Abstract
The oncogenic tyrosine kinase BCR-ABL activates a variety of signaling pathways and plays a causative role in the pathogenesis of chronic myelogenous leukemia (CML); however, the subcellular distribution of this chimeric protein remains controversial. Here, we report that BCR-ABL is localized to stress granules and that its granular localization contributes to BCR-ABL-dependent leukemogenesis. BCR-ABL-positive granules were not colocalized with any markers for membrane-bound organelles but were colocalized with HSP90a, a component of RNA granules. The number of such granules increased with thapsigargin treatment, confirming that the granules were stress granules. Given that treatment with the ABL kinase inhibitor imatinib and elimination of the N-terminal region of BCR-ABL abolished granule formation, kinase activity and the coiled-coil domain are required for granule formation. Whereas wild-type BCR-ABL rescued the growth defect in IL-3-depleted Ba/F3 cells, mutant BCR-ABL lacking the N-terminal region failed to do so. Moreover, forced tetramerization of the N-terminus-deleted mutant could not restore the growth defect, indicating that granule formation, but not tetramerization, through its N-terminus is critical for BCR-ABL-dependent oncogenicity. Our findings together provide new insights into the pathogenesis of CML by BCR-ABL and open a window for developing novel therapeutic strategies for this disease.
Key words: BCR-ABL, subcellular localization, stress granule
Introduction
Chronic myelogenous leukemia (CML) is a myeloproliferative disorder resulting from a reciprocal translocation between chromosomes 9 and 22, which forms the characteristic Philadelphia (Ph1) chromosome. This translocation fuses the breakpoint cluster region (BCR) gene into the juxtaposition of the nonreceptor tyrosine kinase-encoding ABL gene to generate the fusion gene BCR-ABL, a driver mutation in CML. The fusion protein BCR-ABL, a constitutively activated tyrosine kinase, thereby plays a causative role in the pathogenesis of CML through the upregulation of a wide range of signaling pathways. It has been shown that expression of BCR-ABL can transform mouse fibroblast cells, factor-dependent hematopoietic cell lines, and primary bone marrow cells (Ren, 2005). Furthermore, the forced expression of BCR-ABL in mouse bone marrow cells results in a CML-like myeloproliferative disorder (Daley et al., 1990). Such transformation activity of BCR-ABL is completely dependent on its tyrosine kinase activity. In fact, oncogenesis by BCR-ABL was diminished by the introduction of a point mutation in the ATP-binding site of ABL, which inactivates its kinase activity (Zhang and Ren, 1998). Based on these findings, tyrosine kinase inhibitors of ABL have been developed and utilized for CML treatment. For instance, the first generation of the tyrosine kinase inhibitor imatinib has innovated the treatment of CML and changed this disease into being controllable by oral administration (Druker et al., 1996, 2002).
The ABL kinase activity of BCR-ABL is necessary, as described above, but not sufficient for CML pathogenesis. Fusion with the BCR protein provides ABL kinase domains and motifs that are required for constitutive activity, interaction with other downstream signaling pathways, and subsequent oncogenic activities. For example, it was shown that mutant c-ABL activated by SH3 deletion cannot induce myeloproliferative disorder in mice (Gross et al., 1999). A mutant p210 BCR-ABL lacking the N-terminal coiled-coil (CC) oligomerization domain is unable to induce myeloproliferative disorder in mice (He et al., 2002). Phosphorylation of tyrosine at 177 in BCR is also essential for the activation of Ras/extracellular-regulated kinase (ERK) and phosphoinositide 3-kinase (PI3K)/Akt pathways through binding to the SH2 domain of an adaptor protein, growth factor receptor-bound protein 2 (Grb2) (Sattler et al., 2002). However, knowledge about the importance of other domains of BCR-ABL in oncogenic signaling pathways has been limited (Ren, 2005).
The spatiotemporal regulation of signaling molecules is needed to determine the diversity and specificity of intracellular signaling. Although BCR-ABL is predominantly localized in the cytoplasm, the specific subcellular localization of this fusion has also been reported. For example, Wertheim and colleagues demonstrated that BCR-ABL is localized to F-actin through the actin-binding domain in its C-terminus (Wertheim et al., 2003). Another group also claimed that BCR-ABL was localized in the Golgi apparatus (Miroshnychenko et al., 2010). In addition, both endogenous and exogenous BCR-ABL form granule-like structures in either hematopoietic or nonhematopoietic cells (Skourides et al., 1999; Patel et al., 2008).
Recent developments in imaging techniques have enabled us to examine the precise spatiotemporal regulation of intracellular signaling in living cells. We previously developed the Förster resonance energy transfer (FRET)-based biosensor Pickles (phosphorylation indicator of CrkL en substrate) and succeeded in measuring BCR-ABL kinase activity and its drug response in living patient-derived CML cells (Mizutani et al., 2010; Horiguchi et al., 2017). While monitoring kinase activity, we noticed that BCR-ABL is localized in liquid-like granules. Here, we identified that such BCR-ABL-positive granules are stress granules (SGs) and that their formation requires ABL kinase activity and the N-terminal region of BCR. Interestingly, its granular localization is necessary for the oncogenic function of BCR-ABL, demonstrating that a novel functional domain of BCR-ABL promotes the pathogenesis of CML.
Materials and Methods
Reagents and antibodies
Imatinib was a kind gift from Novartis Pharma (Basel, Switzerland). Hoechst 33342 was obtained from Thermo Fisher Scientific (Carlsbad, CA, USA). Cycloheximide (used at a final concentration of 10 μg/ml) and thapsigargin (1 μM) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan) and Sigma-Aldrich (St. Louis, MO, USA), respectively. An anti-c-Abl monoclonal antibody (sc-23) was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). Antibodies against Hsp90α (ab2928) and Dcp1a (ab47811) were purchased from Abcam (Cambridge, UK). Alexa Fluor 488-, 594-, and 647-conjugated goat anti-mouse IgG antibodies and an Alexa Fluor 647-conjugated goat anti-rabbit IgG antibody were obtained from Thermo Fisher Scientific.
