Abstract
Although the co-development of companion diagnostics with molecular targeted drugs is desirable, truly efficient diagnostics are limited to diseases in which chromosomal translocations or overt mutations are clearly correlated with drug efficacy. Moreover, even for such diseases, few methods are available to predict whether drug administration is effective for each individual patient whose disease is expected to respond to the drug(s). We have previously developed a biosensor based on the principle of Förster resonance energy transfer to measure the activity of the tyrosine kinase BCR-ABL and its response to drug treatment in patient-derived chronic myeloid leukemia cells. The biosensor harbors CrkL, one of the major substrates of BCR-ABL, and is therefore named Pickles after phosphorylation indicator of CrkL en substrate. The efficacy of this technique as a clinical test has been demonstrated, but the number of cells available for analysis is limited in a case-dependent manner, owing to the cleavage of the biosensor in patient-derived leukemia cells. Here, we describe an improved biosensor with an amino acid substitution and a nuclear export signal being introduced. Of the two predicted cleavage positions in CrkL, the mutations inhibited one cleavage completely and the other cleavage partially, thus collectively increasing the number of cells available for drug evaluation. This improved version of the biosensor holds promise in the future development of companion diagnostics to predict responses to tyrosine kinase inhibitors in patients with chronic myeloid leukemia.
Introduction
Chronic myeloid leukemia (CML), a hematopoietic malignancy, is characterized by the formation of the Philadelphia chromosome (Ph), which arises via a reciprocal translocation between chromosomes 9 and 22 (Kurzrock et al., 1988). The bcr-abl fusion oncogene generated at the break point encodes a constitutively active tyrosine kinase (Kurzrock et al., 1998) and acts as a causative driver gene in CML through the tyrosine phosphorylation of its substrates, including CrkL and signal transducer and activator of transcription (Groffen et al., 1984; Heisterkamp et al., 1982). In 2001, the tyrosine kinase inhibitor imatinib mesylate (IM) was approved as a first-line therapy for CML (Druker, 2008; Druker et al., 1996). The therapeutic outcomes associated with this agent are better than those of alternative options, which include chemotherapy, interferon, and allogeneic hematopoietic stem cell transplantation. This means that CML is a disease that can be effectively controlled by oral medication (Druker, 2008; Druker et al., 1996).
Although the overall 5-year survival rate of IM-treated patients is approximately 80%, a considerable number of patients do not demonstrate optimal responses to this therapy. In addition, IM resistance is acquired during treatment in certain cases. Resistance is attributable to mutations in the Abl kinase domain in ~50% of cases. In this situation, the second-generation drugs nilotinib (NL) and dasatinib (DS) are available, and their effectiveness is predictable based on the types of mutations present (Weisberg et al., 2007). However, if no mutations are detected, because of either a lack of mutation or the presence of a small population of mutation-positive cells, drug efficacy is determined through the long-term follow-up of alterations in disease status, as determined by blood and bone marrow tests (throughout the treatment period, and then for several months to a year or more). Clinical tests include total blood cell count, detection of Ph by fluorescence in-situ hybridization, and quantification of BCR-ABL mRNA levels by qPCR (Kantarjian et al., 2002; Preudhomme et al., 1999; Tkachuk et al., 1990). Although BCR-ABL mRNA quantification is widely used to monitor disease status and drug efficacy, it reflects the number of tumor cells present and hence only measures current drug treatment outcomes. Therefore, the development of techniques to predict future responsiveness is desirable.
We previously developed the Förster resonance energy transfer (FRET)-based biosensor Pickles (phosphorylation indicator of CrkL en substrate) and successfully and accurately monitored BCR-ABL activity in living patient-derived CML cells (Mizutani et al., 2010). This biosensor enables detection of a small population of drug-resistant cells among a heterogeneous leukemia cell population and prediction of future drug responses for individual patients. However, in certain patient-derived CML cells, the biosensor undergoes cleavage. Because Pickles was designed as an intramolecular FRET biosensor consisting of a pair of donor and acceptor fluorescent proteins (cyan and yellow fluorescent proteins, CFP and YFP), cells must express equal amounts of the donor and acceptor proteins. Therefore, cells expressing “incomplete biosensors” must be excluded, thus limiting the number of available cells for evaluation and assay performance. In this study, we describe improved versions of the Pickles biosensor, including amino acid substitution and a nuclear export signal (NES) introduced to prevent cleavage. This improved biosensor increases the number of cells available for analysis, thus making Pickles a promising companion diagnostic tool to predict future drug responses in CML patients.
Materials and Methods
Reagents and antibodies
IM and NL were kind gifts from Novartis Pharma (Basel, Switzerland), and DS was obtained from Bristol-Myers Squibb (New York, NY, USA). Anti-CrkL mouse monoclonal and anti-phospho CrkL (Y207) rabbit polyclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA), whereas an anti-c-Abl mouse monoclonal antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An anti-FLAG mouse monoclonal antibody was purchased from Agilent Technologies (Palo Alto, CA, USA). Antisera against GFP were kindly provided by Dr. M. Matsuda. A horse radish peroxidase-conjugated goat anti-mouse IgG and a horse radish peroxidase-conjugated goat anti-rabbit IgG were purchased from Invitrogen (Carlsbad, CA, USA) and Sigma-Aldrich (St. Louis, MO, USA), respectively.
Expression plasmids
The expression vectors for the Pickles prototype (2.31) and for BCR-ABL have been reported previously (Mizutani et al., 2010). The primers hCrkL_dEco_F (5'-gcatggaaataggaaCtccaacagttatggg-3') and hCrkL_dEco_R (5'-cccataactgttggaGttcctatttccatgc-3') (capital letters indicate bases that differ from the template) were used to obtain pPickles 2.33, and PCR was performed using a QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s protocol. A silent mutation at the EcoRI site in pPickles 2.33 was introduced to facilitate the following procedures. The amino acid sequences of Pickles 2.31 and 2.33 were therefore identical (see Fig. 1, Table I). Next, using the primers hCrkL_D94A_F (5'-agatccactacctggCcaccaccaccctcat-3') and hCrkL_D94A_R (5'-atgagggtggtggtgGccaggtagtggatct-3'), we performed PCR by using the QuikChange kit and obtained pPickles 2.34, in which D94 in CrkL was substituted by alanine. The coding sequences of Pickles 2.31, 2.33, and 2.34 were cleaved by EcoRI and BglII and subcloned into the EcoRI-BglII site of the pFX vector to obtain pFX-Pickles 2.31, 2.33, and 2.34, respectively. Pickles 2.35 was constructed with the primers hCrkL_E202A_F (cagttatgggatcccagCacctgctcatgcat) and hCrkL_E202A_R (atgcatgagcaggtGctgggatcccataactg) using the QuikChange kit. The coding sequences of CrkL (either wild type or mutant) in pFX-Pickles 2.31, 2.34, and 2.35 were further cleaved by XhoI and NotI and subcloned into the XhoI-NotI site of the pCXN2-FLAG-ECFP vector (Inuzuka et al., 2016) to obtain FLAG 2.31, 2.34 and 2.35.

