Abstract
Glycosylphosphatidylinositol (GPI)-anchored proteins are post-transcriptionally modified with GPI and anchored to the plasma membrane. GPI is attached to nascent proteins in the endoplasmic reticulum by the GPI transamidase complex, which consists of PIGT, PIGK, GPAA1, PIGU, and PIGS. Of these, PIGK is a catalytic subunit that is unstable without PIGT. This study investigated the pathway by which unassembled PIGK not incorporated into the complex is degraded. We showed that unassembled PIGK was degraded via the proteasome-dependent pathway and that Hrd1 (also known as SYVN1), a ubiquitin ligase involved in the endoplasmic reticulum-associated degradation pathway, was responsible for degradation of unassembled PIGK.
Key words: Glycosylphosphatidylinositol, GPI transamidase complex, protein stability, transamidation, ERAD
Introduction
Glycosylphosphatidylinositol (GPI)-anchored proteins (GPI-APs) are post-translationally modified with GPI and anchored to the plasma membrane. GPI-anchoring regulates various cellular molecules including hydrolytic enzymes, adhesion molecules, receptors, protease inhibitors, and complement regulatory proteins. Therefore, defective GPI-APs can cause serious diseases, including inherited GPI deficiency and paroxysmal nocturnal hemoglobinuria (Kinoshita, 2020).
GPI is attached to nascent proteins in the endoplasmic reticulum (ER) by the GPI transamidase complex (GPI-TA). The GPI-TA consists of at least five subunits called PIGT, PIGK, GPAA1, PIGU, and PIGS. PIGK has a catalytic activity. It cleaves the C-terminal GPI signal of the precursor protein and forms an enzyme-substrate complex via a thioester bond (Ohishi et al., 2000). GPAA1 is predicted to form an amide linkage between the newly exposed C-terminal amino acid of the precursor protein and the phosphoethanolamine of GPI (Eisenhaber et al., 2014). GPAA1 and PIGU are suggested to be subunits that recognize GPI (Eisenhaber et al., 2018; Hong et al., 2003; Vainauskas and Menon, 2004). Although PIGS is essential for the GPI modification, its specific role is unknown. PIGT is required for complex assembly and protein stability of GPAA1 and PIGK in mammals (Ohishi et al., 2001). A disulfide bond forms between PIGT and PIGK in mammals (Ohishi et al., 2003) .
Many proteins assemble into complexes. The stoichiometry of the subunits is tightly regulated to suppress proteotoxic effects. (Juszkiewicz and Hegde, 2018) It is unknown how the stoichiometry of the GPI-TA subunits is regulated. We previously revealed that PIGK is destabilized in the absence of any other GPI-TA subunit in Drosophila (Kawaguchi et al., 2019). Similarly, efficient expression of exogenous PIGK requires co-transfection of PIGT in mammals (Ohishi et al., 2001). These findings suggest that the level of PIGK is tightly regulated and that unassembled PIGK not incorporated into the GPI-TA is eliminated. This study investigated the pathway by which unassembled PIGK is degraded in mammalian cells. We revealed that unassembled PIGK is degraded via the Hrd1-dependent ER-associated degradation (ERAD) pathway.
Materials and Methods
Cell culture and DNA transfection
HEK293 cells and variants were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. DNA was transfected using polyethylenimine transfection reagents (Polyscience, Warrington, PA) following the manufacturer’s instructions.
Plasmids
For CRISPR-based gene knockout (KO), BbsI-digested pX330 was ligated with a DNA fragment designed using the CRISPOR website (http://crispor.tefor.net/). The following oligonucleotides were used for gene KO: PIGK-gRNA1-F1, caccGCTCTTGTCCTTCGGCAGCG; PIGK-gRNA1-R1, aaacCGCTGCCGAAGGACAAGAGc; Doa10-gRNA1-F1, caccgTGTAGAGTGTGTCGGTCAGA; Doa10-gRNA1-R1, aaacTCTGACCGACACACTCTACAc; Doa10-gRNA2-F1, caccgATACTGCCAGTACATACACA; Doa10-gRNA2-R1, aaacTGTGTATGTACTGGCAGTATc; Hrd1-gRNA1-F1, caccGGCCAGGGCAATGTTCCGCA; Hrd1-gRNA1-R1, aaacTGCGGAACATTGCCCTGGCc; Hrd1-gRNA2-F1, caccgTGGTCAGGTACACCACAGTG; Hrd1-gRNA2-R1, aaacCACTGTGGTGTACCTGACCAc; gp78-gRNA1-F1, caccgCGTTAGCTGGTCCGGCTCGC; gp78-gRNA1-R1, aaacGCGAGCCGGACCAGCTAACGc; gp78-gRNA2-F1, caccgGCCTCGCGAACGGCGCCCAC; gp78-gRNA2-R1, aaacGTGGGCGCCGTTCGCGAGGCc; TRC8-gRNA1-F1, caccgGCACGATGCAGAACCGGCTT; TRC8-gRNA1-R1, aaacAAGCCGGTTCTGCATCGTGCc; TRC8-gRNA2-F1, caccgGAACAGGGTACCGTACGTCC; TRC8-gRNA2-R1, aaacGGACGTACGGTACCCTGTTCc; RHBDL4-gRNA1-F1, caccgCCCTCTTGATCTCCGTTGCA; RHBDL4-gRNA1-R1, aaacTGCAACGGAGATCAAGAGGGc; RHBDL4-gRNA2-F1, caccgAGCAATGGGAAAACGTACTC; and RHBDL4-gRNA2-R1, aaacGAGTACGTTTTCCCATTGCTc.