Cell culture and transfection
HeLa (CCL-2) and Cos-1 (CRL-1650) cells were obtained from the American Type Culture Collection (Manassas, VA, USA). K562 (RCB1897), Ku812 (RCB0495), MY (RCB1701), WEHI-3 (RCB0035) and Ba/F3-CL1 (RCB4474) cells were purchased from RIKEN CELL BANK (Ibaraki, Japan). ALL/MIK cells are Ph1-positive cell line developed from a patient with acute lymphoblastic leukemia (Okabe et al., 1994). TOM-1 cells are Ph1-positive cell line developed from a patient with acute lymphocytic leukemia (Okabe et al., 1987). K562 cells were maintained in RPMI 1640 (Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS, Thermo Fisher Scientific). Ku812, TOM-1, and ALL/MIK were cultured in RPMI 1640 containing 10% FBS and 1% sodium pyruvate. MY cells were maintained in minimal essential medium (MEM, Sigma-Aldrich) supplemented with 10% FBS and 1% sodium pyruvate. HeLa, Cos-1, and murine interleukin 3 (IL-3)-producing WEHI-3 cells were cultured in Dulbecco’s modified Eagle medium (DMEM, Sigma-Aldrich) supplemented with 10% FBS. Ba/F3-CL1 cells were maintained in RPMI 1640 containing 10% FBS and IL-3-containing WEHI-3-conditioned media. The conditioned medium was recovered from near-confluent WEHI-3 cells, centrifuged for 5 min at 390×g, and cleared by filtration with a 0.22 μm-diameter pore. All cells were maintained under a 5% CO2 humidified atmosphere at 37°C.
The expression vectors were transfected into HeLa, Cos-1, K562, Ku812, and TOM-1 cells with “Max” polyethylenimine (Polysciences, Warrington, PA, USA) according to the manufacturer’s recommendations. Gene transfer into K562, Ku812, TOM-1, and Ba/F3 cells was performed via nucleofection with the use of solution V and programs T-016 (for K562 cells), C-005 (for Ku812 and TOM-1 cells), and X-001 (for Ba/F3 cells) according to the manufacturer’s recommendations (Lonza, Basel, Switzerland).
Plasmids
The expression vectors for BCR-ABL, Pickles 2.34, were described previously (Mizutani et al., 2010; Horiguchi et al., 2017). cDNA for BCR-ABL truncation mutants was generated by PCR with the following primers: BCR_F and BCR_fr1_R, BCR_fr2_F and BCR_fr2_R, BCR_fr3_F and BCR_fr3_R, BCR_fr4_F and BCR_fr4_R, BCR_fr5_F and BCR_fr5_R, BCR-ABL-J_F and BCR-ABL-J_R, ABL_fr1_F and ABL_fr1_R, ABL_fr2_F and ABL_fr2_R, ABL_fr3_F and ABL_fr3_R, ABL_fr4_F and ABL_fr4_R, and ABL_fr5_F and ABL_R. cDNA for BCR-ABL-ΔN was also obtained by PCR with BCR_fr3_F and ABL_R. The resulting PCR products were cleaved by XhoI and NotI and were then subcloned into the XhoI/NotI sites of the pFX-H2B-Venus or the pFX-Venus-Rab5 (Kashiwagi et al., submitted for publication).
cDNAs for FYVE, CD63, LAMP1, and TMEM192 were amplified by PCR with the following primers: FYVE_F and FYVE_R, CD63_F and CD63_R, LAMP1_F and LAMP1_R and TMEM192_F and TMEM192_R. These sequences were then subcloned into the XhoI/NotI sites of the pFX-mCherry vector or the pFX-TFP650 vector (Kashiwagi et al., submitted for publication). mCherry-Sec61 β (# 49155) was obtained from Dr. Gia Voeltz via Addgene (Watertown, MA, USA). Expression vectors for other organelle markers will be described elsewhere (Kashiwagi et al., submitted for publication). The primers used in this study are listed in Table S1.
Microscopic setup
Cells were imaged with IX-83 and IX-81 inverted microscopes (Olympus, Tokyo, Japan) equipped with a BioPoint MAC 6000 filter and shutter control unit (Ludl Electronic Products, Hawthorne, NY, USA), an automated XY-stage (Chuo Precision Industrial, Tokyo, Japan), and a SOLA Light Engine (Lumencor, Beaverton, OR, USA) as an illumination source. UPlanSApo 60×/1.35 oil objective lenses were used. The following excitation and emission filters were used in this study: FF01-387/11-25 and FF02-447/60-25 (Semrock, Rochester, NY, USA) for Hoechst 33342; FF02-438/24 and FF01-483/32 (Semrock) for cyan fluorescent protein (CFP) and its derivatives; BP470-490 and BP510-550 (Olympus) for green fluorescent protein (GFP) derivatives; FF01-500/24-25 and FF01-542/27 (Semrock) for yellow fluorescent protein (YFP) and its derivatives; BP520-550 and BA580IF (Olympus) for red fluorescent protein (RFP) derivatives; and FF02-438/24 and FF01-542/27 (Semrock) for FRET. Confocal images were acquired with an sDISK spinning disk unit (Oxford Instruments, Abingdon, UK) and a Rolera EM-C2 electron multiplying cooled charge-coupled device camera (QImaging, Surrey, BC, Canada), whereas the epifluorescence images were acquired with a Cool SNAP MYO cooled charge-coupled device camera (Photometrics, Tucson, AZ, USA). MetaMorph software (Molecular Devices, San Jose, CA, USA) was used for the control of the microscope and the peripheral equipment. For live-cell imaging, the atmosphere was maintained at 37°C with a Chamlide incubator system (Live Cell Instrument, Seoul, Korea) for both microscopes.