Table I
A series of Pickles biosensors
| Pickles |
Acceptor |
Backbone |
Donor |
Other future |
| 2.31 |
m1Venus |
CrkLt |
cp173 |
|
| 2.33 |
m1Venus |
CrkLt delEcoRI |
cp173 |
|
| 2.34 |
m1Venus |
CrkLt delEcoRI D94A |
cp173 |
|
| 2.34NES |
m1Venus |
CrkLt delEcoRI D94A |
cp173 |
NES |
| 2.35 |
m1Venus |
CrkLt delEcoRI D94A E202A |
cp173 |
|
Key modifications compared with the previous version are underlined. m1 Venus, monomeric Venus (L222K/F224R); CrkLt, truncation mutant of CrkL (a.a. 1–222); NES, nuclear export signal; cp173, a circular permutated ECFP from a.a. 173.
The coding sequence of Venus was amplified by PCR with the primers XbaI-NES-EcoRI-Venus_fw (5'-gtctagacgccatgaatgaattagccttgaaattagcaggtcttgatatcgaattcggatccatggtgagcaagggcgag-3') and XFP-STOP-PspOMI-BglII-rv (5'-ttgggcccggagatctctcgatgttgtggcggatct-3') to obtain a DNA fragment of Venus containing the XbaI site, the NES of protein kinase A inhibitor (PKI, 107-141 a.a), the EcoRI site, the BamHI site at the 5' end and the BglII site at the 3' end. The resulting PCR product was digested by XbaI and BglII and cloned into the XbaI-BglII site of pFX-Pickles 2.31 to obtain pFX-NES-Venus. The coding sequence of Pickles 2.34 was cleaved by EcoRI and BglII and cloned into the EcoRI-BglII site of pFX-NES-Venus to obtain pFX-Pickles 2.34NES.
Patient samples (ethics)
This study was reviewed and approved by the Institutional Review Board of the Hokkaido University Graduate School of Medicine, and all patients provided informed consent before the collection of bone marrow or peripheral blood samples. Bone marrow mononuclear cells were isolated from bone marrow using Lymphoprep (Nycomed, Oslo, Norway), transfected with the expression vectors for Pickles using nucleofection (Amaxa Biosystems, Cologne, Germany; program number T-020 and Solution V), and maintained in RPMI supplemented with 10% fetal bovine serum and 1% (v/v) sodium pyruvate. After 18 to 24 h of transfection, the cells were treated with 2 μM IM or left untreated for 24 h and then subjected to microscopic analysis to determine FRET efficiency (the transfection efficiency obtained was approximately 20%).
Cell culture and transfection
293F cells were purchased from Invitrogen, maintained in Freestyle 293 expression medium (Invitrogen), and transfected using 293fectin (Invitrogen) according to the manufacturer’s protocol. The Ph-positive CML cell line K562 was obtained from Riken and maintained in RPMI 1640 (Sigma-Aldrich) supplemented with 10% (v/v) fetal bovine serum. Gene transfer into K562 cells was performed via nucleofection according to the manufacturer’s recommendations (Amaxa Biosystems).
Fluorescence microscopy
Cells expressing Pickles were cultured in phenol red-free RPMI 1640 (Invitrogen) buffered with 15 mM HEPES (pH 7.4 to avoid CO2 control) and plated on a poly-L-lysine-coated glass base plate (Asahi Techno Glass, Tokyo, Japan). In the experiments in Fig. 6 and Table II, the cells were treated with 2 μM IM, 4 μM NL, and 100 nM DS for 24 h prior to observation. Cell image acquisition was performed as previously described (Mizutani et al., 2010; Ohba et al., 2003). Briefly, the cells were imaged with an IX81 inverted microscope (Olympus, Tokyo, Japan) equipped with an automated XY-stage (Chuo Precision Industrial, Tokyo, Japan), a stage-top incubation chamber at 37°C (Live Cell Instrument, Seoul, Korea), an MAC6000 filter and shutter control unit (Ludl Electronic Products, Hawthorne, NY, USA), and a Cool SNAP MYO cooled charge-coupled device camera (Photometrics, Tucson, AZ, USA) controlled with MetaMorph software (Universal Imaging, West Chester, PA, USA). The filters were obtained from Semrock (Rochester, NY, USA): FF02-438/24-25 excitation and FF01-483/32-25 emission filters for CFP, FF01-504/12-25 excitation and FF01-542/27-25 emission filters for YFP, and FF02-438/24-25 excitation and FF01-542/27-25 emission filters for FRET. An FF458-Di02-25x36 dichroic mirror (Semrock) was used throughout the experiments. The cells were illuminated with a SOLA Light Engine (Lumencor, Beaverton, OR, USA) and imaged through a 60× immersion objective lens (numerical aperture, 1.35). The exposure times for 4×4 binning were 200 ms for CFP, YFP, and FRET images and 50 ms for differential interference contrast images. After background subtraction, the CFP, YFP, and FRET intensities of each cell were exported as comma-separated-value formatted data and imported into JMP Pro software (SAS Institute Inc., Cary, NC, USA, ver. 12). Scatter plots for CFP and YFP intensities with a regression line, 95% confidence interval and 95% prediction interval were plotted using the software. In Fig. 5, CFP/YFP ratio images are shown in intensity-modulated display mode, in which eight colors from red to blue represent the emission ratios, with the intensity of each color indicating the mean intensity of CFP.
Table II
Numbers of cells subjected to analyses
|
|
Ctr |
IM |
NL |
DS |
Mean |
| 2.31 |
Total cell number |
160 |
152 |
189 |
184 |
171.3 |
|
Number of subjected cells |
95 |
118 |
138 |
126 |
119.3 |
|
(%) |
59.4 |
77.6 |
73 |
68.5 |
69.6 |
| 2.34NES |
Total cell number |
190 |
148 |
142 |
151 |
157.8 |
|
Number of subjected cells |
142 |
120 |
118 |
129 |
127.3 |
|
(%) |
74.7 |
81.1 |
83.1 |
85.4 |
81.1 |
|
-fold |
1.26 |
1.05 |
1.14 |
1.25 |
1.17 |
Total cell numbers and the numbers of cells located in the 95% prediction interval were counted, and subjected cell fractions were calculated. Fold changes were also calculated and are shown (see also Fig. 5). Ctr, control (no drug treatment); IM, imatinib mesylate; NL, nirotinib; DS, dasatinib.
