one role of E-cadherin in junction formation is to stabilize cell-cell contact sites by the cell adhesion activity which in turn facilitates the localization of DSP and ZO-1 at the sites. Indeed, the Ecad(ΔCP) transfectants of DLD-1(ΔE) cells showed a clearer linear localization of DSP plaques and ZO-1 at intercellular contact sites than that of DLD-1(ΔE) cells, although Ecad(ΔCP) did not contain the cytoplasmic domain and the transfectants showed weak cell adhesion activity (Fig. 4 and Fig. 5). Moreover, the blebbistatin experiments seem to support the above notion: The blebbistatin treatment of DLD-1(ΔE) cells enhanced the localization of junctional proteins at the intercellular contact sites without E-cadherin (Fig. 6). The mechanism is unclear, but one explanation could be that the inhibition of non-muscle myosin II reduces the cell tension and stabilizes the cell-cell contact sites, thereby increases the localization of junctional proteins.
The present results do not necessarily exclude the possibility that E-cadherin is involved in a signaling pathway that regulates the junction formation. A previous study reported that DLD-1(Δα) cells do not form desmosomes, but TPA facilitated desmosome formation in an E-cadherin dependent manner without α-catenin (van Hengel et al., 1997). The present results confirmed this report and demonstrated further that TPA weakly enhanced the localization of DSP in DLD-1(ΔE) cells and in the double-knockout MDCK cells. However, TPA did not dramatically affect the localization of ZO-1 in DLD-1(ΔE) cells, DLD-1(Δα) cells, or the double-knockout MDCK cells. TPA is known to enhance the formation of desmosomes via the phosphorylation of DSP by protein kinase C (Harmon and Green, 2013), but this mechanism is not known for ZO-1 localization. Therefore, the mechanism of TPA’s effect on ZO-1 localization seems to be different from that on DSP. However, the relationship between the vital role of E-cadherin in junction formation and the TPA effect remains to be elucidated.
Another characteristic feature of epithelial cells is the formation of apico-basal polarity. The juxtamembrane region in the cytoplasmic domain of E-cadherin associates with p120-catenin and is suggested to participate in polarity formation (Nelson et al., 2013) in addition to its regulatory role in the recycling of E-cadherin (Miyashita and Ozawa, 2007). However, the present result did not support the involvement of p120-catenin in apico-basal polarity formation. In fact, E-cadherin knockout cells formed ring-like clusters of ZO-1 and DSP at their apical regions without cell-cell contact, which implies the formation of immature apico-basal polarity (Fig. 6). Furthermore, electron microscopy showed the formation of tight junction at the subapical region (Fig. 7D). Capaldo and Macara (2007) obtained similar conclusion using a knockdown approach. Indeed, Baas et al. (2004) previously reported that LKB1 was necessary and sufficient for the formation of apico-basal polarity even in single cells. Notably, the formation of mature apico-basal polarity never occurred in E-cadherin knockout cells or Ecad(ΔCP) transfectants (Fig. 4), indicating the importance of E-cadherin for the completion of apico-basal polarity.
The role of E-cadherin in the formation of epithelial structure has also been examined at organismal level. Costa et al. (1998) reported that C. elegans expressed a possible ortholog of vertebrate E-cadherin named HMR1 and the protein was dispensable for the formation and maintenance of cell adhesion in embryos by mutation analysis. On the other hand, Bondow et al. (2012) reported conditional knockout of E-cadherin in the mouse intestinal epithelium. The knockout mice formed intestinal epithelium, although the structure was severely damaged. Interestingly, the junctional complexes were evident in the epithelium, although the barrier function was compromised. Furthermore, incomplete apico-basal polarity was formed in the epithelium. Schneider et al. (2010) reported similar conditional knockout experiments. In the present study, a small portion of the Ecad(ΔCP) transfectants and some DLD-1(ΔE) cells in long-term culture showed epithelia-like cell arrangement (Fig. 4E). Hence, our present results using cultured cells seem to be mostly consistent with these results.
In this study, gene editing methods were successfully applied to a study of E-cadherin function in epithelial cells. The results suggest that the formation of basic epithelial structure does not necessarily require fully-functional cadherin, but the formation of mature epithelia requires E-cadherin. E-cadherin may organize the entire process of formation of epithelial structures, possibly through its binding proteins and actin-based cytoskeleton. Moreover, E-cadherin may have acquired the vital function in epithelium during the evolution of vertebrate. Naturally, further studies are required to elucidate the mechanism of epithelial formation with and without E-cadherin, but the present knockout cells may provide a useful model system for further investigation.