Live cell imaging
Cells expressing either fluorescent protein-tagged or epitope-tagged proteins were plated on 35-mm-diameter glass-bottom dishes (AGC Techno Glass, Shizuoka, Japan) coated with poly-L-lysine- (for hematopoietic cells) or collagen-coated (for nonhematopoietic cells). During the observation, cells were maintained in phenol red-free RPMI 1640 (for hematopoietic cells, Thermo Fisher Scientific) or phenol red-free DMEM/F12 (Thermo Fisher Scientific).
For imaging of FRET, cells were subjected to time-lapse, dual-emission microscopy with an interval of every 30 sec. Beginning at 10 min, cells were treated with 2 μM imatinib. To represent FRET efficacy, the FRET/CFP emission ratio was calculated as the quotient of background-subtracted FRET and CFP images and normalized to that at time 0. In Fig. 3, FRET/CFP ratio images are shown in intensity-modulated display mode, in which eight colors from red to blue represent the normalized emission ratios, with the intensity of each color indicating the mean intensity of FRET.
Immunofluorescence
Cells were fixed with 3% paraformaldehyde for 15 min at room temperature. The cells were permeabilized with 0.1% Triton X-100 in PBS for 4 min at room temperature and then incubated with 1% bovine serum albumin and Hoechst 33342 (1 μg/ml). The cells were further incubated with primary antibodies (c-Abl, 1:1,000; Dcp1a, 1:1,000; Hsp90α, 1:200) overnight at 4°C, after which immune complexes were detected by incubation for 1 h at room temperature in the dark with Alexa Fluor 488-, Alexa Fluor 594-, or Alexa Fluor 647-conjugated secondary antibodies (1:300). Images were acquired with an FV10i confocal microscope (Olympus).
Quantification of BCR-ABL-GFP-positive granules
HeLa cells expressing BCR-ABL-GFP were either untreated or treated with 10 μg/ml cycloheximide for 2 h. The cells were then fixed, incubated with Hoechst 33342 and subjected to scanning confocal microscopy. Z-stack images were obtained from top to bottom of the cell with a step of 1 μm and then projected by maximum intensity. The number of granules was automatically counted from the projected images by the MetaMorph plugin “Granularity” within the area that was used for the determination according to the phase contrast images.
Proliferation assay
Forty-eight hours after nucleofection, Ba/F3 cells were transferred into the media, including WEHI-conditioned media and 2 μg/ml puromycin, selected for 7–8 days. Stable cell lines were washed three times with prewarmed phosphate buffered saline (PBS), and 106 live cells were plated in media devoid of IL-3. Viable cell counts were performed daily for 5 days with the trypan blue exclusion test. If necessary, cells were diluted appropriately to <1.0×106/ml.