293F cells (2×106) were seeded in the wells of a 6-well plate and transfected with expression vectors (1 μg each) as indicated in each figure or figure legend. After 48 h, the cells were harvested by centrifugation, resuspended in 200 μl of Tris-buffered saline, diluted in 200 μl of 2× Laemmli sample buffer, and sonicated. After additional centrifugation and incubation at 95°C for 5 min, the cell lysates were subjected to SDS-polyacrylamide gel electrophoresis. The separated proteins were transferred to polyvinylidene difluoride membrane (Millipore, Darmstadt, Germany) and subjected to immunoblot analysis using antibodies indicated in the figure legends. Immune complexes were detected using ECL Western Blotting Detection Reagent (GE Healthcare, Little Chalfont, UK) and an LAS-1000UVmini image analyzer (Fujifilm, Tokyo, Japan).
Flow cytometry
K562 cells expressing Pickles biosensors were incubated in the presence or absence of IM for 24 h. The cells were then harvested and resuspended in the medium used for microscopic observation. CFP and FRET fluorescence intensities at the excitation wavelength of 405 nm were measured by using Flicyme (Mitsui Engineering & Shipbuilding, Tamano, Japan), and the emission ratio of each cell was calculated with FlowJo software (Ashland, OR, USA). The data was fitted with the Hill equation:
y=min+(max–min)/[1+(x/IC50)n], using the least-squares method.
Statistical analysis
In Fig. 5, the mean values (N=10) were compared by the Wilcoxon rank sum test because the data did not exhibit a Gaussian distribution.
Results
Pickles is cleaved in primary CML cells
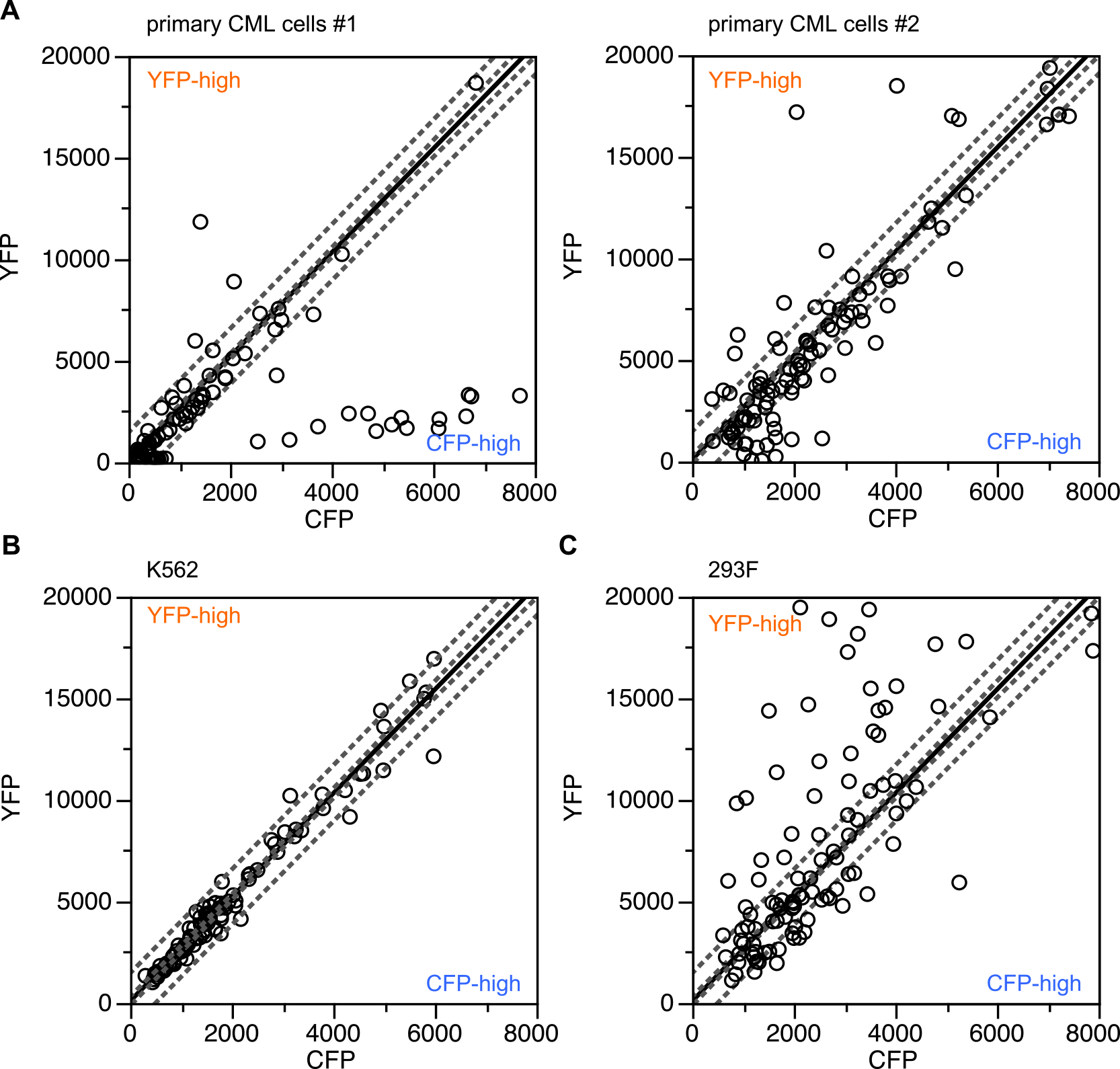
After successfully developing a FRET-based biosensor to evaluate drug responses in CML patients (Mizutani et al., 2010), we conducted a series of clinical tests to assess its significance using patient-derived CML cells (the results of the clinical tests will be reported elsewhere). Future drug responses could be indeed predicted before drug administration in a majority of patients. However, we also observed the generation of “incomplete” Pickles biosensors in primary CML cells in certain cases. One of the major causes of prediction failure was due to a decreased number of analyzed cells by excluding ineligible cells, in which such “incomplete” Pickles biosensors were detected. Because the Pickles biosensor harbors a pair of fluorescent proteins, a monomeric version of Venus (a variant of EYFP) and a circular permutated ECFP (Mizutani et al., 2010) (Fig. 1, Table I), a linear correlation between the fluorescence intensities observed through the YFP and CFP channels is expected, as observed for the FRET-based intramolecular biosensor Raichu (Fig. S1). However, certain patient-derived primary CML cells expressing Pickles occasionally exhibited expression profiles indicative of excess YFP (YFP-high) and excess CFP (CFP-high) (Fig. 2A). These cells were not detected among K562 cells (Fig. 2B); however, only YFP-high cells were observed among 293F cells expressing BCR-ABL (Fig. 2C). Therefore, the emergence of YFP-high and/or CFP-high cells is dependent on cell context.
There are three possible explanations for the presence of “incomplete” biosensors. First, transcription and/or translation are stopped at or started from an unexpected position, thus producing incomplete mRNA or peptide molecules. Second, although the full-length protein is synthesized, one of the fluorescent proteins cannot fold to emit fluorescence. Third, full-length proteins are cleaved and degraded. Because it was reported that the expression of BCR-ABL induces protease expression (Haaß et al., 2012; Patel and Gordon, 2009) and promotes proteasome activity (Crawford et al., 2009; Magill et al., 2004), we tested the possibility of the third scenario. To evaluate this possibility, we introduced a silent mutation into the EcoRI site in CrkL (Pickles 2.33, see also Fig. 1, Table I, Materials and Methods) and then constructed an expression plasmid for the Pickles-derived protein in which Venus was substituted by FLAG such that the N- and C-termini were recognized by different antibodies (FLAG 2.33, Fig. 1). The protein was expressed with or without BCR-ABL in 293F cells and analyzed by immunoblotting. As shown in Fig. 3, in addition to the full-length protein, a fragment of approximately 27 kD was observed when an anti-FLAG antibody was used (arrowhead). This fragment was assumed to be an N-terminal fragment generated by the cleavage between the tyrosine residue that is phosphorylated by BCR-ABL (Y207) and ECFP. Indeed, bands with similar molecular weights were detected by anti-CrkL and anti-pCrkL antibodies, whereas a counterpart fragment of ~27 kD, potentially corresponding to ECFP, was detected by an anti-GFP antibody (Fig. 3, Fig. S2, arrow).

In addition to the cleavage between Y207 and ECFP, there might also be other multiple cleaved sites at the more N-terminal portion of Pickles. First, it was likely to be cleaved on the C-terminal side of the SH2 domain because bands of ~40 kD were observed in blots with anti-pCrkL and anti-GFP antibodies (white triangle). Second, Pickles can also be cleaved at the very N-terminal position of the SH2 domain. Because the estimated recognition sites by anti-FLAG and anti-CrkL antibodies are close to each other, these antibodies should give very similar bands. Although they indeed gave similar band patterns, the relative expression levels between full length and cleaved Pickles were different from each other. Given that band intensities of the small fragment by the anti-FLAG antibody were weaker than that by anti-CrkL, it is possible that some portion of the small fragment lost the antigenicity of FLAG. In fact, there is a putative cleavage site by a serine protease or a metalloproteinase at this position (ARLEMSSA, cleaved by proteases after the underlined residue).
As shown all of the blots in Fig. 3, the cleavage of FLAG 2.33 was enhanced in the presence of BCR-ABL. It might be accounted for by the increased expression of proteases or promoted proteasome activity by BCR-ABL (Crawford et al., 2009; Haaß et al., 2012; Magill et al., 2004; Patel and Gordon, 2009). Together, these data indicate that more than three Pickles fragments are generated through cleavage at the SH2 domain and between Y207 and CFP (Fig. 1). Although smaller 12.5-kD and 14.4-kD fragments should be present, we could not detect these bands under our experimental conditions. This might be partially because the size of these bands themselves are too small to be detected in this assay, or they undergo further cleavage and degradation.
Pickles is cleaved in the SH2 domain of CrkL
We next sought to identify cleavage sites. CrkII, another Crk adaptor protein, is cleaved by metalloproteinases at its SH2 domain and acts as a proapoptotic protein (Austgen et al., 2012). Amino acid sequence comparisons revealed that the sequence around this position was completely conserved between CrkL and CrkII (KIHYLDTTTLI, Fig. 1). Therefore, we substituted D94, a critical residue for metalloproteinase recognition, by alanine. We prepared FLAG 2.34 harboring the D94A mutation and evaluated its expression by immunoblotting. A band of approximately 27-kD emerged after blotting with an anti-CrkL antibody (arrowhead), whereas in the blot with anti-GFP, the band of approximately 40-kD disappeared (white triangle) after the introduction of the D94A mutation (Fig. 4A). As in the case of the result shown in Fig. 2, small fragments less than 15 kD in their size could not be detected. In addition, because there are multiple bands in 25~50 kD-area in the anti-GFP blot, we could not stringently identify the origin of each band. However, the change in the band pattern observed here was, at least, consistent with the change induced by the inhibition of the cleavage at D94. Thus, as predicted, cleavage within the SH2 domain was inhibited by the D94A mutation.
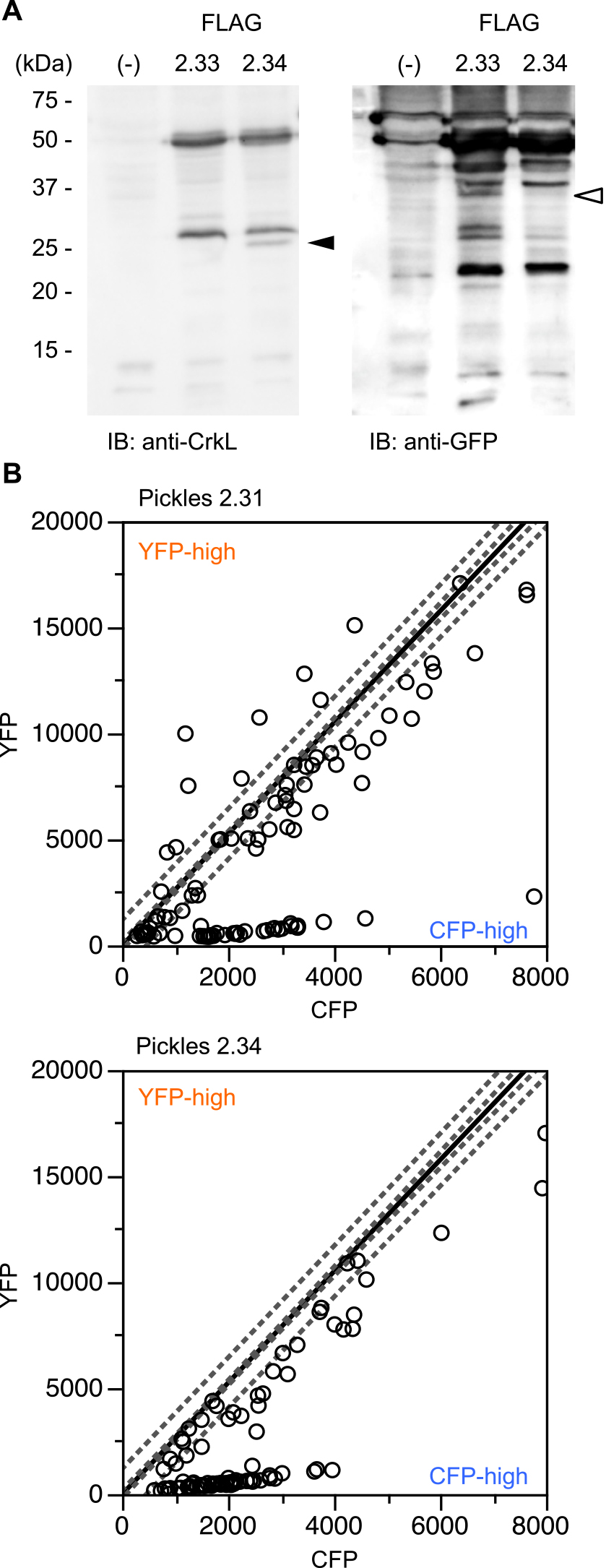
When Pickles 2.34 was introduced into primary CML cells, YFP-high cells disappeared, whereas CFP-high cells remained (Fig. 4B), showing that the emergence of YFP-high cells is attributable to cleavage at D94, whereas CFP-high cells are generated via other mechanisms.
The nuclear localization of the biosensor affects the generation of CFP-high cells
To test whether CFP-high cells were generated by cleavage at the sites surrounding the juncture between CrkL and ECFP, we searched the Pickles sequence for cleavage sites and identified a sequence similar to that surrounding D94 (197-SYGIPEPAHAY-207, E202 is underlined) in the corresponding region. We therefore constructed FLAG 2.35 by introducing the E202A mutation into FLAG 2.34. However, the banding patterns observed in the anti-CrkL blot and the anti-GFP blot were not altered by this mutation (Fig. S3); thus, the cleavage at E202 is dispensable for the generation of fragmented Pickles. Given that the remaining cleavage site was predicted near the tyrosine residue that is phosphorylated by BCR-ABL (Y207, see also Fig. 1), we refrained from further analysis and the introduction of mutations to retain the original properties of the sensor region of the biosensor.
Careful microscopic observation of patient CML cells revealed that distribution of the biosensor in CFP-high cells differed from that in cells in which the relative expression levels of CFP and YFP fell within the expected range (C/Y-equivalent) (Fig. 5A). Specifically, the biosensors primarily localized in the cytoplasm in C/Y-equivalent cells, whereas they localized in both the cytoplasm and the nucleus in CFP-high cells. When the nuclear/cytoplasmic fluorescence intensity ratio was plotted, it was significantly higher in CFP-high cells than in C/Y-equivalent cells (Fig. 5B). To promote cytoplasmic localization of the biosensor, we introduced the NES of PKI into Pickles at its N-terminus (Pickles 2.34NES). As expected, Pickles 2.34NES, compared with the prototype Pickles 2.34, predominantly localized in the cytoplasm (Fig. 5C). The fraction of CFP-high cells in Pickles 2.34NES-expressing cells was decreased compared with the fraction in Pickles 2.34-expressing cells. Although this decrease was modest (from 85% to 72%, Fig. 5D), it resulted in a substantial increase (two-fold, from 15% to 28%) in the number of cells exhibiting equivalent YFP/CFP expression. The YFP/CFP intensity ratio of Pickles 2.34NES was significantly higher than that of Pickles 2.34 (p=0.18, Wilcoxon test). Notably, responsiveness to IM concentrations did not differ significantly among biosensors (Fig. S4). Together, the results indicated that the introduction of two distinct mutations, a point mutation and the addition of NES, improved the biosensor Pickles by altering the fluorescence intensity ratio of CFP and YFP.

The significance of the improved biosensor for practical use was evaluated. In the original protocol developed for Pickles 2.31, cells were selected for further analysis according to the relative fluorescence intensities of CFP and YFP (mean–s.d.<YFP/CFP<mean+s.d., where the mean and s.d. are determined in K562 cells). Here, we compared the properties of Pickles 2.31 and Pickles 2.34NES. Although we observed only a moderate number of CFP-high and YFP-high cells in this case, the number of cells that satisfied the criterion increased from 5% to 25%, regardless of the presence or absence of drug treatment (Table II). The emission ratio was further evaluated at the level of individual cells. According to our reported definition, cells with a FRET/CFP emission ratio higher than 2.04 (indicated by dashed lines in Fig. 6) displayed significantly high BCR-ABL activity (FRET-high) (Mizutani et al., 2010). According to this criterion, the newly developed biosensor detected four FRET-high cells in the control sample, whereas only one cell above 2.04 was detected with the prototype (Fig. 6, left panels). These results are probably attributable to an increase in the number of cells analyzed, thus resulting in increased detectability. In contrast, in the dasatinib-treated sample, a small number of cells were detected on the dashed line by Pickles 2.31, but these cells disappeared in the setting using Pickles 2.34NES. Because this patient exhibited an optimal response to this drug, observation of these cells is not anticipated. The borderline cells emerged because of a bias in the relative expression levels of the donor and acceptor with the prototype, but the emergence of biased cells was suppressed by introduction of the improved biosensor, thus resulting in both increased sensitivity and specificity of the method. Because we sought to identify a small population of drug-resistant cells (0.1–1%) from among approximately one thousand cells, this improvement should greatly facilitate the use of our method in clinical testing.

Discussion
In this study, we developed improved versions of the FRET-biosensor Pickles toward more precise measurement of BCR-ABL activity in living patient-derived CML cells. Unlike other established cell lines, Pickles was cleaved at multiple sites in primary CML cells, producing cells exhibiting CFP-high or YFP-high expression levels. Because such cells must be excluded from analysis, they negatively affect the accurate assessment of drug responses. By introducing a mutation and NES, we successfully prevented intramolecular cleavage of Pickles and completely and partially suppressed the emergence of YFP-high and CFP-high cells, respectively. As revealed by immunoblotting analysis, there are still other cleavage sites in Pickles biosensor; however, given that the fluorescence intensity profile of CFP and YFP was dramatically improved, such small fractions of the cleaved form will not significantly affect the versatility of the biosensor per se and the determination of drug sensitivity in CML cells using Pickles. In the current study, only a single case was available for analysis due to the limitation of the currently approved protocol of our clinical studies, and we could not determine the clinical significance of the new biosensor in the prediction of drug responses. The suppression of YFP-high and CFP-high cells apparently increased the number of cells available for analysis to detect drug-resistant cells and evaluate drug responsiveness, indicating that Pickles 2.34NES must have higher sensitivity and specificity than the prototype. We are designing clinical trials related to CML treatment, in which FRET-based biosensors are involved as a companion diagnostic. The clinical significance will indeed be demonstrated when a relatively large number of cases are analyzed. Given that the Pickles-based method is, to our knowledge, the only means to predict future drug responses before treatment, it represents a step toward the realization of tailored medicine in molecular-targeted CML therapy.
Any proteins or peptides can be cleaved in cells if they harbor protease recognition sites. Indeed, a series of FRET-based biosensors to measure the spatiotemporal activation of caspase-3 and caspase-9 have been developed (Luo et al., 2001; Rehm et al., 2002; Takemoto et al., 2003; Tyas, 2000). In these biosensors, the caspase-3 and caspase-9 cleavage sequences (DEVD and LEHD, respectively) are included in the peptides that link the donor and acceptor fluorescent proteins for FRET, and a cleavage-dependent increase in caspase activity followed by a decrease in FRET efficiency was observed. However, in the case of FRET biosensors that depend on conformational changes, including Pickles, any cleavage should be avoided. Cleavage results in lower FRET efficiency than that expected if the cleaved fragments remain without being degraded. The increased sensitivity and specificity achieved by Pickles 2.34NES is partially attributable to the prevention of cleavage.
Theoretically, FRET efficiency can be calculated by subtracting the spectral bleedthrough of donor and acceptor fluorescence into the FRET channel to correct for differences in their relative expression levels; (Galperin and Sorkin, 2003; Honda et al., 2004; Inuzuka et al., 2009) this calculation is generally used to determine the FRET efficiency of intermolecular FRET biosensors. However, this is not the case for Pickles in CML cells because substantial numbers of cells were estimated to express only donor molecules (the majority of CFP-high cells). Such cells still must be removed from the assessment.
Most K562 cells expressed “full-length” biosensors, while 293F cells only showed up as YFP-high cells, but primary CML cells were the most cleavage-prone among all tested cell lines. Why are proteins easily cleaved and degraded in CML cells? Myeloid cells, particularly granulocytes, contain well-developed lysosomes (the azurophilic granules) and a variety of proteinases, including myeloperoxidase, cathepsin G, elastase, proteinase 3, and defensin. Generally, these enzymes are encapsulated in the granules to avoid self-digestion and never encounter cytosolic proteins. At times, however, when massive amounts of neutrophil elastase are produced in promyelocytes, a small amount of this enzyme escapes normal trafficking controls and encounters cellular protein elsewhere in the cell (Campbell et al., 1989; Lane and Ley, 2003). An example of this is the cleavage of cytosolic or nuclear PML-RARα fusion protein, generated by the t(15;17)(q22;q11.2) translocation associated with acute promyelocytic leukemia (Melnick and Licht, 1999; Pollock et al., 2001), by the neutral serine protease (neutrophil elastase), which is essential for the pathogenesis of this disease (Lane and Ley, 2003). Two closely related serine proteases, Granzyme A (Jans et al., 1998) and Granzyme B (Jans et al., 1996; Trapani et al., 1996), have also been known to enter the nucleus. Therefore, there is a reasonable possibility that “leaked enzymes” cleave Pickles in primary CML cells, and whether Pickles-transfected CML cells become CFP-high or YFP-high is determined by the nature of the dominant leaked enzyme.
Another possibility is that CML cells acquired the cleavage-prone phenotype due to the BCR-ABL protein, in addition to their natural myeloid properties. In fact, separase, a cysteine endopeptidase involved in triggering anaphase and the maintenance of centriole numbers through cohesion hydrolysis, was highly expressed in CML cells (Haaß et al., 2012; Patel and Gordon, 2009).Two putative separase cleavage sites were identified in the CrkL sequence at amino acid positions 67–70 and 139–142 by sequencing analysis. It is therefore possible that Pickles could be cleaved by this protease, which might explain the disappearance of small bands that should be observed after cleavage at D94 by metalloproteinases. However, given that the activity of separase has been reported to be tightly regulated by securin in a cell cycle-dependent manner (Zou et al., 1999), and the D94A mutation could effectively prevent the cleavage at the N-terminal portion of CrkL, cleavage by separase seems unlikely as a primary cause of the generation of fragmented biosensors.
Once Pickles is cleaved by proteases, the resulting fragment eventually leads to degradation by the ubiquitin-proteasome, which might generate CFP-high and YFP-high cells. It was reported that proteasome activity is enhanced in CML cells (Magill et al., 2004), and the activity was correlated with the expression level of BCR-ABL (Crawford et al., 2009). Indeed, CrkL possesses a putative degron sequence that can be recognized as a degradation signal by the E3 ubiquitin ligase SPOP [Speckle-type POZ (Pox virus and Zinc finger) protein] (Guharoy et al., 2016). It might be possible that the cleavage of Pickles triggers changes in posttranslational modifications to switch the degron into E3-binding-competent states, thereby undergoing degradation. This mechanism might partly explain the disappearance of counterpart fragments derived from Pickles cleavage, eventually leading to the generation of YFP-high and CFP-high cells.
Given the intracellular localization of the above-mentioned proteases and proteasomes, cleavage and degradation of Pickles are expected to occur in the cytoplasm. Indeed, because the molecular weights of full length and cleaved Pickles are approximately 81 and 27 kDa, respectively, it is more likely that the cytoplasmic full-length Pickles is cleaved, and the resulting CFP fragments diffuse passively into the nucleus. Nevertheless, Pickles cleavage, specifically generation of CFP-high cells, was unexpectedly inhibited by forced expression in the cytoplasm. This result indicated that Pickles was indeed cleaved or degraded in the nucleus, albeit in part. However, this result does not necessarily negate the possibility of cytoplasmic cleavage/degradation. Indeed, the effect of NES addition on the prevention of cleavage was relatively modest.
Another explanation for the excess expression of CFP might be “leaky scanning” (Kozak, 2002), which allows for 40S ribosomes to skip the initial AUG and start translation at downstream AUG start codons. This occurs in the absence of “strong” Kozak sequences and highly associates with consistent GC-rich leader sequences. Because the Kozak sequence of Pickles mRNA is considered to be strong enough (gccAUGg) and either such GC-rich sequences or AUG codons with strong Kozak sequence can be found at the estimated cleavage site [there is only one methionine codon (gggATGa)], leaky scanning is unlikely to be a major cause of the generation of CFP-high cells.
To our surprise, cellular heterogeneity, regarding expression levels of CFP and YFP, could be observed in not only primary CML cells, but also in the established cell line 293F. In this cell line, although CFP-high cells were never observed, diversity in relative expression levels of YFP to CFP was rich as compared to that in CML-derived K562 cells. Because primary CML cells from chronic phase patients consist of a heterogeneous cell population from a variety of differentiation stages, it might be likely to display heterogeneity in their cell properties. For instance, BCR-ABL expression level and its activity are varied among CML patients and even among cells from a single patient, as described previously (Keating et al., 1994; Mizutani et al., 2010). When we introduced Pickles expression vectors along with those for BCR-ABL, and analyzed FRET efficiency at a single cell level, BCR-ABL activity was not so different among cells. On the other hand, microscopic observation revealed that the 293F cell line consists of cells with a variety of cell sizes (data not shown), and similar phenomena were reported for the parental 293 cells (Graham et al., 1977). Therefore, the heterogeneity in cell properties of 293F cells, including protease/proteasome activity, might account for the variations in the relative expression levels of YFP and CFP.
The barriers to obtain “good” intramolecular FRET-based biosensors remain high, despite accumulating evidence for their rational engineering. Much effort has been devoted to developing new biosensors as well as improved versions of existing biosensors. Recently, a trial to improve biosensor properties by using an optimized backbone for FRET-based biosensors has been reported (Komatsu et al., 2011). In this system (the Eevee system, or extension for enhanced visualization by evading extra-FRET system), the introduction of a long flexible linker reduced the basal FRET efficiency, thereby rendering the FRET biosensors almost completely dependent on the distance between the donor and the acceptor. The Eevee system has successfully improved a variety of biosensors for tyrosine kinases, serine/threonine kinases, and GTPases (Komatsu et al., 2011). However, the long linker, consisting of a SAGG repeat, is a proteinase target in CML cells.
Another attempt to improve biosensors has involved the introduction of circular permutations into one or both of the donor/acceptor fluorescent proteins. This strategy has successfully facilitated the development of excellent biosensors for Ca2+ (Heim and Griesbeck, 2004; Mank et al., 2006) and cGMP (Ohta et al., 2016). Because our prototype Pickles 2.31 already harbors circular permutations in the donor molecule (Mizutani et al., 2010), dramatic improvement by use of this strategy is not expected. FRET biosensors can also be improved by fine-tuning the linker regions within the biosensors to increase the affinity of the sensor and/or ligand region. For example, Cameleon-Nano, which detects small changes in intracellular Ca2+ concentrations at low levels, has been developed with high Ca2+ affinity (Horikawa et al., 2010). Given our knowledge to date, we believe that further improvement of Pickles might be possible via the generation of a mutant SH2 domain, possessing higher affinity for a peptide containing the tyrosine residue phosphorylated by BCR-ABL.
Acknowledgments
We thank A. Miyawaki for the Venus cDNA, J. Groffen for the human CrkL cDNA, D. Baltimore for the BCR-ABL cDNA, M. Matsuda for Raichu cDNA and the anti-GFP antisera, S. Darmanin for critical reading of the manuscript, Novartis Pharma for the IM and NL, Bristol-Myers Squibb for the DS, and A. Kikuchi for technical assistance. During the preparation of this manuscript, our dear laboratory member and a contributor to the development of the prototype biosensor, Ms. Akiko Kaneyasu, passed away. We sincerely pray that her soul rest in peace.
Conflict of interest
Y.O. and T.K. have an issued patent (#5665262), and Y.O. has a pending patent (WO/2014/168143). The other authors have no conflicts of interest.
References
- Austgen, K., Johnson, E.T., Park, T.-J., Curran, T., and Oakes, S.A. 2012. The adaptor protein CRK is a pro-apoptotic transducer of endoplasmic reticulum stress. Nat. Cell Biol., 14: 87–92.
- Campbell, E.J., Silverman, E.K., and Campbell, M.A. 1989. Elastase and cathepsin G of human monocytes. Quantification of cellular content, release in response to stimuli, and heterogeneity in elastase-mediated proteolytic activity. J. Immunol., 143: 2961–2968.
- Crawford, L.J., Windrum, P., Magill, L., Melo, J.V., McCallum, L., McMullin, M.F., Ovaa, H., Walker, B., and Irvine, A.E. 2009. Proteasome proteolytic profile is linked to Bcr-Abl expression. Exp. Hematol., 37: 357–366.
- Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. 1996. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat. Med., 2: 561–566.
- Druker, B.J. 2008. Translation of the Philadelphia chromosome into therapy for CML. Blood, 112: 4808–4817.
- Galperin, E. and Sorkin, A. 2003. Visualization of Rab5 activity in living cells by FRET microscopy and influence of plasma-membrane-targeted Rab5 on clathrin-dependent endocytosis. J. Cell Sci., 116: 4799–4810.
- Graham, F.L., Smiley, J., Russell, W.C., and Nairn, R. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol., 36: 59–74.
- Groffen, J., Stephenson, J.R., Heisterkamp, N., de Klein, A., Bartram, C.R., and Grosveld, G. 1984. Philadelphia chromosomal breakpoints are clustered within a limited region, bcr, on chromosome 22. Cell, 36: 93–99.
- Guharoy, M., Bhowmick, P., Sallam, M., and Tompa, P. 2016. Tripartite degrons confer diversity and specificity on regulated protein degradation in the ubiquitin-proteasome system. Nat. Commun., 7: 10239.
- Haaß, W., Stehle, M., Nittka, S., Giehl, M., Schrotz-King, P., Fabarius, A., Hofmann, W.-K., and Seifarth, W. 2012. The proteolytic activity of separase in BCR-ABL-positive cells is increased by imatinib. PLoS ONE, 7: e42863.
- Heim, N. and Griesbeck, O. 2004. Genetically encoded indicators of cellular calcium dynamics based on troponin C and green fluorescent protein. J. Biol. Chem., 279: 14280–14286.
- Heisterkamp, N., Groffen, J., Stephenson, J.R., Spurr, N.K., Goodfellow, P.N., Solomon, E., Carritt, B., and Bodmer, W.F. 1982. Chromosomal localization of human cellular homologues of two viral oncogenes. Nature, 299: 747–749.
- Honda, K., Yanai, H., Mizutani, T., Negishi, H., Shimada, N., Suzuki, N., Ohba, Y., Takaoka, A., Yeh, W., and Taniguchi, T. 2004. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Nature, 101: 15416–15421.
- Horikawa, K., Yamada, Y., Matsuda, T., Kobayashi, K., Hashimoto, M., Matsu-ura, T., Miyawaki, A., Michikawa, T., Mikoshiba, K., and Nagai, T. 2010. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat. Methods, 7: 729–732.
- Inuzuka, T., Tsuda, M., Kawaguchi, H., and Ohba, Y. 2009. Transcription factor 8 activates R-Ras to regulate angiogenesis. Biochem. Biophys. Res. Commun., 379: 510–513.
