Flagellar-associated Protein FAP85 Is a Microtubule Inner Protein That Stabilizes Microtubules
2018 Volume 43 Issue 1 Pages 1-14

Details
2018 Volume 43 Issue 1 Pages 1-14
Genomics and proteomics studies in Chlamydomonas have revealed that an axoneme is composed of 200–600 types of proteins, including uncharacterized proteins collectively named flagellar-associated proteins (FAPs). Nine FAPs contain the EF-hand motif; however, they have not yet been well characterized. To find components responsible for Chlamydomonas-specific waveform changes coupled with intracellular Ca2+ concentrations, we focused on FAP85, an EF-hand motif-containing FAP specific to Chlamydomonas and its relatives. We cloned the cDNA encoding FAP85, expressed it in Escherichia coli cells, and generated a polyclonal antibody against the expressed protein. Immunoblotting showed that FAP85 was present in every axoneme of several flagellar mutants lacking major axonemal components. Immuno-electron microscopy revealed that anti-FAP85 antibodies were found only on the inner wall of A-tubules of the doublets exposed by N-lauroylsarcosine (Sarkosyl) treatment. The zero-length cross-linker 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) applied to 0.6 M KCl-extracted axonemes generated a 75-kDa complex containing β-tubulin and FAP85. Further characterization of FAP85 and its effects on microtubule dynamics showed that FAP85 binds to tubulin and stabilized microtubules. According to these results, we conclude that FAP85 is a novel member of microtubule-binding proteins, localizing on the inner wall of the A-tubule and stabilizing microtubules.
Key words: Chlamydomonas, flagella, doublet microtubule, microtubule inner proteins
Eukaryotic cilia and flagella possess a conserved scaffold structure known as the 9+2 structure or the axoneme, in which nine peripheral doublet microtubules (DMTs) surround a pair of singlet MTs, and several functional components, e.g. inner/outer dynein arms, radial spokes, and nexin-dynein regulatory complexes are arranged regularly along the DMTs (Ishikawa, 2015). In addition to the major axonemal proteins, proteomics and genomics have revealed the presence of flagellar-associated proteins (FAPs) in the axoneme; the latter have not been functionally characterized to date (Merchant et al., 2007; Pazour et al., 2005).
Precise three-dimensional arrangement of flagellar components, including FAPs, in the axoneme is essential to flagellar functions, e.g. the bend formation and propagation. Only a small number of FAPs have been characterized biochemically and/or biophysically. Recent progress in electron tomography has enabled the elucidation of minor flagellar structures such as proteins within the DMTs, known as microtubule inner proteins (MIPs) (Heuser et al., 2012; Ichikawa et al., 2017; Maheshwari et al., 2015; Nicastro et al., 2011, 2006; Pigino et al., 2012; Sui and Downing, 2006). However, the composition, biochemical properties, and functions of these MIPs have not been well characterized. Various types of MIPs have been described in a variety of organisms, and the divergence in their molecular architecture as well as their structural similarities have been revealed. Specifically, the divergence of these MIPs may contribute to differences in the bending patterns of these organisms, as pointed out by Pigino and colleagues (Pigino et al., 2012).
The high homology of axonemal components with those of humans, and the availability of a large repertoire of axonemal mutants have made Chlamydomonas a useful model system for the investigation of the structure and function of eukaryotic cilia and flagella (Silflow and Lefebvre, 2001). On the other hand, Chlamydomonas shows a unique change in waveform between the ciliary and flagellar types in response to changes in intracellular calcium concentrations (Kamiya and Witman, 1984). With the ciliary-type waveform, the cell swims forward with its flagella beating asymmetrically with an oar-like effective stroke and a surf-casting-like recovery stroke. With the flagellar-type waveform, the cell moves backward with its flagella beating symmetrically like sinusoidal waves. These unique changes led us to examine Chlamydomonas-specific flagellar-associated proteins with EF-hand motifs, which are putative calcium-binding proteins. Several important studies on calcium-mediated regulation of flagellar beating have been performed (DiPetrillo and Smith, 2010; Sakato et al., 2007; Smith and Lefebvre, 1997; Wakabayashi et al., 1997; Yang et al., 2001); however, nine types of FAPs containing EF-hand motifs (FAP85, FAP183, FAP200, FAP225, FAP252, FAP268, FAP272, FAP288, and FAP290) isolated from Chlamydomonas remain un-characterized, due to the lack of a screenable phenotype and probably to functional redundancy amongst them. A Basic Local Alignment Search Tool (BLAST) sequence homology search showed that, among these proteins, FAP85 is specific to Chlamydomonas and its relatives. Thus, we focused on FAP85 as the potential candidate involved in switching the flagellar waveform.
In this study, we found that FAP85 is a MIP, which is localized on the inner wall of the A-tubules of DMTs and stabilizes MTs. We believe our findings indicate that FAP85 is important for the development and stability of flagella in Chlamydomonas.
The strains of Chlamydomonas reinhardtii used in this study are listed in Table I. Cells were grown as previously described (Gorman and Levine, 1965), in Tris-acetic acid-phosphate (TAP) medium with aeration and continuous illumination at 25°C for four days.
| Strain | Missing Structures/Components | References |
|---|---|---|
| oda1 | outer arm dyneins/ODA-DC | (Kamiya and Okamoto, 1985) |
| oda2 | outer arm dyneins | (Mitchell and Rosenbaum, 1985) |
| ida1 | dynein f (dynein I1) | (Kamiya et al., 1991; Myster et al., 1997) |
| ida5 | dynein a, c, d, e | (Kato-Minoura et al., 1997; Kato et al., 1993) |
| ida7 | dynein I1/f | (Perrone et al., 1998) |
| pf2 | dynein b, e (reduced), DRC | (Rupp and Porter, 2003) |
| pf3 (wild type for CNK11) | dynein e, DRC | (Lin et al., 2015) |
| pf14 | radial spokes | (Piperno et al., 1977) |
| pf18 | central pair | (Adams et al., 1981) |
| mbo1 | beak-like projection #5,6 | (Segal et al., 1984) |
Chlamydomonas flagella were isolated using the dibucaine method (Witman et al., 1978). Resultant flagella were suspended and demembranated in HMDEK wash and resuspension solution (30 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 5 mM MgSO4, 1 mM dithiothreitol [DTT], 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid [EGTA], and 50 mM potassium acetate, pH 7.4) containing 0.2% Nonidet P-40 (NP-40, Nacalai Tesque, Kyoto, Japan). The demembranated flagella were washed several times with HMDEK to remove the detergent and finally resuspended in HMEDK.
To obtain axonemes without components soluble in 0.6 M KCl or 0.6 M KI, the demembranated axonemes were suspended in high-salt-HMDE solution (0.6 M KCl or 0.6 M KI, 30 mM HEPES-NaOH, 5 mM MgSO4, 1 mM DTT, 1 mM EGTA, pH 7.4) for 15 min and centrifuged at 21,130×g for 8 min. The high-salt extraction was repeated twice; residual axonemes were obtained as the pellet and used for immunoblotting and the N-lauroylsarcosine (Sarkosyl)-treatment experiments.