Acknowledgments
We thank to Drs. K. Ohbo, M. Ono, and K. Yoshida (Yokohama City University School of Medicine) for their helpful discussion on electron microscopy.
Funding
This study was supported in part by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKENHI #24240062 to S. O.) and from Kwansei Gakuin University (#167AB0177b to S. T. S).
References
- Baas, A.F., Kuipers, J., van der Wel, N.N., Batlle, E., Koerten, H.K., Peters, P.J., and Clevers, H.C. 2004. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell, 116: 457–466.
- Bondow, B.J., Faber, M.L., Wojta, K.J., Walker, E.M., and Battle, M.A. 2012. E-cadherin is required for intestinal morphogenesis in the mouse. Dev. Biol., 371: 1–12.
- Capaldo, C.T. and Macara, I.G. 2007. Depletion of E-Cadherin Disrupts Establishment but Not Maintenance of Cell Junctions in Madin-Darby Canine Kidney Epithelial Cells. Mol. Biol. Cell, 18: 189–200.
- Cermak, T., Doyle, E.L., Christian, M., Wang, L., Zhang, Y., Schmidt, C., Baller, J.A., Somia, N.V., Bogdanove, A.J., and Voytas, D.F. 2011. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res., 39: e82.
- Cong, L., Ran, F.A., Cox, D., Lin, S., Barretto, R., Habib, N., Hsu, P.D., Wu, X., Jiang, W., Marraffini, L.A., and Zhang, F. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science, 339: 819–823.
- Costa, M., Raich, W., Agbunag, C., Leung, B., Hardin, J., and Priess, J.R. 1998. A putative catenin-cadherin system mediates morphogenesis of the Caenorhabditis elegans embryo. J. Cell Biol., 141: 297–308.
- Demlehner, M.P., Schafer, S., Grund, C., and Franke, W.W. 1995. Continual assembly of half-desmosomal structures in the absence of cell contacts and their frustrated endocytosis: a coordinated Sisyphus cycle. J. Cell Biol., 131: 745–760.
- Fujiwara, M., Nagatomo, A., Tsuda, M., Obata, S., Sakuma, T., Yamamoto, Y., and Suzuki, S.T. Desmocollin-2 alone forms functional desmosomal plaques, with the plaque formation requiring the juxtamembrane region and plakophilins. J. Biochem., in press.
- Harmon, R.M. and Green, K.J. 2013. Structural and functional diversity of desmosomes. Cell Commun. Adhes., 20: 171–187.
- Harris, TJ. 2012. An introduction to adherens junctions: from molecular mechanisms to tissue development and disease. Subcell Biochem., 60: 1–5.
- Incontro, S., Asensio, C.S., Edwards, R.H., and Nicoll, R.A. 2014. Efficient, complete deletion of synaptic proteins using CRISPR. Neuron, 83: 1051–1057.
- Kim, H.J., Lee, H.J., Kim, H., Cho, S.W., and Kim, J.S. 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res., 19: 1279–1288.
- Kulinski, J., Besack, D., Oleykowski, C.A., Godwin, A.K., and Yeung, A.T. 2000. CEL I enzymatic mutation detection assay. Biotechniques, 29: 44–46.
- Lambeth, L.S. and Smith, C.A. 2013. Short hairpin RNA-mediated gene silencing. Methods Mol. Biol., 942: 205–232.
- Lewis, J.E., Wahl, J.K., 3rd, Sass, K.M., Jensen, P.J., Johnson, K.R., and Wheelock, M.J. 1997. Cross-talk between adherens junctions and desmosomes depends on plakoglobin. J. Cell Biol., 136: 919–934.
- Meng, W. and Takeichi, M. 2009 Adherens junction: molecular architecture and regulation. Cold Spring Harb. Perspect. Biol., 1: a002899.
- Miyashita, Y. and Ozawa, M. 2007. Increased internalization of p120-uncoupled E-cadherin and a requirement for a dileucine motif in the cytoplasmic domain for endocytosis of the protein. J. Biol. Chem., 282: 11540–11548.
- Nagafuchi, A., Ishihara, S., and Tsukita, S. 1994. The roles of catenins in the cadherin-mediated cell adhesion: functional analysis of E-cadherin-alpha catenin fusion molecules. J. Cell Biol., 127: 235–245.