Cell viability assay
Forty-eight hours after nucleofection, Ba/F3 cells (1.0×106) were washed with prewarmed PBS and resuspended in 1 mL of PBS. The cells were incubated with 1 μL LIVE/DEADTM Fixable Green Dead Cell Stain (Thermo Fisher Scientific) in the dark at room temperature for 30 min. After washing once with PBS, the cells were plated on poly-L-lysine-coated glass-bottom dishes and fixed with 3% paraformaldehyde for 10 min at room temperature. For the detection of BCR-ABL-positive cells, the cells were subjected to immunofluorescence with the use of an anti-c-Abl antibody.
Statistical analyses
The quantitative data are presented as the mean±standard error of the mean (s.e.m.) of at least three independent experiments (unless indicated otherwise) and were compared by one-way analysis of variance (ANOVA) followed by Student’s t-test (parametric test between two conditions). Time series data sets were compared by multivariate analysis of variance (MANOVA) with Bonferroni correction (among multiple conditions). No statistical methods were used to predetermine the sample size. The studies were performed unblinded.
Results
BCR-ABL is localized to cytoplasmic granules
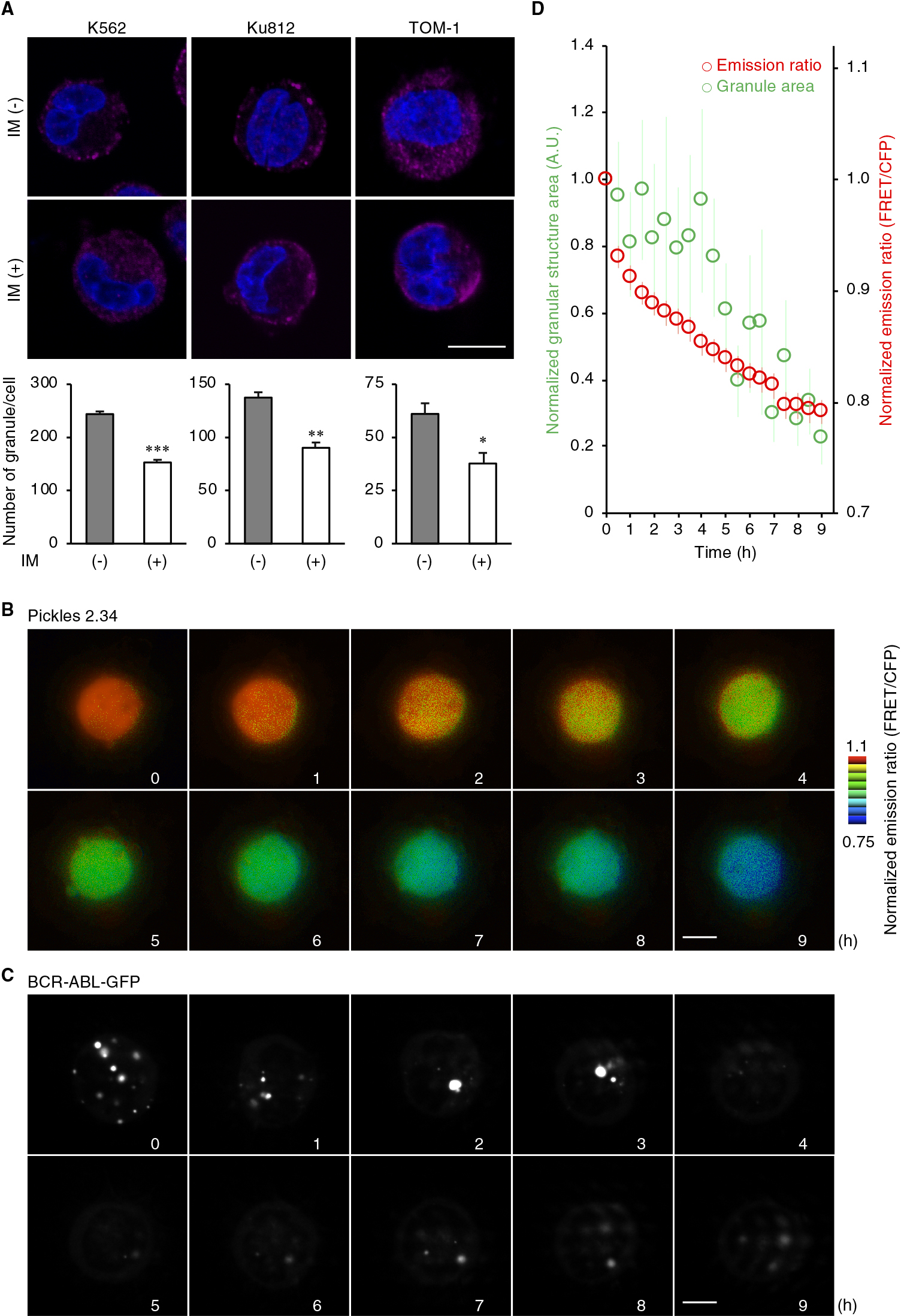
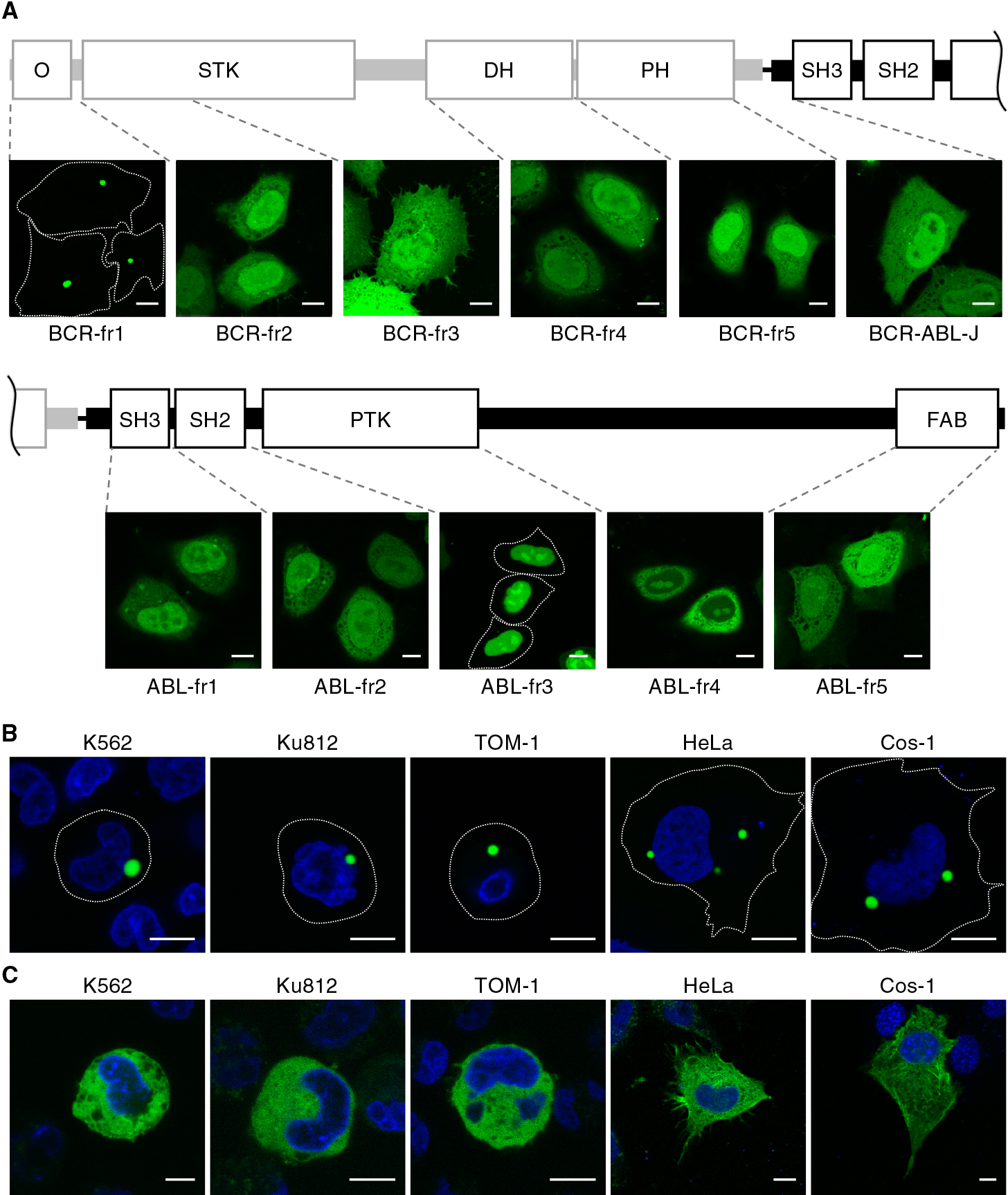
To investigate the subcellular localization of BCR-ABL in Ph1-positive cell lines, K562, Ku812, PALL-2, ALL/MIK, MY, and TOM-1 cells were fixed and subjected to immunofluorescence with the use of an anti-ABL antibody. We found that endogenous BCR-ABL was localized in the cytoplasm and accumulated in cytoplasmic granules in all cell lines (Fig. 1A), consistent with previous reports (Skourides et al., 1999; Patel et al., 2008). Exogenously expressed BCR-ABL-GFP also formed clear cytoplasmic granules not only in the Ph1-positive cells (Fig. 1B) but also in nonhematopoietic cells (Fig. 1C). These results indicated that the formation of granules is due to the intrinsic properties of BCR-ABL but not to cell context-dependent conditions. It has been reported that BCR-ABL-positive granules were not colocalized with endosomal markers, EEA1, Rab7, transferrin, LysoTracker, or caveolin (Skourides et al., 1999), encouraging us to examine in detail the localization of BCR-ABL with the use of a variety of organelle markers (Kashiwagi et al., submitted for publication). Nevertheless, the BCR-ABL-GFP-positive granules were not colocalized with any markers for membrane-bound organelles, which include those for endosomes (early, late, and recycling endosomes), multivesicular bodies, lysosomes, endoplasmic reticulum, and mitochondria (Fig. S1).