Sequencing of the 25-kDa FAP85 by mass spectrometryProteins were separated by SDS-PAGE and stained with Comassie brilliant Blue (CBB-R250). The relevant protein bands were excised from the gel, trypsin-digested and sent to Cosmo Bio (Tokyo, Japan) for matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectrometry, according to the company’s procedures.
Cloning and expression of FAP85 cDNAThe DNA encoding FAP85 was amplified from a Chlamydomonas cDNA library by PCR. The primers used were: FAP85-F, 5' TCG AAG GTA GGC ATA TG ATG TCG TTC TTC GGG CTC 3'; and FAP85-R, 5' CGA CAA GCT TGA ATT CTT ACC CCA GGA AGT GGC CGT 3'. The FAP85 cDNA was inserted into the NdeI/EcoRI-digested pCold-ProS2 vector (Takara Bio, Kusatsu, Japan), originating the ProS2-FAP85 plasmid: this plasmid expressed FAP85 N-terminally fused with a polyhistidine tag followed by the ProS2 tag protein, consisting of the tandem N-terminal domain of Protein S from Myxococcus xanthus, which increases solubility of expressed proteins. For MT binding assays, the Orange Nano-lantern (ONL, Takai et al., 2015) was cloned and inserted in N-terminal of FAP85 plasmid to obtain the orange luminescent protein, ONL-FAP85 (migrating as 85 kDa by SDS-PAGE). The ProS2-FAP85, pCold-ProS2 and ONL-FAP85 plasmids were transformed into BL21 Star (DE3) chemically competent cells (ThermoFisher Scientific, Waltham, MA, USA) and the recombinant proteins they encoded for were expressed according to conventional methods. The expressed proteins were loaded on a Ni-IMAC resin (Bio-Rad, Hercules, CA, USA) and eluted with elution buffer (20 mM Tris-HCl, 250 mM NaCl, 1 mM DTT, and 250 mM imidazole, pH 7.5).
Preparation of an anti-FAP85 antibodyOne milligram of ProS2-FAP85 was prepared and used as antigen for the production of anti-FAP85 antibodies from rabbits by Eurofins Genomics (Tokyo, Japan), according to company’s procedures. The anti-FAP85 antibody was purified from the serum of the rabbits by affinity chromatography, excluding also undesirable antibodies against ProS2.
ImmunoblottingFlagella, KCl extract, KI extract, and residual axonemes were loaded on to a 4–15% SDS-polyacrylamide gel and separated by electrophoresis. The resulting protein bands were then transferred onto a polyvinylidene fluoride (PVDF) membrane using a Bio-Rad mini-gel transfer apparatus, followed by incubation with a blocking buffer (3% skim milk in phosphate-buffered saline with 0.05% Tween-20) for an hour at 20–25°C. The PVDF membrane was incubated for 45 min with the anti-FAP85 antibody. After washing three times with the blocking buffer, the membrane was incubated for 45 min with an alkaline phosphatase-conjugated goat anti-rabbit secondary antibody (Vector Laboratories, Burlingame, CA, USA) for Sarkosyl experiments, and IgG, horseradish peroxidase-conjugated donkey anti-rabbit secondary antibody (GE Healthcare, Tokyo, Japan) for other experiments. The membrane was then washed three times with blocking buffer and stained with the BCIP/NBT phosphatase substrate system (Sera Care Life Sciences, Inc., Milford, MA, USA) or the ECL substrate (Clarity Wester ECL Substrate, Bio-Rad).
Sarkosyl treatment of doublet microtubulesTo prepare A- and B-tubules from DMTs, the axonemes were suspended in 0.2, 0.3, 0.4, and 0.7% Sarkosyl in 10 mM Tris-NaCl, pH 7.8 and incubated for an hour. One of the samples with 0.7% Sarkosyl was treated overnight to dissolve all DMTs. After treatment, the axonemes were collected as pellets upon centrifugation at ~100,000×g for an hour. Supernatants containing the dissolved portions of DMTs were also collected. The pellets were re-suspended in 1 ml of Tris-NaCl and spun down again. Both supernatants and pellets were analyzed with 4–15% SDS-PAGE, followed by immunoblotting analysis. The pellet obtained by treatment with 0.3% Sarkosyl was used for immuno-gold electron microscopy.
Immuno-gold electron microscopy (EM) of doublet microtubulesImmuno-gold electron microscopy of DMTs was performed as previously described (Ikeda et al., 2003) with some modifications. All procedures were performed at 20–25°C and solution exchange was carried out by replacing grids onto the drop of another solution. Carbon-coated, collodion nickel EM grids were placed onto the droplet of 0.1% poly-L-lysine for 5 min, and washed with distilled water. Sarkosyl-treated DMTs were placed onto the grids and incubated for 10 min. The grids were rinsed with a blocking solution (1% bovine serum albumin [BSA] in PBS, pH 7.4) and incubated for an hour with the anti-FAP85 antibody diluted 10 times in the blocking buffer. The grids were then washed with the blocking buffer six times. A 10 nm-gold-conjugated goat anti-rabbit IgG antibody (G7402, SIGMA, Kawasaki, Japan) was applied to the grid, incubated for an hour, and washed out with the blocking buffer. Finally, grids were washed with PBS and distilled water. The specimens were negatively stained with 1% uranyl acetate and observed with a transmission electron microscope (JEM 2000, JEOL, Tokyo, Japan).
Immunoprecipitation of FAP85Immunoprecipitation of FAP85 was carried out using Dynabeads (ThermoFisher Scientific) in accordance with the supplier’s instructions. In brief, Dynabeads conjugated with protein G were incubated for 10 min with 2 μg of anti-FAP85 antibody diluted in PBS-T buffer (10 mM Na-PBS, pH 7.4, with 0.02% Tween-20). The beads were washed and resuspended in PBS-T buffer. A small aliquot of the sample containing the antigen was added and incubated while gently mixing for 15 min. After washing with PBS-T, collected beads were boiled with SDS sample buffer (62.5 mM Tris-HCl, 10% glycerol, 1.25% SDS, 1.25% bromophenol blue, 1.25% 2-mercaptoethanol) for electrophoresis and the immunoblotting.
For preparation of the antigen sample, the axonemes isolated from 20 L of culture of the wild type Chlamydomonas were demembraned, extracted with 0.6 M KCl-HMDE solution and treated with 0.7% Sarkosyl overnight. The supernatant was diluted 10 times with Tris-HCl, pH 7.8, followed by dialysis against PBS to remove Sarkosyl from the sample.
Cross-linking of FAP85 with a zero-length cross-linkerSarkosyl treatment of the DMTs yielded the A-tubule-rich fractions. These fractions were further treated with 0–10 mM 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC, ThermoFisher) as previously described (King et al., 1991). The pellet of A-tubules was resuspended in HMDEK to 1 mg/ml, and an equal volume of 0–20 mM EDC in HMDEK was added to final concentrations of 0–10 mM EDC. This treatment was performed for 1 hour at 20–25°C, and quenched by addition of 50 mM 2-mercaptoethanol. The samples were analyzed by SDS-PAGE and FAP85 and its complex were detected by immunoblotting using the anti-FAP85 antibody.
Microtubule-binding assayThe affinity of FAP85 to MTs was measured as previously described (Butner and Kirschner, 1991), with some modifications. In brief, ONL-FAP85 and tubulin purified from porcine brain (Castoldi and Popov, 2003) were centrifuged with the Beckman TLA 120.2 angle rotor at 100,000 rpm with 355,040×g, for 10 min at 4°C to remove undesirable aggregates (Beckman Coulter, Fullerton, CA, USA). Tubulin was polymerized at 37°C for 30 min at the concentration of 10 mg/ml in BRB80 buffer (80 mM piperazine-N,N'-bis(2-ethanesulfonic acid) [PIPES]-KOH, 1 mM MgCl2, 1 mM EGTA, pH6.8) with 1 mM guanosine triphosphate (GTP), and stabilized with 10 μM taxol. The polymerized MTs were loaded over sucrose cushions (25% sucrose in BRB80, 10 μM taxol) and centrifuged at 150,000×g. The pellet was suspended in BRB80 containing 1mM GTP and 10 μM taxol and serial-diluted with BRB80 containing 1 mM GTP and 10 μM taxol to obtain solutions containing 1, 2, 5, 10, and 20 μM tubulin, which were incubated for 60 min with 0.77 μM ONL-FAP85 in 100 μg/mg BSA. The mixtures were then centrifuged at 355,040×g. Supernatants and pellets were carefully collected and analyzed by SDS-PAGE.
For co-polymerization experiments, 5, 10, 20 and 30 μM tubulin and 0.77 μM of ONL-FAP85 were co-polymerized in BRB80 with 1 mM GTP, 100 μg/ml BSA at 37°C for 30 min and resultant MTs were stabilized with 10 μM taxol. The mixtures were centrifuged in Beckman TLA120.2 angle rotor at 100,000 rpm with 355,040×g for 10 min. The supernatants and the pellets were collected and analyzed by SDS-PAGE as described for the co-sedimentation assay.
Proteins was quantified by densitometry. The percentage of ONL-FAP85 bound to MTs was calculated as the ratio of the density of ONL-FAP85 bands in the supernatant and the pellet. Binding curves were fitted to the experimental data with the equation:
B=Bmax T/(Kd+T),
where B represents the percentage of bound FAP85, T is the concentration of tubulin, Bmax is the maximum percentage of FAP85 that can bind to the MTs and Kd is the dissociation constant.
In vitro assembly and disassembly of microtubulesTubulin polymerization was monitored as turbidity at 350 nm, using a Synergy H1 microplate reader (BioTek, Tokyo, Japan). The solution to analyze was added into a 384-well black glass bottom plate. The change in the absorbance at 350 nm (Abs350nm) was measured at 37°C for 120 min; this duration was sufficient for polymerization of most of the tubulin without evaporation of the solution. The total volume of the solution was 50 μl. The tubulin solutions (5–35 μM) in BRB80 and 1 mM GTP were incubated for 5 min on ice. Next, 2 μl of 70 μM FAP85 in 250 mM KCl in HMDE (final concentrations, 2.8 μM of FAP85 and 10 mM KCl), or of 250 mM KCl in HMDE (final concentration, 10 mM KCl), were added and samples were incubated for 5 min and then analyzed in the microplate reader.
The turbidity of pre-polymerized MTs was monitored at 350 nm using a DU 800 spectrophotometer (Beckman Coulter). Hundred microliters of 30 μM tubulin, with and without 2.8 μM FAP85, were polymerized at 37°C for 30 min. Microtubules were immediately moved to a cuvette; then, depolymerization was induced by cooling the cuvette to 15°C, and monitored for 5 min. Abs350nm, stable during the first minute of incubation, subsequently and progressively decreased, indicating the depolymerization of the MTs. The absorbance values were fitted to a linear equation. The slope of the curve obtained, which was defined as time (min) versus ΔAbs350nm, represented the depolymerization rate (Johnson and Borisy, 1977).
FAP85 has previously been identified in proteomics studies of Chlamydomonas flagella (Pazour et al., 2005), and included in the National Center for Biotechnology Information database (NCBI: https://www.ncbi.nlm.nih.gov/). FAP85 is predicted to be 197 amino acids in length and with a molecular mass of 22.2 kDa. InterPro annotation shows that, FAP85 has an EF-hand motif near its center, which is characteristic of calcium-binding proteins (Fig. 1A). BLAST searches showed that FAP85 homologs of unknown function, including hypothetical proteins are present in Volvocine algae such as Volvox, and Gonium (E value < 1 e–105), and also found in Micromonas (E value < 2e–21) and Monoraphidium (E value < 2e–13).

Characterization of FAP85. (A) Amino-acid sequence of FAP85 and predicted EF-hand motif (underlined, identified by InterPro analysis). No coiled-coil regions were predicted. The scale indicates the positions of amino acid residues. (B) SDS-PAGE (upper panel) and immunoblot (lower panel, lanes blotted from the SDS-PAGE) of flagella and their extracts; the anti-FAP85 antibody detected a single band with the expected molecular weight of FAP85. FAP85 was not detected in the 0.6 M KCl extract; however, it was observed in the residual axonemes. The treatment of the axonemes with 0.6 M KI resulted in complete extraction of FAP85 in the supernatant. (C) FAP85 was present in various types of flagellar mutants lacking major axonemal components. Prior to electrophoresis, flagella from each mutant were demembraned and extracted with 0.6 M KCl. Protein loading was standardized based on tubulin concentrations determined using densitometry of the CBB staining. Relatively lower content of FAP85 in pf18 axonemes was due to the variation of the transfer efficiency of the blotting.
We cloned and sequenced the cDNA corresponding to the 22-kDa polypeptide and confirmed that it is identical to the sequence recorded as FAP85 in GenBankTM/EBI (accession number, XM_001695284). To increase the solubility of the protein, the cDNA of FAP85 was fused to the ProS2 protein, which increases solubility of expressed polypeptides. Protease treatment of the recombinant protein liberated FAP85 that was insoluble and formed aggregates. Consequently, we used recombinant FAP85 as a ProS2-conjugate.
The association of Ca2+ with this recombinant ProS2-FAP85 was examined using isothermal titration calorimetry (Supplementary Fig. S1). The ITC supports that FAP85 has a single EF-hand motif. As ProS2 has two Ca2+ binding sites, recombinant ProS2-FAP85 possesses a total of three Ca2+-binding sites. By analyzing data, including ProS2 data, we estimated the Kd of the Ca2+-binding site of FAP85 to be 1 – 8×10–8 M. The Kd was far from the range of Ca2+ concentrations that occur during the waveform changes (pCa4 - pCa5). The results suggest that FAP85 is bound to Ca2+ under physiological conditions and may not be directly involved in the waveform changes.
A polyclonal rabbit antibody against ProS2-FAP85 recognizes native FAP85After extraction of the dynein arms with 0.6 M KCl, the pellet obtained from wild-type axonemes was analyzed by SDS-PAGE followed by immunoblotting. The anti-ProS2-FAP85 antibody (whose generation is described in Materials and Methods) detected a single band of the expected size (~25 kDa). However, upon further extraction with 0.6 M KI, FAP85 was solubilized (Fig. 1B).
Immune-precipitates obtained using the anti-ProS2-FAP85 antibody were subjected to peptide mass fingerprinting by MALDI-TOF mass spectrometry, which identified the single band detected by the anti-ProS2-FAP85 antibody as FAP85. Thus, we concluded that the anti-ProS2-FAP85 antibody we generated was capable of recognizing the native FAP85 protein (hereinafter, the anti-ProS2-FAP85 antibody is referred to as the anti-FAP85 antibody).
Analysis of FAP85 in mutant axonemesThe presence and localization of the FAP85 in the axoneme were examined with the anti-FAP85 antibody in mutants missing major axonemal components (Table I).
First, we treated the demembraned axonemes with 0.6 M KCl to remove soluble components from the axonemes and analyzed the residual axonemes by SDS-PAGE and by immunoblotting. Our data indicate that the anti-FAP85 antibody recognizes a prominent band of 23 kDa also in mutant axonemes (Fig. 1C). Although quantification of protein abundance by immunoblotting is difficult because of the poor linearity to protein content and large variation of transfer efficiency, no significant difference in the amount of the FAP85 with respect to the amount of tubulin was observed between these axonemes. Because FAP85 was found in the residual axonemes of each mutant examined, this protein was suggested to be localized on DMTs.
Solubilization of DMTs with SarkosylImmunofluorescence microscopy was carried out in Chlamydomonas cells; however, the antibody we generated did not show significant immune-reactivity towards axonemes of wild-type cells (Supplementary Fig. S2). Based on the results of the western blots and immunofluorescence assays described above, we hypothesized that FAP85 is located in a region of the DMTs not accessible to the antibody. To expose the epitopes of FAP85 and determine the localization of FAP85 on DMTs, we extensively solubilized axonemes of pf14 (a mutant missing the radial-spokes, Piperno et al., 1977) with Sarkosyl. This allowed us to observe DMTs protofilaments in detail. To do so, the demembranated axonemes of pf14 were pre-treated with 0.6 M KCl to remove soluble components from DMTs. At concentrations of Sarkosyl>0.3%, most B-tubules were dissolved while the A-tubules remained almost intact: centrifugation would separate into B-tubules, in the supernatant, from A-tubules, in the pellet (Witman et al., 1972).
Treatment of axonemes with 0.2% Sarkosyl showed that proteins over 250 kDa were dissolved in the supernatant; however, most of the tubulin and FAP85 protein remained in the pellet (Fig. 2A). Increasing the Sarkosyl concentration to 0.3% resulted in partial dissolution of the DMTs; however, most of the FAP85 remained in the pellet. The supernatants of 0.4% and 0.7% Sarkosyl-treated axonemes contained proteins similar to those found in the supernatant of 0.3% Sarkosyl-treated axonemes. However, when the concentration of Sarkosyl was increased to 0.7%, DMTs and FAP85 were completely dissolved in the supernatant following overnight treatment. To examine the morphology of DMTs in the pellet as a function of the concentrations of Sarkosyl, we dispersed and stained the pellet with uranyl acetate and observed it using an electron microscope (Fig. 2B, C, D).

Extraction of FAP85 from the doublet microtubules (DMTs) via treatment with 0.2–0.7% Sarkosyl. (A) Upon treatment with increasing concentrations of Sarkosyl, FAP85 was extracted from axonemes of the mutant pf14 that lacks radial spokes. Increasing the concentration of Sarkosyl facilitated solubilization of FAP85 in the supernatant. Overnight treatment with 0.7% Sarkosyl (0.7 o/n) resulted in complete dissolution of FAP85 in the supernatant. (B–D) Negatively stained DMTs in the pellet of Sarkosyl-treated axonemes were observed by transmission electron microscopy (bars: left panels, 200 nm; right panels, 50 nm). (B) Treatment with 0.3% Sarkosyl partially dissolved the DMTs. (C) Treatment with 0.7% Sarkosyl, completely dissolved the B-tubules, and left the A-tubules partially dissolved; the inner walls of the A-tubules were exposed in some regions and protofilaments were partially disrupted. (D) Overnight treatment with 0.7% Sarkosyl resulted in the formation of Sarkosyl-insoluble “ribbon” structures. (E) The line plots summarize the electron microscopic observations of the fractions of disrupted A-tubules (▲: mean of three experiments; △: individual experiments), B-tubules (●: mean of three experiments; ○: individual experiments), and the amount of FAP85 detected in the supernatant by immunoblotting (■: mean of three experiments; □: individual experiments). The increase in the amount of dissolved FAP85 corresponded to the increase in the proportion of disrupted A-tubules.
In the presence of 0.2% Sarkosyl, the pellet contained a significant amount of DMTs. Increasing the Sarkosyl concentration to 0.3% resulted in dissolution of the B-tubules; however, the singlet A-tubules remained (doublets [A- and B-tubules]: A-tubule=24:76). The pellets of 0.4% and 0.7% Sarkosyl-treated axonemes contained numerous singlet A-tubules without B-tubules. Furthermore, A-tubules were cleaved into the ribbon structure by 0.7% Sarkosyl (Norrander et al., 2000) (Fig. 2C and D). The ribbon structures were identified via their characteristic features, by negative staining: these structures were thick filaments composed of three protofilaments, whose details, however, could not be clearly observed (Supplementary Fig. S3).
Extensive long-term treatment of the axonemes with 0.7% Sarkosyl dissolved the DMTs and generated three protein bands upon SDS-PAGE of the pellet: tubulin and two proteins with molecular weight of ~70 and ~50 kDa; the latter was distinct from tubulin. According to a previous study (Ikeda et al., 2003), the ~70-kDa protein was Rib72, and the ~50-kDa protein Rib43. Electron microscopic observations of these axoneme samples showed that only ribbon structures were present in the pellet (Fig. 2D). Under long-term 0.7% Sarkosyl treatment, FAP85 was found in the supernatant but not in the pellet.
Comparison between the immunoblotting results of FAP85 and the electron microscopic observations of Sarkosyl-treated axonemes showed that FAP85 emerged in the supernatant concomitantly with the disassembly of A-tubules only (Fig. 2E). This observation suggested that FAP85 is associated tightly with the A-tubules. Furthermore, the treatment of axonemes with 0.6 M KCl followed by 0.3–0.7% Sarkosyl removed almost all of axonemal proteins from DMTs (Fig. 2B, C). Even under these conditions, FAP85 was detected in the pellet containing A-tubules. Therefore, the emergence of FAP85 in the supernatant coincided with the disassembly of A-tubules. These observations additionally suggest that the binding of FAP85 to the DMTs is independent of axonemal components such as dyneins, radial spokes, and the central apparatus.
Immuno-gold electron microscopy of FAP85 on DMTsIn order to investigate the presence of FAP85 on the A-tubules, we carried out immuno-gold electron microscopy of the Sarkosyl-treated axonemes (Fig. 3). These were first incubated with the anti-FAP85 antibody, and subsequently treated with a gold-conjugated secondary antibody. Rabbit IgGs obtained from the serum of a non-immunized animal were used as negative control.

Immunogold electron microscopy of 0.3% Sarkosyl-treated axonemes with anti-FAP85 antibody and gold-conjugated secondary antibodies. The anti-FAP85 antibody did not frequently label intact A-tubules; however, A-tubules with loss of structural integrity (white arrowhead) were labeled: gold particles were found only where the inner wall of the A-tubule was exposed. (A) A-tubule of DMTs showed both closed and opened parts along the long axis (black and white arrowhead, respectively). No gold particles were observed on the intact A-tubules. (B) A magnified image of the DMTs, with the inner walls were exposed (indicated as “opened”). The gold particles were often found on the exposed portion. (C) Another field of the view showed in (A), confirming that the gold particles were frequently found on the exposed inner wall of the A-tubules. (D) A schematic drawing of the positions of gold particles and the doublet MT exposing the inner walls of the A-tubule shown in (C); bar in (A–C)=200 nm. (E) The number of gold particles in the opened region per unit area was significantly higher than that in the closed regions and that in the opened region treated with IgGs isolated from non-immunized rabbits (**: p<0.01, *: p<0.05, χ2-test).
In the presence of 0.3% Sarkosyl, gold particles were found in partially dissolved axoneme only on regions where the inner wall of the A-tubules was exposed (Fig. 3A). The A-tubules with exposed inner wall were identified according to the width of the tubule: in the negatively stained samples, an inner-wall-exposed A-tubule is wider than an intact one, and a larger number of protofilaments may be recognized in the A-tubule than in an intact one (Fig. 3B, C, D). The number of gold particles in the exposed region per unit area was significantly higher than that in the intact regions (Fig. 3E, Table II). These results suggest that FAP85 is localized only on the inner wall of the A-tubules. The immuno-gold particles were rarely observed on the ribbon structures, which were easily identified with negative staining; however, these particles occurred on protofilaments juxtaposed with the ribbon structure (Supplementary Fig. S3A).
| Number of particles per μm2 (Total number of particles) | Un-assigned | Total | |||
|---|---|---|---|---|---|
| On axonemes | Background | ||||
| Opened region | Closed region | ||||
| Anti-FAP85 | 17.1**,* (542) | 1.4 (268) | 1.1 (598) | 87 | 1495 |
| Non-immune rabbit IgG | 5.2 (108) | 1.6 (193) | 0.5 (289) | 30 | 620 |
The gold particles observed on the specimens were categorized into four groups: particles within a distance of 23 nm (the expected distance occupied by primary and secondary antibody complexes) from the edge of the DMTs, further divided into opened and closed groups; particles farther than 23 nm were defined as background, and particles positioned near the edge of the field of view or over unrecognizable materials were unassigned. The number of gold particles per unit length in the exposed region was significantly higher than that in the intact regions (p<0.01, by χ2 test) and in the exposed region of samples treated with control antibody from non-immunized rabbits (p<0.05, by χ2 test). The total number of gold particles in the anti-FAP85 sample was also higher than that in normal rabbit IgG samples. The total number of particles were determined in three independent experiments.
** compared to the number of gold particles per μm2 in the closed region (p<0.01 by χ2-test).
* compared to the number of non-immunized-gold particles per μm2 in the opened region (p<0.05 by χ2-test).
Since the gold particles conjugated to the secondary antibody exhibited various angles of binding to the antigen, these particles were distributed within a 23-nm radius around a fixed point (Norrander et al., 2000), on which the antigen presumably exists. Thus, the precise position of the FAP85 antigens could not be determined. However, a line could be drawn wherein we minimized the sum of the squared distances between the gold particles (the best-fit least square line in Supplementary Fig. S3A). Since this line was almost parallel to the protofilament in A-tubule, we considered FAP85 to be attached to the inner wall of A-tubule along a protofilament. Furthermore, the pairwise distance measured between each possible pair of particles was distributed as a multimodal distribution with peaks at integral multiples of ~50 nm (Supplementary Fig. S3B, C).
In some cases of partially dissolved axonemes, nine DMTs originating from the same axoneme were found. In this case, immuno-gold particles were found to be labeled on all DMTs with exposed inner walls of A-tubules (Supplementary Fig. S4). The results indicate that FAP85 does not localize in any specific doublet.
Direct interaction between FAP85 and tubulinTo examine whether native FAP85 directly binds to tubulin or other proteins, we carried out immunoprecipitation of Sarkosyl-treated axonemes with the anti-FAP85 antibody (Fig. 4A). First, the axonemes were completely dissolved by treatment with 0.7% Sarkosyl overnight and dialyzed against PBS for removal of Sarkosyl from the solubilized proteins. Additionally, the dialysis was expected to facilitate re-binding of FAP85 to its binding partners. The FAP85 immunocomplexes were precipitated with the anti-FAP85 antibody and separated by SDS-PAGE. They contained two proteins of ~25 and ~50 kDa proteins: MALDI-TOF mass spectrometry identified them as FAP85 and β-tubulin, respectively. No significant trace of the α-tubulin fragments was found in the MALDI-TOF mass spectra. These data suggest that FAP85 is capable of directly binding to β-tubulin in the A-tubules.

Binding of FAP85 to tubulin and microtubules. (A) Immunoprecipitation of 0.7% Sarkosyl-dissolved axonemes with the anti-FAP85 antibody demonstrated that FAP85 binds to a protein of ~50 kDa (star), identified as β-tubulin by MALDI-TOF mass spectrometry. (B) The zero-length chemical crosslinker EDC, used on Sarkosyl-treated axonemes produced a ~75- and ~150-kDa protein bands as detected by immunoblotting against the anti-FAP85 antibody, in addition to the FAP85 band (white arrowhead). These two bands were identified as β-tubulin-FAP85 complexes by MALDI-TOF mass spectrometry.
Chemical crosslinking provides a direct method for identifying both transient and stable interactions of proteins. Therefore, we used the heterobifunctional, water-soluble, zero-length carbodiimide crosslinker EDC to crosslink FAP85 and its binding partners. The formation of crosslinks between FAP85 and other proteins would be a direct and convincing evidence of their close proximity/interaction.
Sarkosyl-treated A-tubules were cross-linked with EDC. The reaction was terminated by addition of an excess of 2-mercaptoethanol. EDC-treated proteins were separated by electrophoresis, and cross-linked FAP85 was detected by immunoblotting with the anti-FAP85 antibody (Fig. 4B). The untreated sample showed a single band corresponding to FAP85 (Fig. 4B, lane 1). With the increase of the EDC concentrations, two bands (~75 and ~150 kDa) emerged in addition to the FAP85-band. MALDI-TOF mass spectrometry indicated they were contained peptides derived from β-tubulin and FAP85. This result, combined with immunoblotting data, was consistent with the results from the immunoprecipitation assays, and indicated that the ~75-kDa band was formed by crosslinking of β-tubulin (~50 kDa) and FAP85 (~22 kDa); the ~150-kDa band was formed by crosslinking of two β-tubulin-FAP85 complexes that were in close proximity. Although no significant trace of the α-tubulin fragments was found in the MALDI-TOF mass spectra we could not exclude the possibilities that the ~150-kDa complex was composed of α-tubulin, β-tubulin, and FAP85 or composed of two β-tubulin and FAP85.
Binding affinity of FAP85 to microtubules in vitroThe in vitro interactions of FAP85 with tubulin and/or MTs were examined using recombinant ONL-FAP85 and tubulin purified from porcine brains. Because ProS2-FAP85 (~50 kDa) and tubulin (~55 kDa) have similar molecular weights, they cannot be distinguished after SDS-PAGE. Therefore, we used ONL-FAP85 (85 kDa) instead of ProS2-FAP85. ONL-FAP85 and taxol-stabilized MTs or tubulin mixed at various ratios were incubated, and spun down; the proteins were then separated by SDS-PAGE. The density of ONL-FAP85 bands was quantified and the ratio of the amount of bound FAP85 was calculated (Fig. 5A, B). The same procedure was applied to a solution containing only ONL-FAP85: a small amount of ONL-FAP85 precipitate (~5% of the total amount of ONL-FAP85) was generated. Therefore, we concluded that FAP85 directly binds the MTs.
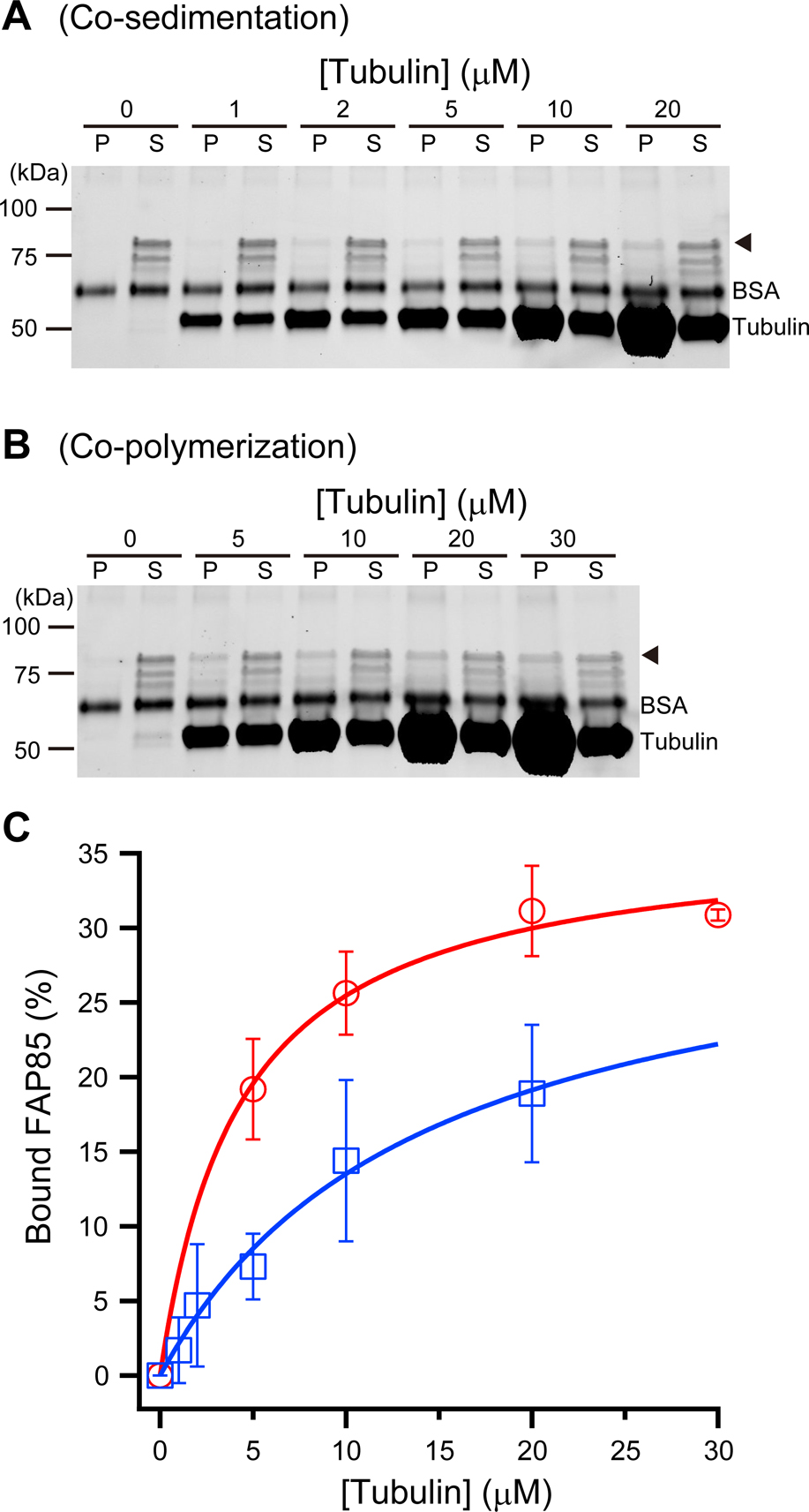
FAP85 microtubule-binding assay. (A) Co-sedimentation of ONL-FAP85 and taxol-stabilized MTs at various concentrations of MTs. ONL-FAP85 (~85 kDa) is indicated by the arrowhead. The band just below ONL-FAP85, which might indicate the degradation/cleavage of ONL-FAP85. (B) Co-polymerization of tubulin with ONL-FAP85. Tubulin at various concentrations was polymerized in the presence of ONL-FAP85. At higher tubulin concentrations, a significant amount of ONL-FAP co-precipitated with the MTs. To indicate the FAP85 bands clearly, the contrast of the gel images was linearly enhanced in (A) and (B). The images of tubulin bands were thus saturated due to the large difference in concentrations between tubulin and FAP85. (C) Percentages of ONL-FAP85 bound to MTs plotted against the tubulin concentrations. The equation, B=Bmax T/(Kd+T) was used to fit the data. Error bars represent the standard deviation (S.D., n=3). The red curve is the best fit for the data from co-polymerization experiments. Below the critical concentration of tubulin (<5 μM), no pellet was found after the centrifugation. The blue curve is the best fit for the data from the co-sedimentation experiments. For co-polymerization experiments, Bmax=36.4±1.4, Kd=4.3±0.6 μM and for co-sedimentation experiments, Bmax=32.7±4.3 and Kd=14.2±3.5 μM.
To determine the binding affinity of FAP85, we analyzed the amount of ONL-FAP85 (0.77 μM) depleted from the supernatant with increasing concentrations of stabilized MTs in co-sedimentation assays (Fig. 5C). The dissociation constant (Kd) of FAP85 was estimated to be 14.2±3.5 μM (n=3). Strikingly, in co-polymerization experiments in which tubulin was polymerized in the presence of FAP85, we observed rapid depletion of FAP85 even at concentrations just above the critical concentration of tubulin, indicating that, during tubulin polymerization, FAP85 interacts with the binding sites of the inner wall of MTs (Kd=4.3±0.6 μM, Fig. 5B, C).
According to the parameters determined by the fit, the stoichiometry of FAP85 to tubulin was about 1:31 in co-polymerization experiments. Assuming that the number of protofilaments in the A-tubule is thirteen, the stoichiometry suggests that two~three tubulin dimers (16~24 nm) interact with one FAP85, which is consistent to a 16nm-structural repeat of MIP1a (Ichikawa et al., 2017).
Binding of FAP85 to microtubules altered their dynamicsFirst, to examine whether the expressed ProS2-FAP85 can interact with tubulin, we performed immunoprecipitation with the anti-ProS2 antibody against the ProS2-FAP85-tubulin mixture and confirmed that recombinant ProS2-FAP85 interacts with tubulin (Supplementary Fig. S5). Next, to examine the effects of FAP85 on MT dynamics, we assembled and disassembled MTs in vitro, with or without ProS2-FAP85, and monitored the turbidity of the MT solution at 350 nm (Fig. 6). The MTs without FAP85 were depolymerized almost completely within the observation period. However, the turbidity of MTs solutions with FAP85 decreased about 0.7 times more slowly than that of the solution without FAP85, and remained higher even after prolonged cold incubation. The depolymerization rates measured were 0.11±0.01 ΔAbs350nm/min (tubulin only) and 0.07±0.01 ΔAbs350nm/min (tubulin with FAP85). These data indicate that FAP85 stabilizes MTs against cold-induced disassembly (Fig. 6A).

Effect of FAP85 on polymerization and depolymerization dynamics of tubulin. Tubulin polymerization and depolymerization were monitored as the change in turbidity at 350 nm (Abs350nm). (A) The depolymerization rate (ΔAbs350nm/min) was calculated in the range in which the turbidity decreased linearly (dotted lines). The bar graph indicated the depolymerization rates with (blue line) and without (red line) FAP85, which were 0.071±0.011 min–1 and 0.113±0.014 min–1, respectively (n=3, *: p<0.05, Student’s t-test). (B) Polymerization of tubulin at various concentrations in the presence (blue lines) or the absence (red lines) of FAP85. The polymerization was initiated by heating the tubulin solution in the micro-plate to 37°C and was monitored for 120 min. (C) Within the range of tubulin concentrations from 5 to 25 μM, the addition of FAP85 resulted in faster polymerization of tubulin into MTs (blue lines compared to red ones). (D) At concentrations of 30 and 35 μM tubulin, polymerization occurred too rapidly to identify FAP85 effect. However, the initial delay in the increasing absorbance was shorter in the presence of FAP85 (blue lines) than without (red lines). (E and F) The critical concentration of tubulin polymerization was calculated from the plot of Abs350nm at 120 min after the initiation of polymerization. The linear fits to the data obtained for 20, 25, 30, and 35 μM tubulin provide the critical concentrations of tubulin in the presence or absence of FAP85, which were 11.2±1.8 μM and 14.3±1.3 μM, respectively (mean±the standard deviation [S.D.], n=3, *: p<0.05, Student’s t-test).
On the other hand, polymerization of tubulin was initiated by incubation at 37°C (Fig. 6B). Within the range of tubulin concentrations used (5 to 25 μM), the addition of FAP85 (2.8 μM) resulted in substantially faster polymerization of tubulin into MTs (Fig. 6C). At higher concentrations of tubulin (30 and 35 μM), polymerization was so rapid that the acceleration of the polymerization by FAP85 was not obvious. In these conditions, however, the initial delay in increasing absorbance became shorter in samples with FAP85 than in those without (Fig. 6D). To verify MT formation, we observed the resulting solutions with dark-field microscopy and negative staining electron microscopy and confirmed the formation of MTs but no formation of significant bundles in the presence of FAP85 (data not shown) .
These in vitro results suggest that FAP85 plays a role in MT dynamics by reducing the critical concentration for tubulin polymerization. The critical concentration was calculated from the plot of absorbance at 350 nm, 120 min following initiation of polymerization, as a function of tubulin concentration (Fig. 6E). The linear fits to the data for 20 to 35 μM tubulin provided the critical concentrations with or without FAP85: these were 14.3±1.3 μM (n=3), and 11.4±1.8 μM (n=3), respectively (Fig. 6F).
In this study, we cloned the cDNA of FAP85 and expressed it as a ProS2-FAP85 fusion protein in E. coli cells. We generated an anti-ProS2-FAP85 antibody, demonstrated it recognizes native FAP85 with high specificity and used it for localization and biochemical characterization of FAP85. Biochemical studies of FAP85 showed that the protein binds directly to MTs, stabilizing them. Immuno-EM showed that FAP85 localizes on the inner wall of the A-tubule of DMTs. Therefore, we concluded that FAP85 is a MIP. Specifically, FAP85 capability of stabilizing MTs and slight enhancing tubulin polymerization suggests that FAP85 might play a similar role in vivo, during flagellar assembly.
FAP85 is a component of MIP1a, characteristic of ChlamydomonasAdvanced EM, particularly cryo-EM and cryo-electron tomography, have revealed structures associated with the inner wall of DMTs; filamentous tektin runs along the ribbon structure of the doublets and other globular or filamentous proteins periodically arranged on the inner wall of the DMTs (Linck et al., 2014). These proteins are now known as MIPs. Recently, Ichikawa and his colleagues performed cryo-EM combined with single particle analysis of Tetrahymena cilia, and demonstrated the presence of various types of MIPs and their interactions with tubulin dimers at sub-nanometer-resolutions (Ichikawa et al., 2017). On the basis of the localization of MIPs on the protofilaments, the authors claimed that these proteins play important roles in building DMTs and are involved in their assembly.
Comparison of the axonemes from various types of organisms has enabled the categorization of MIPs into several types such as MIP1a, MIP1b, MIP2a, MIP2b, MIP3a, MIP3b and MIP4 (Pigino et al., 2012). Numerous MIPs are well conserved in different organisms; however, organisms-specific MIPs have also been reported. The axonemal components are arranged on DMTs as axial repeats that are multiples of the 8-nm tubulin-dimer repeat. Major axonemal components, such as the dynein arms and radial spokes, have 96-nm structural repeats that represent the fundamental functional unit (Bui et al., 2008; Toba et al., 2015). Previous studies have shown that MIPs possess such structural repeats: MIP1a and MIP1b make an array alternatingly and possess repeats with 16-nm spacing; those of MIP2a exhibit 16- and 32-nm spacing; those of MIP2b have 48-nm spacing; those of MIP3a and MIP3b have 16-nm spacing; and those of MIP4 exhibit 48-nm spacing.
Immuno-gold particles binding to the anti-FAP85 antibodies were found along a line parallel to the protofilaments in the A-tubule (Fig. 7 and Supplementary Fig. S3A). The pairwise distance between gold particles was measured and the distribution of the distance was examined and broad distribution with multimodal peaks, which emerged at integral multiples of ~50 nm, was observed (Supplementary Fig. S3C).

Localization of FAP85 on the A-tubule of Sarkosyl-treated doublet microtubules. (A) Immuno-gold labeling of the MT disrupted by Sarkosyl treatment and protofilament ribbons (R) that have been negatively stained with uranyl acetate. The least square method applied on the gold-particle positions provided the line (red line), on which FAP85 molecules were presumably located. The line was almost parallel to the protofilament and there were at least seven protofilaments between the line and the edge of the ribbon structure. The A-tubule was opened (an) and partially dissolved indicated by arrows. The opened A-tubule was wider than intact A-tubule (an+1). Scale bar, 50 nm. (B) Intensity profile of the disrupted A-tubule along the dashed-line in (A). Each peak corresponded to a protofilament. The ribbon corresponded to the region with large gray values with few details. In this electron micrograph (A), the least square line was located on the 7th protofilament (arrow). (C) Diagram illustrating the assignment of numbers to the thirteen protofilaments of the A tubule of the DMT and localization of FAP85 on the basis of electron microgram (A). The protofilament numbering was done according to Witman et al. (1972) and Linck et al. (2014), assuming that the 8th protofilament was first dissolved by Sarkosyl treatment and the A-tubule was cleft between 7th and 9th protofilaments. The stable ribbon corresponds to the protofilaments 11th-12th-13th, according to Ichikawa et al. (2017). In the electron micrograph (A), immune-gold particles were localized around the antigen (red circle). (D) Diagram illustrating the localization of FAP85 (red circle) in a DMT. On the basis of the observation on the Sarkosyl-treated A-tubule in (A), the position of FAP85 overlapped with MIP1a (the yellow blob) but not MIP1b (the green blob). Modified after Pigino et al., 2012.
Furthermore, the location of FAP85 in the A-tubule was estimated by immune-EM. The typical example was shown in Fig. 7. The gold particles were not found on the ribbon structure, but on or near the protofilaments juxtaposed five to seven protofilaments away from the edge of the ribbon (Fig. 7 and Supplementary Fig. S3A). This observation suggests that FAP85 is located closer to protofilaments 5th to 7th of the A-tubule (in this paper, we follow the numbering of protofilaments used in Ichikawa et al., 2017).
MIP1 is composed of a large unit, MIP1a, and a small unit, MIP1b, each of which has a periodicity of 16 nm. In addition, MIP1a of Chlamydomonas flagella has three slightly different structures that repeat with a 48-nm periodicity (Pigino et al., 2012). This feature has also been observed in sea urchin sperm, but not in Tetrahymena. The stoichiometry of FAP85 to tubulin we biochemically determined suggests that two~three tubulin dimers (16~24 nm) interact with one FAP85, which is consistent with a 16-nm structural repeat of MIP1a. A 50-nm structural repeat of FAP85 revealed by immunoelectron microscopy is explained by the triplet MIP1a complex, which could occupy two adjacent binding sites of FAP85 but not the rest where the FAP85 can bind. Three slightly different structures of MIP1a with a 48-nm periodicity would reflect ~50-nm structural repeat of FAP85 in situ.
In addition, MIP1a of Chlamydomonas was found to be larger than that of sea urchin sperm, and had an obvious structure bridging protofilaments 5th to 7th of A-tubules (these correspond to protofilaments 10th to 8th in the study from Pigino and colleagues [Pigino et al., 2012]). Therefore, comparing the localization and the periodicity of FAP85 on the A-tubule with that of the previously described MIP1a, we concluded that FAP85 is a component of MIP1a.
FAP85 stabilizes DMTsAlthough MIPs have been reported as components of DMTs, their composition, biochemical properties, and functions have not been characterized to date. FAP85 is therefore the first MIP to be biochemically characterized. Previous studies suggest that MIPs may contribute to the stability of the DMTs, as the doublets do not fragment even when cilia/flagella beat with a frequency of dozens of hertz. Schaedel and colleagues (Schaedel et al., 2015) used polymerized MTs and a microfluidic device to show that the stiffness of MTs decreased each time they were folded by flow. These findings are similar to other cases of material fatigue; the concentration of mechanical stress on pre-existing defects further decreases MT stiffness. Although damaged MTs were recovered by recruiting new tubulin dimers into their lattice in vitro (Schaedel et al., 2015), this is not considered to occur in the axoneme as recovery takes tens of seconds. Therefore, the stability of tubulin-tubulin interaction is important in axonemes to prevent MT breakage caused by beating. The observed effects of FAP85 on tubulin polymerization and depolymerization imply that this protein plays roles in stabilization of the MTs.
Furthermore, various types of MIPs have been described in a variety of organisms, and divergence of the molecular architecture of MIPs as well as structural similarities have been reported in these organisms. The divergence of the MIPs may lead to differences in the bending patterns of these organisms (Pigino et al., 2012), and FAP85 may contribute to such differences by altering stiffness or other mechanical properties of DMTs. In order to elucidate the precise roles of FAP85, further experiments on flagellar mechanics and structure are needed.
We are grateful to Drs. Akane Furuta, Ken’ya Furuta, Takayuki Torisawa, Hideaki Kojima and Yoshinobu Mineyuki for their indispensable advice, support with the research, and their critical reading of the manuscript and insightful discussions. Technical support was kindly provided by Ms. Maki Yoshio, Ms. Misako Amino, and Ms. Yumi Nakajima. This work was supported by a Fellowship from the Leading Graduate School of PicoBiology at the University of Hyogo (J.K.), funding from the Takeda Science Foundation (K.O.), and Grant-in-Aid for Scientific Research, the Japan Society for the Promotion of Science (JSPS, grant numbers 26440089 and 17K07376 to K.O.).
The authors declare that they have no conflicts of interest with the contents of this article.
J.K. and K.O. designed and performed experiments. Both analyzed and interpreted data, and wrote the manuscript.