Wild-type and C329S mutant Hrd1 and PIGK cDNA fragments were integrated into the pIRES2-3xFLAG-ZsGreen1 and pIRES2-3xMyc-ZsGreen1 vectors, respectively, using an InFusion HD cloning kit (Takara Bio, Shiga, Japan). Hrd1 cDNA fragments were amplified from pcDNA6-Hrd1-WT/C329S-myc-His, which were kindly provided by Masayuki Kaneko (Hiroshima University, Japan). PIGK cDNA fragments were amplified from cDNA derived from SH-SY5Y cells.
Generation of KO cell lines
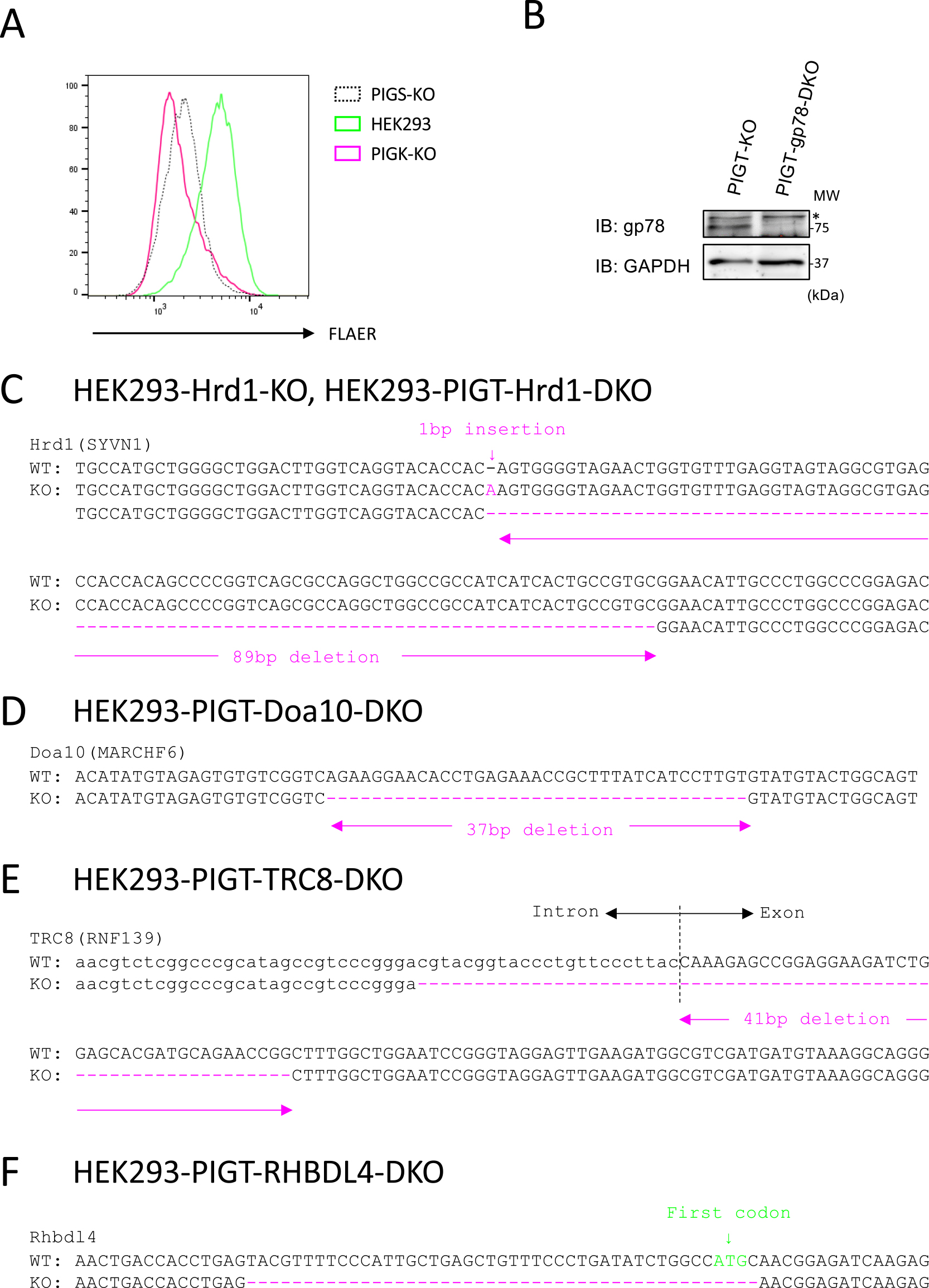
To generate gene KO cell lines, HEK293 and HEK293-CD16-PIGT-KO cells were transiently transfected with one or two pX330 plasmids bearing the sgRNA sequence targeting each gene, and clonal KO cells were obtained. KO of PIGK was validated by flow cytometry to confirm the loss of GPI-AP expression using the fluorescent-labeled inactive toxin aerolysin (FLAER)-Alexa 488 probe. KO of Hrd1, gp78, TRC8, and Doa10 was validated by PCR to confirm a large deletion of each gene. KO of gp78 was validated by immunoblotting to confirm the loss of gp78 protein.
Immunoblotting
Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, and 1 mM EDTA) supplemented with a protease inhibitor cocktail (Nacalai Tesque, Kyoto, Japan), and subjected to SDS-PAGE following standard procedures. Proteins were transferred to PVDF membranes (Merck Millipore, Burlington, MA), which were then blocked with phosphate-buffered saline (PBS) containing 2% skimmed milk and incubated overnight with a primary antibody. The primary antibodies used were an anti-PIGK antibody (1:2000; rabbit; ab201693; Abcam, Cambridge, MA), an anti-myc antibody (1:2000; rabbit; A-14; Santa Cruz Biotechnology, Dallas, TX), an anti-FLAG antibody (1:2000; mouse; 1E6; Wako Biochemicals, Osaka, Japan), an anti-gp78 antibody (1:2000; rabbit; #9590; Cell Signaling Technology, Beverly, MA), and an anti-GAPDH antibody (1:10000; rabbit; NB100-56875; Novus Biologicals, Littleton, CO). After three washes for 5 min with PBS containing 0.05% Tween 20, the primary antibody was detected with an HRP-conjugated anti-mouse IgG or anti-rabbit IgG antibody (1:10000; Jackson ImmunoResearch, West Grove, PA). Signals were visualized using Clarity Western ECL Substrate (Bio-Rad, Irvine, CA) and the ImageQuant LAS 4000 mini chemiluminescence detection system (GE Healthcare, Menlo Park, CA).
Flow cytometry
HEK293 cells stably expressing PIGK-myc were harvested with trypsin/EDTA and fixed with phosphate buffer containing 2% paraformaldehyde. Fixed cells were permeabilized with PBS containing 0.05% Triton X-100 and stained with an anti-myc antibody (1:2000; mouse; PL14; MBL, Nagoya, Japan) for 15 min on ice. HEK293 cells transiently expressing PIGK-myc were harvested with trypsin/EDTA and stained with an anti-CD59 antibody (1:200; mouse; H19; BD Biosciences, San Jose, CA). After incubation, cells were washed twice with FACS buffer (PBS containing 1% bovine serum albumin and 0.1% NaN3) and stained with APC-conjugated anti-mouse IgG for 15 min on ice. Finally, cells were washed twice with FACS buffer and analyzed with a FACSVerse flow cytometer (BD Biosciences). The resulting data were analyzed with FlowJo software (BD Biosciences).
Reverse-transcription quantitative PCR (RT-qPCR)
Total RNA was extracted from cells using an RNeasy Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Superscript III reverse transcriptase (Invitrogen; Thermo Fisher Scientific, Waltham, MA) and oligo(dT) primers were used for reverse transcription. Real-time PCR was performed using QuantStudio 12K Flex (Applied Biosystems; Thermo Fisher Scientific, Waltham, MA) and Power SYBR Green (Applied Biosystems). The amount of amplified transcript was calculated using the relative standard curve method and normalized against GAPDH as an internal control. The following primers were used: PIGK-qPCR-F1, CGTGGCCGCTAGTCATATCG; PIGK-qPCR-R1, CGGGATGTACACACCAGAACA; hGAPDH-qPCR-F1, TGCACCACCAACTGCTTAGC; and hGAPDH-qPCR-R1, GGCATGGACTGTGGTCATGAG.