- Nelson, W.J., Dickinson, D.J., and Weis, W.I. 2013. Roles of cadherins and catenins in cell-cell adhesion and peithelilal cell polarity. Prog. Mol. Biol. Transl. Sci., 16: 3–23.
- Obata, S. and Usukura, J. 1992. Morphogenesis of the photoreceptor outer segment during postnatal development in the mouse (BALB/c) retina. Cell Tissue Res., 269: 39–48.
- Ohkubo, T. and Ozawa, M. 1999. p120(ctn) binds to the membrane-proximal region of the E-cadherin cytoplasmic domain and is involved in modulation of adhesion activity. J. Biol. Chem., 274: 21409–21415.
- Ozaki, C., Obata, S., Yamanaka, H., Tominaga, S., and Suzuki, S.T. 2010. The extracellular domains of E- and N-cadherin determine the scattered punctate localization in epithelial cells and the cytoplasmic domains modulate the localization. J. Biochem., 147: 415–425.
- Ozawa, M., Ringwald, M., and Kemler, R. 1990. Uvomorulin-catenin complex formation is regulated by a specific domain in the cytoplasmic region of the cell adhesion molecule. Proc. Natl. Acad. Sci. USA, 87: 4246–4250.
- Peng, Y., Clark, K.J., Campbell, J.M., Panetta, M.R., Guo, Y., and Ekker, S.C. 2014. Making designer mutants in model organisms. Development, 141: 4042–4054.
- Pieters, T., van Hengel, J., and van Roy, F. 2012. Functions of p120ctn in development and disease. Front. Biosci. (Landmark Ed.), 17: 760–783.
- Sakuma, T., Hosoi, S., Woltjen, K., Suzuki, K., Kashiwagi, K., Wada, H., Ochiai, H., Miyamoto, T., Kawai, N., Sasakura, Y., Matsuura, S., Okada, Y., Kawahara, A., Hayashi, S., and Yamamoto, T. 2013a. Efficient TALEN construction and evaluation methods for human cell and animal applications. Genes Cells, 18: 315–326.
- Sakuma, T., Ochiai, H., Kaneko, T., Mashimo, T., Tokumasu, D., Sakane, Y., Suzuki, K., Miyamoto, T., Sakamoto, N., Matsuura, S., and Yamamoto, T. 2013b. Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Scientific Reports, 3: 3379.
- Schneider, M.R., Dahlhoff, M., Horst, D., Hirschi, B., Trulzsch, K., Muller-Hocker, J., Vogelmann, R., Allgauer, M., Gerhard, M., Steininger, S., Wolf, E., and Kolligs, F. 2010. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS One, 5: e14325.
- Stroud, D.A., Formosa, L.E., Wijeyeratne, X.W., Nguyen, T.N., and Ryan, M.T. 2013. Gene knockout using transcription activator-like effector nucleases (TALENs) reveals that human NDUFA9 protein is essential for stabilizing the junction between membrane and matrix arms of complex I. J. Biol. Chem., 288: 1685–1690.
- Tai, K., Kubota, M., Shiono, K., Tokutsu, H., and Suzuki, S.T. 2010. Adhesion properties and retinofugal expression of chicken protocadherin-19. Brain Res., 1344: 13–24.
- van Hengel, J., Gohon, L., Bruyneel, E., Vermeulen, S., Cornelissen, M., Mareel, M., and von Roy, F. 1997. Protein kinase C activation upregulates intercellular adhesion of a-catenin-negative human colon cancer cell variants via induction of desmosomes. J. Cell Biol., 137: 1103–1116.
- Van Itallie, C.M. and Anderson, J.M. 2014. Architecture of tight junctions and principles of molecular composition. Semin. Cell Dev. Biol., 36: 157–165.
- Watabe-Uchida, M., Uchida, N., Imamura, Y., Nagafuchi, A., Fujimoto, K., Uemura, T., Vermeulen, S., van Roy, F., Adamson, E.D., and Takeichi, M. 1998. alpha-Catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J. Cell Biol., 142: 847–857.
- Yang, J., Zhang, W., Evans, P.M., Chen, X., He, X., and Liu, C. 2006. Adenomatous polyposis coli (APC) differentially regulates beta-catenin phosphorylation and ubiquitination in colon cancer cells. J. Biol. Chem., 281: 17751–17757.