During live cell imaging of BCR-ABL-GFP, we noticed that the BCR-ABL-positive granules exhibited liquid-like behavior, i.e., fusion and separation (Fig. S2). It was reported that the heat shock protein Hsp90, one of the components of cytoplasmic RNA granules, including P-bodies (PBs) and SGs, interacts with BCR-ABL (An et al., 2000; Matsumoto et al., 2011; Pare et al., 2009). Indeed, treatment with the translation inhibitor cycloheximide, which prevents RNA granule assembly through retention of messenger ribonucleoprotein polysomes (Mollet et al., 2008), decreased the number of BCR-ABL-positive granules (Fig. 2A), confirming that BCR-ABL forms RNA granules. To further determine whether such RNA granules are PBs or SGs, the cells expressing BCR-ABL-GFP were fixed and subjected to immunostaining with the use of antibodies against HSP90α and the mRNA-decapping enzyme Dcp1a, a component of PBs. Endogenous Hsp90α accumulated in granular structures with BCR-ABL-GFP in HeLa cells, whereas it was localized diffusely in the cytoplasm in control, GFP-expressing cells (Fig. 2B). In addition, the colocalization of BCR-ABL-GFP and Hsp90α at the granular structures was also confirmed in K562 cells (Fig. S3A). In contrast, Dcp1a formed granular structures even in the absence of BCR-ABL, but such granules were not colocalized with BCR-ABL-GFP-positive granules (Fig. S3B). Moreover, thapsigargin treatment, which induces ER stress and subsequent SG formation (Thomas et al., 2009), increased the number of BCR-ABL-GFP-positive granules (Fig. 2C). These results together suggest that BCR-ABL facilitates the formation of SGs and accumulates therein.

It was previously reported that the kinase-deficient mutation (K1176R) abolishes the vesicle-like structure of BCR-ABL (Skourides et al., 1999). In addition, another oncogenic fusion protein, nucleophosmin-anaplastic lymphoma kinase (NPM-ALK), was reported to form mRNA-containing cytoplasmic granules in a manner dependent on the kinase activity of ALK (Fawal et al., 2006, 2011). We therefore compared the subcellular distribution of BCR-ABL in the presence or absence of the BCR-ABL tyrosine kinase inhibitor imatinib (Druker et al., 1996). In the Ph1-positive cells, the number of BCR-ABL-positive granules was reduced by imatinib treatment (Fig. 3A). To provide more direct evidence for the relationship between the dynamics of kinase activity and granular localization, we utilized FRET-based biosensor Pickles to monitor BCR-ABL kinase activity in living cells (Mizutani et al., 2010; Horiguchi et al., 2017). Time-lapse microscopic observation revealed that imatinib inhibited the kinase activity immediately, followed by the disappearance of BCR-ABL-positive granules (Fig. 3B–D), indicating that BCR-ABL kinase activity is required for granule formation.
To determine the domain responsible for granule formation, we next constructed a series of truncation mutants of BCR-ABL (Fig. 4A). Among the mutants, only the N-terminal fragment of BCR (BCR-fr1) clearly showed subcellular granules. This granular localization of BCR-fr1 was observed in both Ph1-positive and nonhematopoietic cells (Fig. 4B). Consistent with this finding, BCR-ABL lacking the N-terminal region of BCR (hereafter BCR-ABL-ΔN) was diffusely localized in the cytoplasm (Fig. 4C). These results suggested that the N-terminal region, namely, the CC domain, of BCR is involved in granule formation. Other mutants were localized diffusely in the cytoplasm and/or the nucleus (Fig. 4A).
Although we demonstrated that BCR-ABL kinase activity is required for granular formation, BCR-fr1 lacking the kinase domain displayed granular localization. This encouraged us to examined in detail the subcellular localization of this fragment. Immunofluorescence analysis showed that BCR-fr1-positive granules were not colocalized with endogenous Hsp90α in both Ph1-positive and nonhematopoietic cells (Fig. S4A). Consistent with this, the number of BCR-fr1-positive granules was not reduced by imatinib treatment (Fig. S4B, C). Nevertheless, BCR-fr1 and full-length BCR-ABL were found to be colocalized in the granules (Fig. S4D). Interestingly, the full length of BCR-ABL was no longer colocalized with HSP90α in the presence of BCR-fr1. These results suggested that BCR-fr1, including the CC domain possesses oligomerizing activity and is necessary for granule formation, but not sufficient to form stress granules in the absence other domains of BCR-ABL such as the kinase domain.
Cytosolic granule formation by BCR-ABL is essential for the proliferation of Ba/F3 cells
It was reported that the N-terminal CC domain of BCR is required for the formation of homotetrameric BCR-ABL and its malignant transformation activity (Maru and Witte, 1991; Pendergast et al., 1991; McWhirter et al., 1993), although the mutants lacking this domain still possess tyrosine kinase activity (He et al., 2002). To clarify whether the oligomerization of BCR-ABL is necessary for granule formation, we utilized DsRed. DsRed forms an obligatory tetramer, which is required for the generation of red fluorescence (Sacchetti et al., 2002). Therefore, tagging with DsRed promotes tetramerization of a covalently linked protein of interest. Moreover, the tetramerization can be confirmed by the emission of red fluorescence. Under these assumptions, whereas DsRed-tagged full-length BCR-ABL accumulated in cytosolic granules, DsRed-BCR-ABL-ΔN was diffusely localized in the cytoplasm (Fig. S5A). Thus, the oligomerization of BCR-ABL is not sufficient for granule formation.
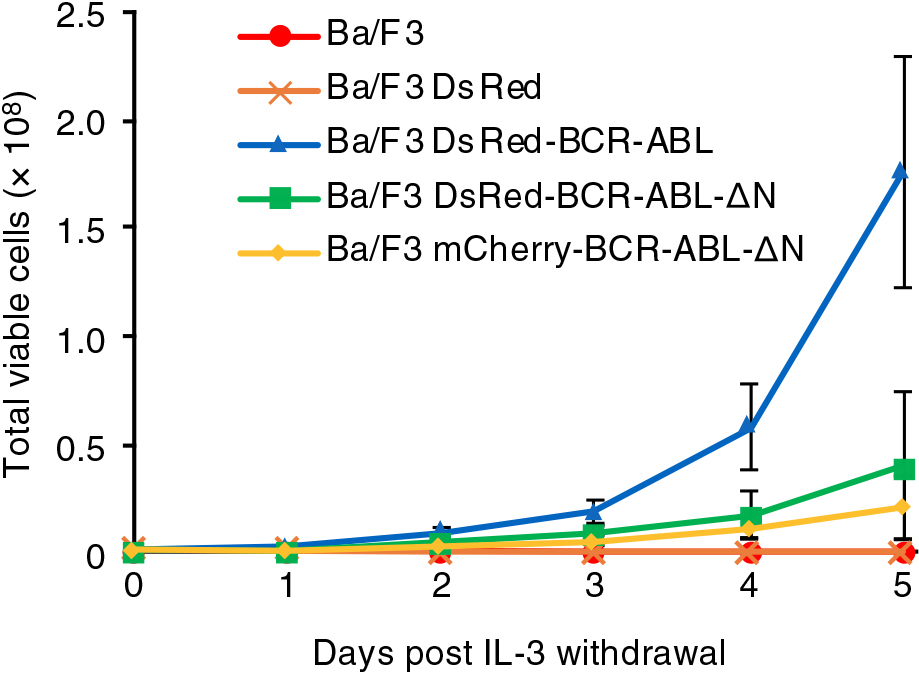
Finally, we examined whether granular localization is involved in the transformation activity of BCR-ABL. To this end, the lymphoid cell line Ba/F3 was utilized. Proliferation of this cell line requires IL-3, the depletion of which resulted in cell growth arrest (Mathey-Prevot et al., 1986, Fig. 5). Under this condition, whereas DsRed-BCR-ABL expression compensated for the growth defect, the expression of mCherry-BCR-ABL-ΔN, which is supposed to be in a monomer form, failed to do so (Fig. 5). Interestingly, DsRed-BCR-ABL-ΔN expression did not restore the defect (Fig. 5). It might be worth noting that the expression of DsRed-tagged BCR-ABL displayed no negative impact on viability of Ba/F3 cells (Fig. S5B). Therefore, given that kinase activity and tetramerization, but not granule forming activity, of DsRed-BCR-ABL-ΔN was assumed to be conserved, these results indicated that the granular localization of BCR-ABL is necessary for its leukemogenic activity.
Discussion
In this study, we characterized cytosolic BCR-ABL-positive granules and demonstrated their significance for oncogenic activities. Such granules displayed liquid-like behavior and indeed colocalized with a marker for membrane-less compartments, SGs. Further investigations revealed that the formation of BCR-ABL-positive granules requires ABL kinase activity and the N-terminal region of BCR. Although it was previously reported that both endogenous and exogenous BCR-ABL form granular structures in CML cells and other cell lines, the subcellular localization of the granules has been controversial (Skourides et al., 1999; Patel et al., 2008). Indeed, no endosomal markers (e.g., markers for early or late endosomes) were colocalized with the BCR-ABL-positive granules. We have demonstrated that the BCR-ABL-positive granules are SGs; however, the molecular mechanism through which this fusion protein is localized to SGs has yet to be determined. In general, the biomolecular condensates, such as nucleoli, SGs, and PBs, are organized by liquid-liquid phase separation driven by interactions of multivalent molecules that harbor multiple elements for intra- or intermolecular interactions (Banani et al., 2017). BCR-ABL is predicted to have the intrinsically disordered regions (IDRs), which provide the weak multivalency to promote phase separation (Banani et al., 2017), in both BCR and ABL (1–417 and 1392–1916 of BCR-ABL) by the web-server-based disorder predictor IUPred (Maru, 2012). Therefore, it might be possible that these IDR domains are involved in granule formation by BCR-ABL. Another possibility might be the involvement of autophosphorylation of the Y177 residue in ABL kinase activity. Phosphorylated Y177 recruits Grb2, which participates in the phase separation of signaling molecules through multivalent interactions (Su et al., 2016).
Similar to our findings on BCR-ABL, the chimeric protein NPM-ALK, a driver gene mutation for anaplastic large cell lymphoma, is also localized to cytoplasmic granules in a manner dependent on ALK kinase activity (Fawal et al., 2006; Honorat et al., 2006). These granules are identified as novel mRNA-containing cytoplasmic granules (ALK granules; AGs) and are distinct from PBs or SGs (Fawal et al., 2011). Similar to our finding on BCR-ABL, AG formation requires ALK kinase activity (Fawal et al., 2006), and NPM is involved in the phase separation through multivalent interactions (Banani et al., 2017). However, it remains unclear whether and how formation of AGs contributes to ALK-mediated oncogenicity.
Non-membrane-bound compartments (e.g., nucleoli, SGs, and PBs) concentrate proteins and nucleic acids at discrete intracellular sites. This condensation could participate in fine-tuning of a variety of cellular processes (e.g., RNA metabolism, ribosome biogenesis, the DNA damage response, and signal transduction) through either the upregulation of reaction specificity and kinetics or inhibition by sequestration of relevant molecules (Banani et al., 2017). For example, the DEAD-box helicase Ded1, a conserved component of SGs that promotes SG formation, represses translation of mRNAs accumulated in SGs (Hilliker et al., 2011). In another example, Dishevelled 2 (Dvl2), a component of canonical and noncanonical Wnt pathways, accumulates in cellular granules (Schwarz-Romond et al., 2007). The formation of Dvl2-positive granules might be required for activation of Wnt signaling because a mutant form of Dvl2 deficient in granule formation exhibits a dominant-negative effect on the Wnt response (Schwarz-Romond et al., 2007). In the case of BCR-ABL, RNA metabolism and/or BCR-ABL activation in SGs is likely to be involved in tumorigenicity. In fact, it has been reported that the mRNA translation of the tumor suppressor breast cancer susceptibility gene 1 (BRCA1) is repressed via BCR-ABL-mediated SG formation in CML cells (Podszywalow-Bartnicka et al., 2014). Additionally, a number of downstream signaling proteins (Grb2, p85 subunit of PI3K, SHC-transforming protein 1, E3 ubiquitin-protein ligase Cbl and CrkL) were found to be localized to the BCR-ABL-positive granules (Skourides et al., 1999). Namely, given that the proportion of CrkL, a major substrate for leukemogenesis by BCR-ABL, is phosphorylated in the granules (Patel et al., 2008), the concentration of the kinase and substrate (BCR-ABL and CrkL in this case) might facilitate excess phosphorylation of substrate and thereby promote leukemogenic activity.
In conclusion, we revealed that the phase separation of BCR-ABL contributes to its oncogenic function and that granule formation requires ABL kinase activity and the N-terminal region of BCR. Our findings suggest a novel mechanism for the oncogenicity of BCR-ABL: i.e., the N-terminal domain of BCR contributes oncogenicity by SG localization as well as by its oligomerization ability. Future studies may provide new therapeutic strategies based on the inhibition of BCR-ABL-positive granule formation.
Acknowledgments
We thank A. Miyawaki for the Venus cDNA, J. Groffen for the human CrkL cDNA, D. Baltimore for the BCR-ABL cDNA, Novartis Pharma for the IM, and A. Kikuchi for technical assistance. This work was supported in part by Grants-in-Aid from the Japan Society for the Promotion of Science (17H04016 and 19K22506 to Y.O.; 16H06227 to Y.F.) and from the Ministry of Education, Culture, Sports, Science and Technology (18H04850 and 19H05411 to Y.O.; 19H04823 to Y.F.), as well as by grants from the Canon Foundation, the Akiyama Life Science Foundation, the Nakatani Foundation, the Takeda Science Foundation, the Japan Research Foundation for Clinical Pharmacology, and the Tokyo Biochemical Research Foundation.
Conflict of interest
Y.O. and T.K. have an issued patent (#5665262), and Y.O. also has an issued patent (#6473080). The other authors have no conflicts of interest.
References
- An, W.G., Schulte, T.W., and Neckers, L.M. 2000. The heat shock protein 90 antagonist geldanamycin alters chaperone association with p210bcr-abl and v-src proteins before their degradation by the proteasome. Cell Growth Differ., 11: 355–360.
- Banani, S.F., Lee, H.O., Hyman, A.A., and Rosen, M.K. 2017. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol., 18: 285–298.
- Daley, G.Q., Van Etten, R.A., and Baltimore, D. 1990. Induction of chronic myelogenous leukemia in mice by the P210 bcr/abl gene of the Philadelphia chromosome. Science, 247: 824–830.
- Druker, B.J., Tamura, S., Buchdunger, E., Ohno, S., Segal, G.M., Fanning, S., Zimmermann, J., and Lydon, N.B. 1996. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr–Abl positive cells. Nat. Med., 2: 561–566.
- Druker, B.J., O’brien, S.G., Cortes, J., and Radich, J. 2002. Chronic myelogenous leukemia. Hematology, 2002: 111–135.
- Fawal, M., Jean-Jean, O., Vanzo, N., and Morello, D. 2011. Novel mRNA-containing cytoplasmic granules in ALK-transformed cells. Mol. Biol. Cell, 22: 726–735.
- Fawal, M., Armstrong, F., Ollier, S., Dupont, H., Touriol, C., Monsarrat, B., Delsol, G., Payrastre, B., and Morello, D. 2006. A “liaison dangereuse” between AUF1/hnRNPD and the oncogenic tyrosine kinase NPM-ALK. Blood, 108: 2780–2788.
- Gross, A.W., Zhang, X., and Ren, R. 1999. Bcr-Abl with an SH3 deletion retains the ability to induce a myeloproliferative disease in mice, yet c-Abl activated by an SH3 deletion induces only lymphoid malignancy. Mol. Cell. Biol., 19: 6918–6928.
- He, Y., Wertheim, J.A., Xu, L., Miller, J.P., Karnell, F.G., Choi, J.K., Ren, R., and Pear, W.S. 2002. The coiled-coil domain and Tyr177 of bcr are required to induce a murine chronic myelogenous leukemia-like disease by bcr/abl. Blood, 99: 2957–2968.
- Hilliker, A., Gao, Z., Jankowsky, E., and Parker, R. 2011. The DEAD-Box Protein Ded1 Modulates Translation by the Formation and Resolution of an eIF4F-mRNA Complex. Mol. Cell, 43: 962–972.
- Honorat, J.F., Ragab, A., Lamant, L., Delsol, G., and Ragab-Thomas, J. 2006. SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood, 107: 4130–4138.
- Horiguchi, M., Fujioka, M., Kondo, T., Fujioka, Y., Li, X., Horiuchi, K., Satoh, A.O., Nepal, P., Nishide, S., Nanbo, A., et al. 2017. Improved FRET biosensor for the measurement of BCR-ABL activity in chronic myeloid leukemia cells. Cell Struct. Funct., 42: 15–26.
- Maru, Y. and Witte, O.N. 1991. The BCR gene encodes a novel serine/threonine kinase activity within a single exon. Cell, 67: 459–468.
- Maru, Y. 2012. Molecular biology of chronic myeloid leukemia. Cancer Sci., 103: 1601–1610.
- Mathey-Prevot, B., Nabel, G., Palacios, R., and Baltimore, D. 1986. Abelson virus abrogation of interleukin-3 dependence in a lymphoid cell line. Mol. Cell. Biol., 6: 4133–4135.
- Matsumoto, K., Minami, M., Shinozaki, F., Suzuki, Y., Abe, K., Zenno, S., Matsumoto, S., and Minami, Y. 2011. Hsp90 is involved in the formation of P-bodies and stress granules. Biochem. Biophys. Res. Commun., 407: 720–724.
- McWhirter, J.R., Galasso, D.L., and Wang, J.Y. 1993. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol. Cell. Biol., 13: 7587–7595.
- Miroshnychenko, D., Dubrovska, A., Maliuta, S., Telegeev, G., and Aspenström, P. 2010. Novel role of pleckstrin homology domain of the Bcr-Abl protein: Analysis of protein–protein and protein–lipid interactions. Exp. Cell Res. , 316: 530–542.
- Mizutani, T., Kondo, T., Darmanin, S., Tsuda, M., Tanaka, S., Tobiume, M., Asaka, M., and Ohba, Y. 2010. A novel FRET-based biosensor for the measurement of BCR-ABL activity and its response to drugs in living cells. Clin. Cancer Res., 16: 3964–3975.
- Mollet, S., Cougot, N., Wilczynska, A., Dautry, F., Kress, M., Bertrand, E., and Weil, D. 2008. Translationally repressed mRNA transiently cycles through stress granules during stress. Mol. Biol. Cell, 19: 4469–4479.
- Okabe, M., Matsushima, S., Morioka, M., Kobayashi, M., Abe, S., Sakurada, K., Kakinuma, M., and Miyazaki, T. 1987. Establishment and characterization of a cell line, TOM-1, derived from a patient with Philadelphia chromosome-positive acute lymphocytic leukemia. Blood, 69: 990–998.
- Okabe, M., Kunieda, Y., Itaya, T., Kurosawa, M., Sakurada, K., Miyazaki, T., Higa, T., Kasai, M., Kodama, S., Maekawa, I., et al. 1994. Establishment and characterization of a new Ph1 positive ALL cell line (ALL/MIK) presenting bcr gene rearrangement on bcr-2 and ALL-Type bcr/abl transcript: Suggestion of in vitro differentiation to monocytoid lineage. Leuk. Lymphoma, 12: 287–296.
- Pare, J.M., Tahbaz, N., López-Orozco, J., LaPointe, P., Lasko, P., and Hobman, T.C. 2009. Hsp90 regulates the function of Argonaute 2 and its recruitment to stress granules and P-bodies. Mol. Biol. Cell, 20: 3273–3284.
- Patel, H., Marley, S.B., Greener, L., and Gordon, M.Y. 2008. Subcellular distribution of p210BCR-ABL in CML cell lines and primary CD34+ CML cells. Leukemia, 22: 559–571.
- Pendergast, A.M., Muller, A.J., Havlik, M.H., Maru, Y., and Witte, O.N. 1991. BCR sequences essential for transformation by the BCR-ABL oncogene bind to the ABL SH2 regulatory domain in a non-phosphotyrosine-dependent manner. Cell, 66: 161–171.
- Podszywalow-Bartnicka, P., Wolczyk, M., Kusio-Kobialka, M., Wolanin, K., Skowronek, K., Nieborowska-Skorska, M., Dasgupta, Y., Skorski, T., and Piwocka, K. 2014. Downregulation of brca1 protein in bcr-abl1 leukemia cells depends on stress-triggered tiar-mediated suppression of translation. Cell Cycle, 13: 3727–3741.
- Ren, R. 2005. Mechanisms of BCR-ABL in the pathogenesis of chronic myelogenous leukaemia. Nat. Rev. Cancer, 5: 172–183.
- Sacchetti, A., Subramaniam, V., Jovin, T.M., and Alberti, S. 2002. Oligomerization of DsRed is required for the generation of a functional red fluorescent chromophore. FEBS Lett., 525: 13–19.
- Sattler, M., Mohi, M.G., Pride, Y.B., Quinnan, L.R., Malouf, N.A., Podar, K., Gesbert, F., Iwasaki, H., Li, S., Van Etten, R.A., et al. 2002. Critical role for Gab2 in transformation by BCR/ABL. Cancer Cell, 1: 479–492.
- Schwarz-Romond, T., Fiedler, M., Shibata, N., Butler, P.J.G., Kikuchi, A., Higuchi, Y., and Bienz, M. 2007. The DIX domain of Dishevelled confers Wnt signaling by dynamic polymerization. Nat. Struct. Mol. Biol., 14: 484–492.
- Skourides, P.A., Perera, S.A., and Ren, R. 1999. Polarized distribution of Bcr-Abl in migrating myeloid cells and co-localization of Bcr-Abl and its target proteins. Oncogene, 18: 1165–1176.
- Su, X., Ditlev, J.A., Hui, E., Xing, W., Banjade, S., Okrut, J., King, D.S., Taunton, J., Rosen, M.K., and Vale, R.D. 2016. Phase separation of signaling molecules promotes T cell receptor signal transduction. Science, 352: 595–599.
- Thomas, M.G., Martinez Tosar, L.J., Desbats, M.A., Leishman, C.C., and Boccaccio, G.L. 2009. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. J. Cell Sci., 122: 563–573.
- Wertheim, J.A., Perera, S.A., Hammer, D.A., Ren, R., Boettiger, D., and Pear, W.S. 2003. Localization of BCR-ABL to F-actin regulates cell adhesion but does not attenuate CML development. Blood, 102: 2220–2228.
- Zhang, X. and Ren, R. 1998. Bcr-abl efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: A novel model for chronic myelogenous leukemia. Blood, 92: 3829–3840.