Results
PIGT is required for PIGK expression
A previous study showed that expression of exogenous PIGK is inefficient and is enhanced by co-transfection of PIGT (Ohishi et al., 2001). We sought to confirm that expression of endogenous PIGK requires PIGT. We knocked out PIGK in HEK293 cells. To validate the establishment of PIGK-KO cells, we stained these cells with FLAER, which binds specifically to GPI-APs. The level of GPI-APs on the surface of PIGK-KO cells was decreased and was similar to that on the surface of PIGS-KO cells (Fig. 1A). Lysates of parental, PIGT-KO, and PIGK-KO cells were subjected to immunoblotting with an anti-PIGK antibody (Fig. 2A). The band that migrated at approximately 45 kDa in parental cells was absent in PIGK-KO cells, suggesting that it represents endogenous PIGK. KO of PIGT reduced expression of PIGK to a similar level as that in PIGK-KO cells. This result suggests that expression of endogenous PIGK requires PIGT.
Unassembled PIGK is degraded in a proteasome-dependent manner
Next, we investigated the pathway by which unassembled PIGK is degraded. We first established HEK293 cells stably expressing PIGK tagged with myc (HEK293-PIGK-myc). We hypothesized that excess PIGK is degraded via the proteasome- or lysosome-dependent pathway. To investigate this, we treated HEK293-PIGK-myc cells with MG132 and Bafilomycin, which inhibit proteasome activity and lysosome acidification, respectively. The cells were permeabilized, stained with an anti-myc antibody, and subjected to flow cytometry. Treatment with MG132, but not with Bafilomycin, increased the level of PIGK-myc (Fig. 2B, C). These results indicate that unassembled PIGK is degraded in a proteasome-dependent manner.
PIGK is degraded in a Hrd1-dependent manner in PIGT-KO cells
We next investigated which E3 ubiquitin ligase is responsible for degradation of PIGK dissociated from PIGT. The GPI-TA mediates GPI anchoring in the ER; therefore, we hypothesized that unassembled PIGK is degraded via the ERAD machinery. We focused on four well-characterized ubiquitin ligases that are part of the ERAD machinery, namely, Hrd1 (also known as SYVN1), gp78 (also known as AMFR), TRC8 (also known as RNF139), and Doa10 (also known as MARCHF6) (Leto et al., 2019; Stefanovic-Barrett et al., 2018). We knocked out these four genes in PIGT-KO cells (Fig. 1B–E). These cells were subjected to immunoblotting with an anti-PIGK antibody. KO of Hrd1, but not of gp78, TRC8, or Doa10, restored expression of PIGK in PIGT-KO cells (Fig. 3A).

A recent study revealed that the intramembrane protease RHBDL4 is required for ERAD-dependent degradation of the unassembled STT3A subunit of the oligosaccharyltransferase complex (Knopf et al., 2020). Therefore, we knocked out RHBDL4 in PIGT-KO cells (Fig. 1F). KO of RHBDL4 did not restore expression of PIGK in PIGT-KO cells (Fig. 3B).
To exclude the possibility that an off-target effect is responsible for the restoration of PIGK expression, we established PIGT-Hrd1-double knockout (DKO) cells stably expressing wild-type (WT) and inactive mutant (C329S) Hrd1. Expression of WT Hrd1, but not of C329S Hrd1, reduced expression of PIGK in PIGT-Hrd1-DKO cells (Fig. 3C). These results confirm that the ubiquitin-ligase activity of Hrd1 is required to reduce expression of PIGK in PIGT-KO cells.
To investigate whether KO of Hrd1 affects mRNA expression of PIGK, we quantified PIGK mRNA expression in parental, Hrd1-KO, PIGT-KO, and PIGT-Hrd1-DKO cells (Fig. 3D). mRNA expression of PIGK was comparable in parental, Hrd1-KO, and PIGT-Hrd1-DKO cells. PIGT-KO cells expressed a slightly higher level of PIGK mRNA. These results suggest that KO of Hrd1 restored expression of PIGK in PIGT-KO cells at the protein level.
To investigate whether Hrd1 regulates the expression level of PIGK in cells expressing PIGT, lysates of parental and Hrd1-KO cells were subjected to immunoblotting with an anti-PIGK antibody (Fig. 3E). