Abstract
Accumulation of unfolded/misfolded proteins in the endoplasmic reticulum (ER) activates the unfolded protein response (UPR). The ATF6 pathway is one of the three major pathways in vertebrates. Although ATF6, a transmembrane-type glycoprotein in the ER, functions as a UPR sensor/transducer, it is an unstable protein with a half-life of approximately 2 h and is constitutively subjected to the ER-associated degradation system with the location of the misfolded part in the ER lumen (ERAD-L). ERAD-L substrates are delivered to the cytosol through the retrotranslocon, which likely contains HRD1 (E3), gp78 (E3), SEL1L (a partner of HRD1), Derlin1/2/3 and Herp1/2. We previously showed that ATF6 represents a novel transmembrane-type ERAD-L substrate requiring both EDEM1/2/3-mediated mannose trimming and SEL1L. Here, by constructing and analyzing chicken DT40 cells deficient in various components of the retrotranslocon, we show that degradation of ATF6 requires Derlin2 or Derlin3 and that Derlin2 and Derlin3 are redundant for ERAD-L of ATF6. We further show that degradation of ATF6 requires Herp1 or Herp2 and that Herp1 and Herp2 are redundant for ERAD-L of ATF6. Furthermore, by investigating five more ERAD-L substrates, we show that SEL1L-dependent substrates require Derlin2/3 and Herp1/2 regardless of their soluble or transmembrane nature. Our results suggest that ERAD-L substrates take several routes to the cytosol. The HRD1-engaged route 1 requires SEL1L, Derlin2 or Derlin3, and Herp1 or Herp2, whereas the HRD1-engaged route 2 does not require them functionally. It remains to be determined whether the latter requires Derlin1 and whether these two routes are compositionally distinct.
Key words: endoplasmic reticulum, proteasome, protein degradation, protein misfolding, ubiquitin
Introduction
The endoplasmic reticulum (ER) provides an optimum environment for the maturation of secretory and transmembrane proteins. It controls the quality of these proteins by two distinct mechanisms, namely productive folding, which is assisted by ER-localized molecular chaperones and folding enzymes; and ER-associated degradation (ERAD), in which proteins unfolded or misfolded even after engagement of ER chaperones are recognized, delivered to the cytosol, ubiquitinated by an E3 ligase and finally degraded by the proteasome. Under a variety of conditions collectively termed ER stress, however, the ER quality control system is perturbed, resulting in the accumulation of unfolded or misfolded proteins in the ER. This in turn activates the unfolded protein response (UPR), which leads to restoration of the homeostasis of the ER (Mori, 2000; Walter and Ron, 2011).
Analysis of the ERAD mechanism in the budding yeast Saccharomyces cerevisiae has identified three separate pathways which handle ERAD substrates differentially depending on the legion in which they act, namely ERAD-L (in the ER lumen), ERAD-M (inside the ER membrane), and ERAD-C (the cytosolic side of a transmembrane protein) (Xie and Ng, 2010). We have been interested in the mechanisms of recognition, delivery and retrotranslocation of ERAD-L substrates in higher eukaryotes. In yeast, it is well known that ERAD-L substrates are retrotranslocated back to the cytosol through a large membranous protein complex containing Hrd1p (E3 ligase), Hrd3p (a partner of Hrd1p), Der1p and Usa1p. In contrast, analysis of the retrotranslocation mechanism in mammals is not straightforward, because most of their components exist redundantly (Hoseki et al., 2010; Smith et al., 2011); for example, HRD1 and gp78 for Hrd1p, Derlin1/2/3 for Der1p, and Herp1/2 for Usa1p. As an exception, SEL1L is the only mammalian homolog of Hrd3p. In response to earlier technical difficulties in gene knockout analysis in mammals, unlike in yeast, we introduced chicken DT40 cells, which have high homologous recombination efficiency, for analysis of ERAD mechanism in higher eukaryotes. Our knockout experiments in DT40 cells confirmed previous findings made by knockdown experiments in mammalian cells that SEL1L is required for ERAD-Ls—the degradation of misfolded soluble (luminal) proteins—but not for ERAD-Lm, the degradation of misfolded transmembrane proteins, which demonstrates its reliability (Ninagawa et al., 2011).
Among three UPR sensors/transducers (IRE1, PERK and ATF6) ubiquitously present in the ER of vertebrates, only ATF6 turns over rapidly, with a half-life of 1.5–2 h in human HeLa and HCT116 cells as well as in chicken DT40 cells (Haze et al., 1999; Horimoto et al., 2013; Ninagawa et al., 2014, 2015). Our utilization of ATF6 as an ERAD-L substrate influenced our understanding of recognition and delivery mechanisms in higher eukaryotes. Namely, we showed that ATF6, a type II transmembrane-type glycoprotein in the ER, is a novel ERAD-Lm substrate which requires both SEL1L and mannose trimming for degradation (Horimoto et al., 2013). Using ATF6, we have succeeded in the unambiguous identification of enzymes responsible for two-step mannose trimming, which is required for recognition of ERAD-L substrates: EDEM2 converts Man9 to Man8 and EDEM1/3 converts Man8 to Man7 in both chicken DT40 and human HCT116 cells (Ninagawa et al., 2014).
Here, we investigated which membranous protein is required for ERAD-Lm of ATF6 among redundantly existing Derlin1/2/3 and Herp1/2, in addition to SEL1L, and whether the requirements of each is distinguished by SEL1L dependency.
Materials and Methods
Cell Culture and Transfection- DT40 cells were cultured at a density of 1×105–1×106 cells per ml in RPMI1640 medium supplemented with 10% fetal bovine serum, 1% chicken serum, and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) at 39.5°C in a humidified 5% CO2/95% air atmosphere. DT40 cells were transfected by electroporation using a Microporator (Digital Bio) with two pulses at 1,500 V for 15 msec according to the manufacturer’s instructions. Neomycin (2 mg/ml), Puromycin (0.5 μg/ml), Histidinol (1 mg/ml), Mycophenolic acid (15 μg/ml), and Blasticidin S (16.6 μg/ml) were used for the selection and maintenance of drug-resistant clones. Cycloheximide was purchased from Sigma-Aldrich.
For determination of doubling time, DT40 cells of various genotypes were seeded at 1×105 cells/ml and their cell numbers were counted every 24 hours in duplicate.
Statistical analysis was conducted using Student’s t-test. *p<0.05 and **p<0.01 for all figures.
Construction of plasmids- Recombinant DNA techniques were performed according to standard procedures (Sambrook et al., 1989) and the integrity of all constructed plasmids was confirmed by extensive sequencing analyses. Chicken Derlin2 and Derlin3 cDNA were obtained by RT-PCR using total RNA prepared from wild type (WT) DT40 cells and the following paired primers: 5’-GGAATTCGGATGGCGTACCAGACCTTCCG-3’ and 5’-CCGCTCGAGTTAGCCTCCAAGACGTTGAC-3’ for gDerlin2, and 5’-GGAATTCGGATGGCCTACCAGGGCTTGGC-3’ and 5’-CCGCTCGAGTTACAGTGGACGCTGCTGCT-3’ for gDerlin3, respectively. These cDNA were inserted between the EcoRI and XhoI sites of pCMV-Myc vector (Clontech) to create pCMV-myc-gDerlin2 and pCMV-myc-gDerlin3, in which the c-myc tag is attached to the N-terminus of gDerlin2 or gDerlin3. Chicken Herp1 and Herp2 cDNA were obtained by RT-PCR using total RNA prepared from WT DT40 cells and the following paired primers: 5’-GGTGAATTCGGCGGAGGATCTGAGTCTGTC-3’ and 5’-GGTGGATCCTCAGTTGCGGGTCAGCCTGG-3’ for gHerp1, and 5’-GGTGAATTCGGAGCAGGGCGTGGTGGGCCA-3’ and 5’-GATGGATCCTTAATTGCCAACCTGTGGAG-3’ for gHerp2, respectively. These cDNA were inserted between the EcoRI and BamHI sites of p3xflag-CMV-14 vector (Sigma) to create pCMV-gHerp1-Flag and pCMV-gHerp2-Flag, in which the Flag tag is attached to the C-terminus of gHerp1 or hHerp2.
Quantitative RT-PCR- Quantitative RT-PCR was performed using the SYBR Green method (Applied Biosystems) using a pair of primers, 5’-AGGCTTCTGAGAACACCATC-3’ and 5’-CCTCAGGGAGGGGGTTATAG-3’ for gDerlin2 mRNA, 5’-GGCTTCTGAGAACACCATCTG-3’ and 5’-TTCTTCTGGCAAGGGATTGT-3’ for gDerlin3 mRNA, or 5’-CTGATGCCCCCATGTTTGTG-3’ and 5’-GCACGATGCATTGCTGACA-3’ for gGAPDH mRNA. 1,000, 10,000, and 100,000 molecules of plasmid carrying the respective gene were used as standards.
Northern Blot Hybridization- Total RNA was prepared from cultured cells by the acid guanidinium/phenol/chloroform method using ISOGEN (Nippon Gene). 5 μg each of total RNA were subjected to 1.0% agarose gel electrophoresis containing 2.2 M formaldehyde and transferred to a Hybond-N+ membrane (GE Healthcare). Digoxigenin–labeled cDNA probes were amplified by PCR using the primers 5’-CGAGAATTCCCATACGCGCGCCATGGCG-3’ and 5’-GTGGGTACCTCTGGGAGAGGAGCTGGGGGT-3’ for gHerp1, or 5’-CAGGAATTCGCCCGGCAGCATGGAGCAGG-3’ and 5’-GAACTAGTGCCTGGCCTGGAAATTGGTTGC-3’ for gHerp2 according to the manufacturer’s instructions (Roche) and hybridized with RNA blotted on a membrane. Subsequent reaction with anti-digoxigenin antibody (Roche) and treatment with chemiluminescent detection reagent CDP-Star (GE Healthcare) were performed according to the manufacturers’ specifications. Chemiluminescence was visualized using an LAS-3000mini LuminoImage analyzer (Fuji Film).
Construction of gDerlin2 targeting vector- The 2.7-kb fragment of the gDerlin2 gene used for the 3’ arm was amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-CGGGATCCGAGGACGTCTTTCCCAATCA-3’ and 5’-CCATCGATGAGTGGTCTTTCCTGAGCCC-3’, and then subcloned between the ClaI and BamHI sites of pBluescript II KS (+) vector to create the pBluescript-3’ arm (gDerlin2). The 2.7-kb fragment of the gDerlin2 gene used for the 5’ arm was amplified similarly using the primers 5’-GACTAGTTGCCCCACCTTCACAAGTTC-3’ and 5’-GACTAGTCCTGCAGGTACTCCTGCCGG-3’, and then subcloned into the SpeI sites of the pBluescript-3’ arms (gDerlin2) to create the pBluescript-5’-3’ arms (gDerlin2). The puromycin-resistant gene flanked by loxP sites and designated Puro-loxP or the neomycin-resistant gene flanked by loxP sites and designated Neo-loxP was subcloned into the BamHI site of the pBluescript-5’-3’ arms (gDerlin2) to create pKO-gDerlin2-puromycin or pKO-gDerlin2-neomycin, respectively. The histidinol-resistance (histidinol dehydrogenase [HisD]) gene was also subcloned into the BamHI site of the pBluescript-5’-3’ arms (gDerlin2) to create pKO-gDerlin2-histidinol. These constructs were transfected into DT40 cells by electroporation after linearization using NotI.
Construction of gDerlin3 targeting vector- The 3.7-kb fragment of the gDerlin3 gene used for the 3’ arm was amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-CCATCGATCCCCCTCCCTGAGGATCATC-3’ and 5’-ACGCGTCGACGGAGAAGAAGATCAGAGACC-3’, and then subcloned between the SalI and ClaI sites of pBluescript II KS (+) vector to create the pBluescript-3’ arm (gDerlin3). The 3.6-kb fragment of the gDerlin3 gene used for the 5’ arm was amplified similarly using the primers 5’-ATAAGAATGCGGCCGCCTTGTAGCTAGTGAGGCTG-3’ and 5’-ATAAGAATGCGGCCGCCCAAAAAAGAGGAAGTTG-3’, and then subcloned into the NotI sites of the pBluescript-3’ arms (gDerlin3) to create the pBluescript-5’-3’ arms (gDerlin3). Puro-loxP or the blasticidin S-resistant gene flanked by loxP sites designated Bsr-loxP or was subcloned into the BamHI site of the pBluescript-5’-3’ arms (gDerlin3) to create pKO-gDerlin3-puromycin or pKO-gDerlin3-blasticidin S, respectively. These constructs were transfected into DT40 cells by electroporation after linearization using KpnI.
Construction of gHerp1 targeting vector 1- The 1.7-kb fragment of the gHerp1 gene used for the 5’ arm was amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-CACGGGGCCGCCCATAAATCAG-3’ and 5’-GCTACTAGTCAACTTCTGGTCCTCCTCGGC-3’, and then subcloned between the SacI and SpeI sites of pBluescript II KS (+) vector to create the pBluescript-5’ arm (gHerp1). The 1.6-kb fragment of the gHerp1 gene used for the 3’ arm was amplified similarly using the primers 5’-CATATCGATTCCTCCTGGTTATGGGTGGCA-3’ and 5’-CAAGTCGACCAGTTGCGGGTCAGCCTGGG-3’, and then subcloned between the ClaI and SalI sites of the pBluescript-5’ arm (gHerp1) to create the pBluescript-5’-3’arms (gHerp1). Neo-loxP was subcloned into the BamHI site of the pBluescript-5’-3’ arms (gHerp1) to create pKO-gHerp1-neomycin. This construct was transfected into DT40 cells by electroporation after linearization using SalI.
Construction of gHerp1 targeting vector 2- The 3.0-kb fragment of the gHerp1 gene used for the 5’ arm was amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-CGGCCACGGGGGATTTTCACC-3’ and 5’-GGACTAGTACATGCTTCGGCTGAGCAGCTT-3’, and then subcloned between the NotI and SpeI sites of pBluescript II KS (+) vector to create the pBluescript-5’ arm (gHerp1’). The 2.4-kb fragment of the gHerp1 gene used for the 3’ arm was amplified similarly using the primers 5’-GAGATCGATGCTTCAACAGATCTATGCAAGGCAA-3’ and 5’-CAAGTCGACCAGTTGCGGGTCAGCCTGGG-3’, and then subcloned between the ClaI and SalI sites of the pBluescript-5’ arm (gHerp1’) to create the pBluescript-5’-3’ arms (gHerp1’). Puro-loxP was subcloned into the BamHI site of the pBluescript-5’-3’ arms (gHerp1’) to create pKO-gHerp1-puromycin. This construct was transfected into DT40 cells by electroporation after linearization using NotI.
Construction of gHerp2 targeting vector- The 3.0-kb fragment of the gHerp2 gene used for the 5’ arm was amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-GAGCGGCCGCGTGACTGGCAGGACTACGGC-3’ and 5’-GAACTAGTGCCTGGCCTGGAAATTGGTTGC-3’, and then subcloned between the NotI and SpeI sites of pBluescript II KS (+) vector to create the pBluescript-5’ arm (gHerp2). The 2.5-kb fragment of the gHerp2 gene used for the 3’ arm was amplified similarly using the primers 5’-GAAATCGATGGGAGCCATGCTACTGGTTTAC-3’ and 5’-GGGGTACCTGAAAGCTGACAGCAGCACCGT-3’, and then subcloned between the ClaI and KpnI sites of the pBluescript-5’ arms (gHerp2) to create the pBluescript-5’-3’ arms (gHerp2). Bsr-loxP was subcloned into the BamHI site of the pBluescript-5’-3’ arms (gHerp2) to create pKO-gHerp2-blasticidin S. The mycophenolic acid-resistance gene, designated E. coli xanthine-guanine phosphoribosyltransferase [EcoGPT], or HisD was also subcloned into the BamHI site of the pBluescript-5’-3’ arms (gHerp2) to create pKO-gHerp2-mycophenolic acid or pKO-gHerp2-histidinol, respectively. These constructs were transfected into DT40 cells by electroporation after linearization using NotI.
Southern Blot Hybridization- Southern blot hybridization was performed according to standard procedures (Sambrook et al., 1989) as described previously (Ninagawa et al., 2011). Specific probes were amplified by PCR from WT DT40 cell genomic DNA using the primers 5’-GCTCTAGAGGCATGCTGCTTCGTGAAGC-3’ and 5’-CCATCGATGACACCAGTGCTGCTTGGAG-3’ for gDerlin2, 5’-CCATGTCTTCAAGTTCCTCTG-3’ and 5’-GAACAAATTGCCAGGGCTCC-3’ for gDerlin3, 5’-GCTTTGTGGAACGTCGGTGAGC-3’ and 5’-TCCACTGAGGTCACTGCCCC-3’ for gHerp1, or 5’-GGGACTTGTGAGCAAGCTAAGGG-3’ and 5’-TGCTTGCAAGATGTAGCAACTGT-3’ for gHerp2. These probes were labeled with digoxigenin. Subsequent reaction with anti-digoxigenin antibody (Roche) and treatment with the chemiluminescent detection reagent CDP-star (GE Healthcare) were performed according to the manufacturer’s specifications. Chemiluminescence was visualized using an LAS-3000mini LuminoImage analyzer (Fuji Film).
RT-PCR- Total RNA prepared from WT DT40 cells or various KO cells (~5×106 cells) by the acid guanidinium/phenol/chloroform method using ISOGEN (Nippon Gene) was converted to cDNA using Moloney murine leukemia virus reverse transcriptase (Invitrogen) and random primers. The full-length open reading frame of gDerlin2, gDerlin3, gHerp1, or gHerp2 was amplified using PrimeSTAR HS DNA polymerase (Takara Bio Inc.) and a pair of primers, namely 5’-GGAATTCGGATGGCGTACCAGACCTTCCG-3’ and 5’-CCGCTCGAGTTAGCCTCCAAGACGTTGAC-3’ for gDerlin2, 5’-GGAATTCGGATGGCCTACCAGGGGTGGC-3’ and 5’-CCGCTCGAGTTACAGTGGACGCTGCTGCT-3’ for gDerlin3, 5’-CGAGAATTCCCATACGCGCGCCATGGCG-3’ and 5’-CACGGATCCGCGTTGCGGGTCAGCCTGG-3’for gHerp1, or 5’-CAGGAATTCGCCCGGCAGCATGGAGCAGG-3’ and 5’-CATGGATCCGCATTGCCAACCTGTGGAGGC-3’ for gHerp2.
Immunological Techniques- Immunoblotting analysis was carried out according to the standard procedure (Sambrook et al., 1989) as described previously (Ninagawa et al., 2011). Approximately 2×106 cells were collected by centrifugation at 3,000 rpm for 2 min, washed with PBS, suspended in Laemmli’s sample buffer and then boiled for 5 min. Samples were subjected to SDS-PAGE. Chemiluminescence obtained using Western Blotting Luminol Reagent (Santa Cruz Biotechnology) was detected using an LAS-3000mini LuminoImage analyzer (Fuji Film). Rabbit anti-chicken ATF6 and rabbit anti-chicken XBP1 antibody were raised previously (Horimoto et al., 2013). Mouse anti-Flag M2 antibody was obtained from Sigma-Aldrich; rabbit anti-HA antibody from Recenttec; anti-β-actin antibody from Wako Pure Chemical Industries; and mouse anti-KDEL antibody, anti-myc-tag antibody, and rabbit anti-Derlin2 antibody from Medical and Biological Laboratories.
Results
Construction and characterization of DT40 cells deficient in Derlin1/2/3 or Herp1/2
Both chicken and human express Derlin1/2/3 as homologues of yeast Der1p (Lilley and Ploegh, 2004; Oda et al., 2006; Ye et al., 2004), which span the ER membrane multiple times, most likely six times (Greenblatt et al., 2011) (Fig. 1A). The constitutively expressed level of mRNA encoding chicken Derlin3 (gDerlin3) was twice as much as that encoding chicken Derlin2 (gDerlin2) (Fig. 1C). Both mRNAs are inducible by ER stress, and the inducibility of gDerlin3 mRNA is much greater than that of gDerlin2 mRNA (Fig. 1C), as in mammalian cells (Adachi et al., 2008). Both chicken and human express Herp1/2 containing a ubiquitin-like domain (UBL) as functional homologues of yeast Usa1p, which associate with the ER membrane with a hairpin structure through their membrane-bound regions (Fig. 1B). Chicken Herp1 (gHerp1) but not Herp2 (gHerp2) is highly inducible by ER stress, (Fig. 1D), as in the case of mammalian Herp1/2 (Huang et al., 2014; Kokame et al., 2000).

We tried to knockout the gDerlin1, gDerlin2 and gDerlin3 genes separately in DT40 cells by conventional methods (Fig. 2A and Fig. 3A). Clones in which a drug-resistance gene was incorporated were selected (Fig. 2B and Fig. 3B) and homologous recombination of the respective gene was confirmed by Southern blotting (Fig. 2C and Fig. 3C). We obtained two independent lines each for gDerlin2-KO and gDerlin3-KO cells, in which respective the mRNA was not expressed (Fig. 2D and Fig. 3D), from 86 and 64 drug-resistant clones, respectively. Unfortunately, we failed to identify gDerlin1-KO cells among 474 drug-resistance clones, indicating that KO of gDerlin1 in DT 40 cells is lethal, as in mice (Eura et al., 2012).

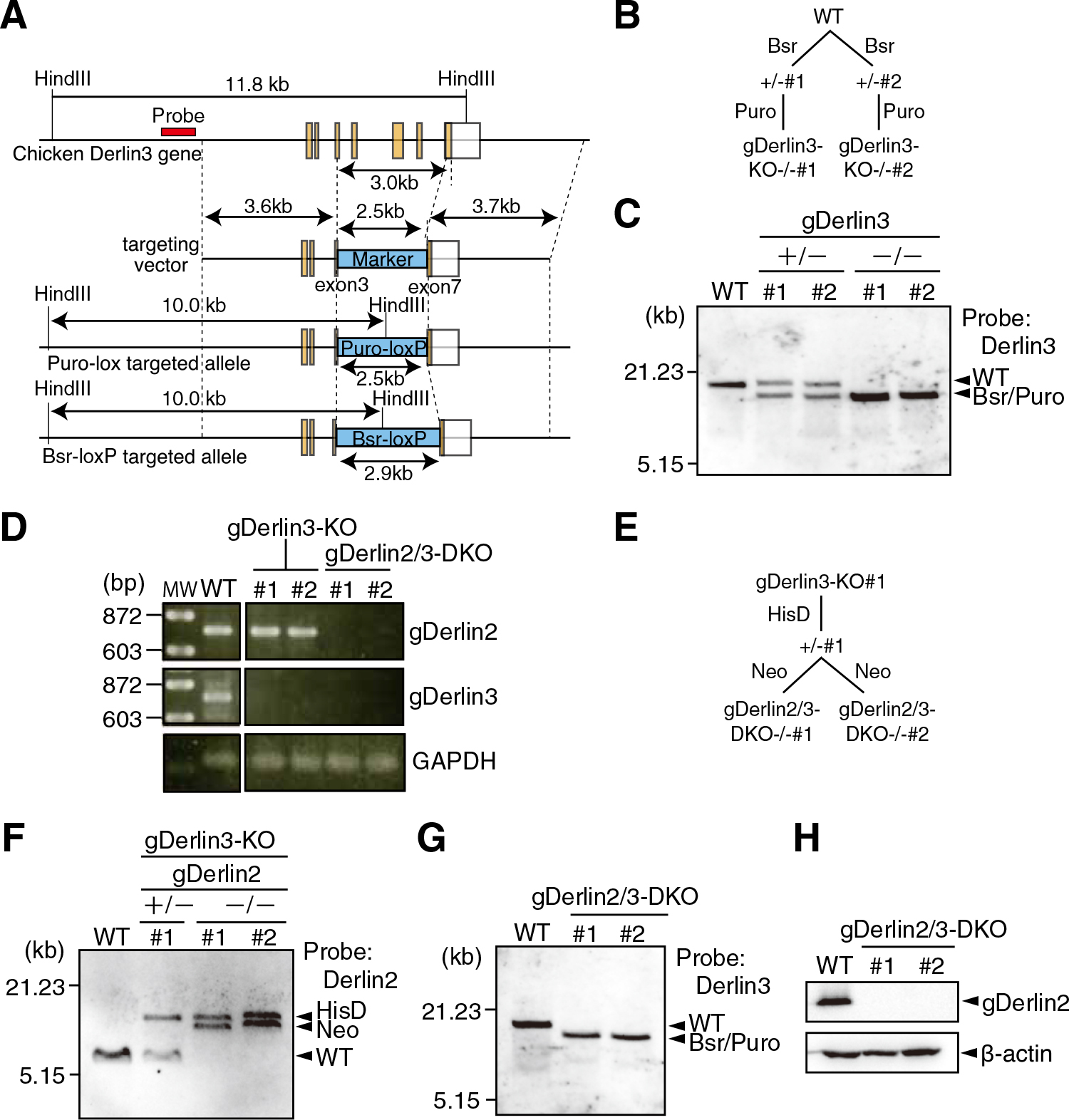
Immunoblotting showed that gDerlin2 protein was not detected in the two lines of gDerlin2-KO cells (Fig. 2E). As commercially available anti-Derlin3 antibodies reacted with human Derlin3 but not with chicken Derlin3, we could not check whether gDerlin3 protein was not in fact expressed in gDerlin3-KO cells. Because gDerlin2 is more homologous to gDerlin3 than to gDerlin1 (Fig. 1A), we knocked out the gDerlin2 gene in gDerlin3-KO cells to obtain gDerlin2/3-double KO (DKO) cells (Fig. 3D-3H) to determine whether gDerlin2/3 are simply redundant or they have differential functions.
We also knocked out the gHerp1 and gHerp2 genes separately in DT40 cells by conventional methods (Fig. 4A and 4B). It should be noted that the gHerp2 gene is encoded in chromosome 2, which is in trisomy in DT40 cells. Clones in which a drug-resistance gene was incorporated were selected (Fig. 4C and 4D) and homologous recombination of the respective gene was confirmed by Southern blotting (Fig. 4F and 4G). We obtained three and four independent clones for gHerp1-KO and gHerp2-KO cells, respectively, in which respective mRNA was not expressed (Fig. 4H), from 111 and 72 drug-resistant clones, respectively. We further knocked out the gHerp2 gene in gHerp1-KO cells, and obtained two independent clones of gHerp1/2-DKO cells from 48 drug-resistant clones (Fig. 4E-4H) to determine whether gHerp1/2 are simply redundant or function differently.

Growth rates of gDerlin2-KO, gDerlin3-KO, gDerlin2/3-DKO, gHerp1-KO, gHerp2-KO and gHerp1/2-DKO cells were comparable to that of WT cells (Fig. 5A and 5B). gDerlin2/3-DKO cells and gHerp1/2-DKO cells but not four single KO cells were constitutively ER stressed as evidenced by constitutive expression of the spliced form of XBP1, pXBP1(S), which indicates activation of the IRE1 pathway (Fig. 5C), and constitutive elevation of the levels of two major ER chaperones, BiP and GRP94, which indicates activation of the ATF6 pathway (Fig. 5D), as in the case of WT cells treated with tunicamycin, which causes ER stress by inhibiting protein N-glycosylation in the ER.

When gDerlin2-KO, gDerlin3-KO and gDerlin2/3-DKO cells were cultured with a low concentration of tunicamycin (20 ng/ml), only gDerlin2/3-DKO cells showed a defect in growth (Fig. 6A). When these three types of cells were treated with a high concentration of tunicamycin (2 μg/ml) for 8 h, washed out, and then incubated in the absence tunicamycin, gDerlin2-KO cells grew as fast as WT cells, whereas gDerlin3-KO cells grew more slowly than WT cells, indicating that gDerlin3 is highly inducible during tunicamycin treatment and exhibited a potent protective effect on the cell, whereas gDerlin2 is only mildly inducible during tunicamycin treatment and protected the cell less efficiently. DT40 cells could not recover from ER stress in the complete absence of gDerlin2 and gDerlin3 (Fig. 6C).

Similarly, when gHerp1-KO, gHerp2-KO and gHerp1/2-DKO cells were cultured with a low concentration of tunicamycin (20 ng/ml), only gHerp1/2-DKO cells showed growth retardation (Fig. 6B). When these three types of cells were treated with a high concentration of tunicamycin (2 μg/ml) for 8 h, washed out, and then incubated in the absence tunicamycin, gHerp2-KO cells grew as fast as WT cells, whereas gHerp1-KO cells grew much more slowly than WT cells, indicating that gHerp1 is highly inducible during tunicamycin treatment and exhibited a potent protective effect on the cell, whereas constitutively expressed gHerp2 protected the cell much less efficiently. DT40 cells could not recover from ER stress in the complete absence of gHerp1 and gHerp2 (Fig. 6D).
Effect of deleting Derlin2/3 or Herp1/2 on degradation of ERAD-L substrates
We determined the degradation rate of endogenous ATF6 (gATF6), a SEL1L-dependent ERAD-Lm substrate, in DT40 cells of various genotypes by cycloheximide chase experiments. As we reported previously (Horimoto et al., 2013), gATF6 was degraded with a half-life of approximately 2 h in WT DT40 cells, whereas gATF6 was markedly stabilized in gSEL1L-KO DT40 cells (Fig. 7A and 7B). Although gATF6 was degraded in gDerlin2-KO or gDerlin3-KO cells as quickly as in WT cells, gATF6 was stabilized in gDerlin2/3-DKO cells as effectively as in gSEL1L-KO cells, indicating that gDerlin2 and gDerlin3 have redundant function in the degradation of gATF6 (Fig. 7A). Similarly, although gATF6 was degraded in gHerp1-KO or gHerp2-KO cells as quickly as in WT cells, gATF6 was stabilized in gHerp1/2-DKO cells as effectively as in gSEL1L-KO cells, indicating that gHerp1 and gHerp2 have redundant function in the degradation of gATF6 (Fig. 7B).

We next determined the degradation rates of various ERAD-Lm substrates, whose degradation does not require SEL1L (Bernasconi et al., 2010; Horimoto et al., 2013), by cycloheximide chase experiments. Results showed that hCD147-myc, mCD3-δ-HA, mTCR-α-Flag and hBACE457-HA were degraded in gDerlin2/3-DKO cells and in gHerp1/2-DKO cells as quickly as in WT cells (Fig. 8), no matter whether their degradation was HRD1-dependent or not (Table I), in marked contrast to the case of gATF6 (Fig. 7).
Table I
| |
Soluble |
Transmembrane |
| |
hBACE457Δ |
gATF6 |
hCD147 |
mTCR-α |
hBACE457 |
mCD3-δ |
| HRD1 |
Yes(1) |
Yes(2) |
Yes(3) |
Yes(4) |
Yes(1) |
No(1) |
| gp78 |
No(1) |
not determined |
No(3) |
Yes(5) |
Yes(1) |
Yes(1) |
| SEL1L |
Yes(1) |
Yes(6) |
No(6) |
No(6) |
No(6) |
No(1) |
| Derlin2/3 |
Yes |
Yes |
No |
No |
No |
No |
| Herp1/2 |
Yes |
Yes |
No |
No |
No |
No |
We finally determined the degradation rate of hBACEΔ457-HA, an ERAD-Ls substrate, whose degradation requires SEL1L (Fig. 9A and 9B), by cycloheximide chase experiments. Results showed that hBACEΔ457-HA was stabilized in gDerlin2/3-DKO cells and in gHerp1/2-DKO cells (Bernasconi et al., 2010) (Fig. 9A and 9B), similarly to the case of gATF6. It should be noted that the introduction of gDerlin2 or gDerlin3 into gDerlin2/3-DKO cells restored degradation capability, as expected (Fig. 9C).
Discussion
Previous studies on Derlin2/3 and Herp1/2 focused on their direct interaction with the E3 ligase HRD1 and their role in the mechanism of HRD1-mediated ERAD (Huang et al., 2013, 2014; Kny et al., 2011; Schulze et al., 2005). Here, by constructing and analyzing various KO cells, we unraveled a novel combination in ERAD-L in higher eukaryotes in regard to the requirement of membranous proteins for the retrotranslocation. It was previously thought that the degradation mechanism of ERAD-Ls substrates differs from that of ERAD-Lm substrates. However, our results suggest that ERAD-L substrates can be categorized by their SEL1L dependency, regardless of their soluble or transmembrane nature and regardless of their dependency on HRD1 or gp78 (Table I), because only SEL1L-dependent ERAD-L substrates require Derlin2 or Derlin3 and Herp1 or Herp2. We consider that ERAD-L substrates take several routes to the cytosol (Fig. 10). The HRD1-engaged route 1 requires SEL1L, Derlin2 or Derlin3, and Herp1 or Herp2, whereas the HRD1-engaged route 2 and gp78-engaged route 3 do not require them functionally. It remains to be determined whether or not the route 2 and route 3 require Derlin1 because we could not obtain Derlin1-KO DT40 cells.

It also remains to be determined whether or not the retrotranslocon in the route 2 contains SEL1L, Derlin2/3 and Herp1/2. It is well known that HRD1 (Hrd1p in yeast) and SEL1L (Hrd3p in yeast) form a stoichiometric complex in both yeast and mammals. Interestingly, Hrd3p is required for the stabilization of Hrd1p (Gardner et al., 2000; Plemper et al., 1999), whereas HRD1 is required for the stabilization of SEL1L (Iida et al., 2011). Therefore, HRD1 can function independently from SEL1L in mammals. Recently, Hampton and his colleagues succeeded in the stabilization of Hrd1p in the absence of Hrd3p by simultaneously deleting the UBL domain of Usa1p (yeast homologue of Herp1/2), which is required for autodegradation of Hrd1p in the absence of Hrd3p (Vashistha et al., 2016). Using this Usa1p-ΔUBL/ΔHrd3p strain they showed that Hrd3p is required for ubiquitination of ERAD substrate by Hrd1p. Thus, Hrd3p is involved in ERAD much more positively than previously thought. Based on these results and the finding that the route 2 does not require SEL1L functionally, we consider that the retrotranslocon in the route 2 does not contain SEL1L, but appreciate that this must be demonstrated biochemically. We are currently constructing Derlin1-KO, Derlin2-KO and Derlin3-KO in the human colorectal carcinoma-derived HCT116 diploid cells line (Ochiai et al., 2014), as we did for EDEM1, EDEM2 and EDEM3 using an innovative genome editing technique (Ninagawa et al., 2014). Complex formation would be much more easily analyzed in human cells than in chicken cells because of the availability of various antibodies. In the route 1 it is likely that SEL1L directly recognizes ERAD-Ls substrates and ATF6 using its Sel1-like tetratricopeptide repeats (Ninagawa et al., 2011). In the route 2 it is possible that HRD1 directly recognizes SEL1L-independent ERAD-Lm substrates. Further extensive studies are necessary to understand substrate recognition mechanisms.
We previously showed that knockdown of Derlin2 alone or Derlin3 alone blocked degradation of ERAD-Ls substrate effectively (Oda et al., 2006). However, we now consider that this blockade resulted from their off-target effects, because our current knockout experiments clearly showed that Derlin2 and Derlin3 are redundant for SEL1L-dependent ERAD-L, consistent with the markedly high similarity between Derlin2 and Derlin3 compared with Derlin1. It was previously shown that knockdown of Derlin2 alone blocked degradation of ERAD-Ls substrates much more efficiently than that of Derlin3 alone (Huang et al., 2013), which may reflect the difference in their abundance in HEK293T cells employed for the analysis. In contrast, it was previously shown that double knockdown of Herp1 and Herp2 blocked degradation of ERAD-Ls substrates much more efficiently than single knockdown of Herp1 or Herp2 (Huang et al., 2014), consistent with our findings that Herp1 and Herp2 are redundant for ERAD-L of ATF6 and hBACE457Δ.
ATF6 functions as one of the three major UPR sensors/transducers in vertebrates. Upon ER stress, ATF6 is translocated from the ER to the Golgi apparatus, where it is cleaved sequentially by the two proteases Site-1 and Site-2 proteases. As a result, its cytosolic fragment containing the basic leucine zipper and transcriptional activation domains is liberated from the membrane, and translocated into the nucleus to activate transcription directly (Mori, 2003). Target genes conserved from the yeast UPR to mammalian UPR encode ER chaperones for refolding of unfolded/misfolded proteins accumulated in the ER. Interestingly, transcriptional induction of ER chaperones in response to ER stress is mediated by the IRE1 pathway, in which IRE1-mediated unconventional splicing of XBP1 mRNA (HAC1 mRNA in yeast) produces an active transcription factor pXBP1(S) [pHAC1(S) in yeast] containing the basic leucine zipper and transcriptional activation domains, in yeast (Chapman and Walter, 1997; Kawahara et al., 1997), worm (Shen et al., 2005) and fly (Hollien and Weissman, 2006). In contrast, transcriptional induction of ER chaperones in response to ER stress is mediated by the ATF6 pathway in medaka fish (Ishikawa et al., 2013), chicken (data not shown) and mice (Yamamoto et al., 2007). We consider that this switch from IRE1 to ATF6 that has occurred during evolution is associated with the difference in their activation speed; activation of ATF6 is more rapid than activation of XBP1, because ATF6 is activated by ER stress-induced transport and cleavage of preexisting ATF6 whereas activation of XBP1 requires ER stress-induced splicing plus translation of spliced XBP1 mRNA (Mori, 2009).
We speculate that ATF6 needs to be unstable to ensure a prompt and effective response to ER stress, because the level of ATF6 in the ER decreases after each ER stress-induced relocation from the ER to the Golgi apparatus. Because ATF6 is a transcription factor but not a signaling molecule, ATF6-mediated output cannot be amplified, unlike that of IRE1 which is an enzyme. If ATF6 were a stable protein, after response to initial ER stress, the cell would be unable to respond promptly and effectively to the next ER stress for a considerable time due to the slow synthesis rate of ATF6 until sufficient ATF6 is supplied. Importantly, however, because ATF6 is indeed an unstable protein with a half life of approximately only 2 h in vertebrates, it is constantly being refreshed and a certain amount of ATF6 is always maintained to respond to ER stress promptly and effectively at any time.
We previously showed that the luminal region confers SEL1L-dependency to gATF6, and that the degradation rate of gATF6 mutants lacking such a region (half-life more than 3 h) was much slower than that of full-length gATF6 (half-life less than 2 h) (Horimoto et al., 2013). SEL1L-dependent degradation therefore ensures rapid turnover of ATF6. The tight coupling of SEL1L with Derlin2/3 and Herp1/2 we unraveled for the first time in this present report further strengthens this important regulation of ATF6.
Acknowledgments
We thank Ms. Kaoru Miyagawa for her technical and secretarial assistance. This work was financially supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (16K18538 to S.N., 15K18529 and 17K15116 to T.I., 15K06996 to T.O., 26291040 and 17H01432 to K.M.) and by a grant from Takeda Science Foundation (to T.O.).
References
- Adachi, Y., Yamamoto, K., Okada, T., Yoshida, H., Harada, A., and Mori, K. 2008. ATF6 is a Transcription Factor Specializing in the Regulation of Quality Control Proteins in the Endoplasmic Reticulum. Cell Struct. Funct., 33: 75–89.
- Bernasconi, R., Galli, C., Calanca, V., Nakajima, T., and Molinari, M. 2010. Stringent requirement for HRD1, SEL1L, and OS-9/XTP3-B for disposal of ERAD-LS substrates. J. Cell Biol., 188: 223–235.
- Chapman, R.E. and Walter, P. 1997. Translational attenuation mediated by an mRNA intron. Curr. Biol., 7: 850–859.
- Chen, B., Mariano, J., Tsai, Y.C., Chan, A.H., Cohen, M., and Weissman, A.M. 2006. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl. Acad. Sci. USA, 103: 341–346.
- Eura, Y., Yanamoto, H., Arai, Y., Okuda, T., Miyata, T., and Kokame, K. 2012. Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS One, 7: e34298.
- Fonseca, S.G., Ishigaki, S., Oslowski, C.M., Lu, S., Lipson, K.L., Ghosh, R., Hayashi, E., Ishihara, H., Oka, Y., Permutt, M.A., and Urano, F. 2010. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Invest., 120: 744–755.
- Gardner, R.G., Swarbrick, G.M., Bays, N.W., Cronin, S.R., Wilhovsky, S., Seelig, L., Kim, C., and Hampton, R.Y. 2000. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J. Cell Biol., 151: 69–82.
- Greenblatt, E.J., Olzmann, J.A., and Kopito, R.R. 2011. Derlin-1 is a rhomboid pseudoprotease required for the dislocation of mutant alpha-1 antitrypsin from the endoplasmic reticulum. Nat. Struct. Mol. Biol., 18: 1147–1152.
- Haze, K., Yoshida, H., Yanagi, H., Yura, T., and Mori, K. 1999. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell, 10: 3787–3799.
- Hollien, J. and Weissman, J.S. 2006. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science, 313: 104–107.
- Horimoto, S., Ninagawa, S., Okada, T., Koba, H., Sugimoto, T., Kamiya, Y., Kato, K., Takeda, S., and Mori, K. 2013. The unfolded protein response transducer ATF6 represents a novel transmembrane-type endoplasmic reticulum-associated degradation substrate requiring both mannose trimming and SEL1L protein. J. Biol. Chem., 288: 31517–31527.
- Hoseki, J., Ushioda, R., and Nagata, K.. 2010. Mechanism and components of endoplasmic reticulum-associated degradation. J. Biochem. (Tokyo), 147: 19–25.
- Huang, C.H., Hsiao, H.T., Chu, Y.R., Ye, Y., and Chen, X. 2013. Derlin2 protein facilitates HRD1-mediated retro-translocation of sonic hedgehog at the endoplasmic reticulum. J. Biol. Chem., 288: 25330–25339.
- Huang, C.H., Chu, Y.R., Ye, Y., and Chen, X. 2014. Role of HERP and a HERP-related protein in HRD1-dependent protein degradation at the endoplasmic reticulum. J. Biol. Chem., 289: 4444–4454.
- Iida, Y., Fujimori, T., Okawa, K., Nagata, K., Wada, I., and Hosokawa, N. 2011. SEL1L critically determines the stability of the HRD1-SEL1L ERAD complex to optimize the degradation kinetics of ERAD substrates. J. Biol. Chem., 286: 16929–16939.
- Ishikawa, T., Okada, T., Ishikawa-Fujiwara, T., Todo, T., Kamei, Y., Shigenobu, S., Tanaka, M., Saito, T.L., Yoshimura, J., Morishita, S., Toyoda, A., Sakaki, Y., Taniguchi, Y., Takeda, S., and Mori, K. 2013. ATF6alpha/beta-mediated adjustment of ER chaperone levels is essential for development of the notochord in medaka fish. Mol. Biol. Cell, 24: 1387–1395.
- Ishikura, S., Weissman, A.M., and Bonifacino, J.S. 2010. Serine residues in the cytosolic tail of the T-cell antigen receptor alpha-chain mediate ubiquitination and endoplasmic reticulum-associated degradation of the unassembled protein. J. Biol. Chem., 285: 23916–23924.
- Kawahara, T., Yanagi, H., Yura, T., and Mori, K. 1997. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol. Biol. Cell, 8: 1845–1862.
- Kny, M., Standera, S., Hartmann-Petersen, R., Kloetzel, P.M., and Seeger, M. 2011. Herp regulates Hrd1-mediated ubiquitylation in a ubiquitin-like domain-dependent manner. J. Biol. Chem., 286: 5151–5156.
- Kokame, K., Agarwala, K.L., Kato, H., and Miyata, T. 2000. Herp, a new ubiquitin-like membrane protein induced by endoplasmic reticulum stress. J. Biol. Chem., 275: 32846–32853.
- Lilley, B.N. and Ploegh, H.L. 2004. A membrane protein required for dislocation of misfolded proteins from the ER. Nature, 429: 834–840.
- Mori, K. 2000. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell, 101: 451–454.
- Mori, K. 2003. Frame switch splicing and regulated intramembrane proteolysis: key words to understand the unfolded protein response. Traffic, 4: 519–528.
- Mori, K. 2009. Signalling pathways in the unfolded protein response: development from yeast to mammals. J. Biochem., 146: 743–750.
- Ninagawa, S., Okada, T., Takeda, S., and Mori, K. 2011. SEL1L is required for endoplasmic reticulum-associated degradation of misfolded luminal proteins but not transmembrane proteins in chicken DT40 cell line. Cell Struct. Funct., 36: 187–195.
- Ninagawa, S., Okada, T., Sumitomo, Y., Kamiya, Y., Kato, K., Horimoto, S., Ishikawa, T., Takeda, S., Sakuma, T., Yamamoto, T., and Mori, K. 2014. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol., 206: 347–356.
- Ninagawa, S., Okada, T., Sumitomo, Y., Horimoto, S., Sugimoto, T., Ishikawa, T., Takeda, S., Yamamoto, T., Suzuki, T., Kamiya, Y., Kato, K., and Mori, K. 2015. Forcible destruction of severely misfolded mammalian glycoproteins by the non-glycoprotein ERAD pathway. J. Cell Biol., 211: 775–784.
- Ochiai, H., Miyamoto, T., Kanai, A., Hosoba, K., Sakuma, T., Kudo, Y., Asami, K., Ogawa, A., Watanabe, A., Kajii, T., Yamamoto, T., and Matsuura, S. 2014. TALEN-mediated single-base-pair editing identification of an intergenic mutation upstream of BUB1B as causative of PCS (MVA) syndrome. Proc. Natl. Acad. Sci. USA, 111: 1461–1466.
- Oda, Y., Okada, T., Yoshida, H., Kaufman, R.J., Nagata, K., and Mori, K. 2006. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol., 172: 383–393.
- Plemper, R.K., Bordallo, J., Deak, P.M., Taxis, C., Hitt, R., and Wolf, D.H. 1999. Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci., 112: 4123–4134.
- Sambrook, J., Fritsch, E.F., and Maniatis, T. 1989. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
- Schulze, A., Standera, S., Buerger, E., Kikkert, M., van Voorden, S., Wiertz, E., Koning, F., Kloetzel, P.M., and Seeger, M. 2005. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol., 354: 1021–1027.
- Shen, X., Ellis, R.E., Sakaki, K., and Kaufman, R.J. 2005. Genetic interactions due to constitutive and inducible gene regulation mediated by the unfolded protein response in C. elegans. PLoS Genet., 1: 355–368.
- Smith, M.H., Ploegh, H.L., and Weissman, J.S. 2011. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science, 334: 1086–1090.
- Tyler, R.E., Pearce, M.M., Shaler, T.A., Olzmann, J.A., Greenblatt, E.J., and Kopito, R.R. 2012. Unassembled CD147 is an endogenous endoplasmic reticulum-associated degradation substrate. Mol. Biol. Cell, 23: 4668–4678.
- Vashistha, N., Neal, S.E., Singh, A., Carroll, S.M., and Hampton, R.Y. 2016. Direct and essential function for Hrd3 in ER-associated degradation. Proc. Natl. Acad. Sci. USA, 113: 5934–5939.
- Walter, P. and Ron, D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science, 334: 1081–1086.
- Xie, W. and Ng, D.T. 2010. ERAD substrate recognition in budding yeast. Semin. Cell Dev. Biol., 21: 533–539.
- Yamamoto, K., Sato, T., Matsui, T., Sato, M., Okada, T., Yoshida, H., Harada, A., and Mori, K. 2007. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6α and XBP1. Dev. Cell, 13: 365–376.
- Ye, Y., Shibata, Y., Yun, C., Ron, D., and Rapoport, T.A. 2004. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature, 429: 841–847.