- Inuzuka, T., Fujioka, Y., Tsuda, M., Fujioka, M., Satoh, A.O., Horiuchi, K., Nishide, S., Nanbo, A., Tanaka, S., and Ohba, Y. 2016. Attenuation of ligand-induced activation of angiotensin II type 1 receptor signaling by the type 2 receptor via protein kinase C. Sci. Rep., 6: 21613.
- Jans, D.A., Jans, P., Briggs, L.J., Sutton, V., and Trapani, J.A. 1996. Nuclear transport of granzyme B (fragmentin-2). Dependence of perforin in vivo and cytosolic factors in vitro. J. Biol. Chem., 271: 30781–30789.
- Jans, D.A., Briggs, L.J., Jans, P., Froelich, C.J., Parasivam, G., Kumar, S., Sutton, V.R., and Trapani, J.A. 1998. Nuclear targeting of the serine protease granzyme A (fragmentin-1). J. Cell Sci., 111: 2645–2654.
- Kantarjian, H., Sawyers, C., Hochhaus, A., Guilhot, F., Schiffer, C., Gambacorti- Passerini, C., Niederwieser, D., Resta, D., Capdeville, R., Zoellner, U., et al. 2002. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. New Eng. J. Med., 346: 645–652.
- Keating, A., Wang, X.H., and Laraya, P. 1994. Variable transcription of BCR-ABL by Ph+ cells arising from hematopoietic progenitors in chronic myeloid leukemia. Blood, 83: 1744–1749.
- Komatsu, N., Aoki, K., Yamada, M., Yukinaga, H., Fujita, Y., Kamioka, Y., and Matsuda, M. 2011. Development of an optimized backbone of FRET biosensors for kinases and GTPases. Mol. Biol. Cell, 22: 4647–4656.
- Kozak, M. 2002. Pushing the limits of the scanning mechanism for initiation of translation. Gene, 299: 1–34.
- Kurzrock, R., Gutterman, J.U., and Talpaz, M. 1988. The molecular genetics of Philadelphia chromosome-positive leukemias. New Eng. J. Med., 319: 990–998.
- Kurzrock, R., Estrov, Z., Kantarjian, H., and Talpaz, M. 1998. Conversion of interferon-induced, long-term cytogenetic remissions in chronic myelogenous leukemia to polymerase chain reaction negativity. J. Clin. Oncol., 16: 1526–1531.
- Lane, A.A. and Ley, T.J. 2003. Neutrophil elastase cleaves PML-RARalpha and is important for the development of acute promyelocytic leukemia in mice. Cell, 115: 305–318.
- Luo, K.Q., Yu, V.C., Pu, Y., and Chang, D.C. 2001. Application of the fluorescence resonance energy transfer method for studying the dynamics of caspase-3 activation during UV-induced apoptosis in living HeLa cells. Biochem. Biophys. Res. Commun., 283: 1054–1060.
- Magill, L., Lynas, J., Morris, T.C.M., Walker, B., and Irvine, A.E. 2004. Proteasome proteolytic activity in hematopoietic cells from patients with chronic myeloid leukemia and multiple myeloma. Haematologica, 89: 1428–1433.
- Mank, M., Reiff, D.F., Heim, N., Friedrich, M.W., Borst, A., and Griesbeck, O. 2006. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys. J., 90: 1790–1796.
- Melnick, A. and Licht, J.D. 1999. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood, 93: 3167–3215.
- Mizutani, T., Kondo, T., Darmanin, S., Tsuda, M., Tanaka, S., Tobiume, M., Asaka, M., and Ohba, Y. 2010. A novel FRET-based biosensor for the measurement of BCR-ABL activity and its response to drugs in living cells. Clin. Cancer Res., 16: 3964–3975.
- Ohba, Y., Kurokawa, K., and Matsuda, M. 2003. Mechanism of the spatio-temporal regulation of Ras and Rap1. EMBO J., 22: 859–869.
- Ohta, Y., Kamagata, T., Mukai, A., Takada, S., Nagai, T., and Horikawa, K. 2016. Nontrivial Effect of the Color-Exchange of a Donor/Acceptor Pair in the Engineering of Förster Resonance Energy Transfer (FRET)-Based Indicators. ACS Chem. Biol., 11: 1816–1822.
- Patel, H. and Gordon, M.Y. 2009. Abnormal centrosome-centriole cycle in chronic myeloid leukaemia? Br. J. Haematol., 146: 408–417.
- Pollock, J.L., Westervelt, P., Walter, M.J., Lane, A.A., and Ley, T.J. 2001. Mouse models of acute promyelocytic leukemia. Curr. Opin. Hematol., 8: 206–211.
- Preudhomme, C., Revillion, F., Merlat, A., Hornez, L., Roumier, C., Duflos-Grardel, N., Jouet, J.P., Cosson, A., Peyrat, J.P., and Fenaux, P. 1999. Detection of BCR-ABL transcripts in chronic myeloid leukemia (CML) using a “real time” quantitative RT-PCR assay. Leukemia, 13: 957–964.
- Rehm, M., Dussmann, H., Janicke, R.U., Tavare, J.M., Kogel, D., and Prehn, J.H.M. 2002. Single-cell fluorescence resonance energy transfer analysis demonstrates that caspase activation during apoptosis is a rapid process. Role of caspase-3. J. Biol. Chem., 277: 24506–24514.
- Takemoto, K., Nagai, T., Miyawaki, A., and Miura, M. 2003. Spatio-temporal activation of caspase revealed by indicator that is insensitive to environmental effects. J. Cell Biol., 160: 235–243.
- Tkachuk, D.C., Westbrook, C.A., Andreeff, M., Donlon, T.A., Cleary, M.L., Suryanarayan, K., Homge, M., Redner, A., Gray, J., and Pinkel, D. 1990. Detection of bcr-abl fusion in chronic myelogeneous leukemia by in situ hybridization. Science, 250: 559–562.
- Trapani, J.A., Browne, K.A., Smyth, M.J., and Jans, D.A. 1996. Localization of granzyme B in the nucleus. A putative role in the mechanism of cytotoxic lymphocyte-mediated apoptosis. J. Biol. Chem., 271: 4127–4133.
- Tyas, L. 2000. Rapid caspase-3 activation during apoptosis revealed using fluorescence-resonance energy transfer. EMBO Reports, 1: 266–270.
- Weisberg, E., Manley, P.W., Cowan-Jacob, S.W., Hochhaus, A., and Griffin, J.D. 2007. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer, 7: 345–356.
- Zou, H., McGarry, T.J., Bernal, T., and Kirschner, M.W. 1999. Identification of a vertebrate sister-chromatid separation inhibitor involved in transformation and tumorigenesis. Science, 285: 418–422.
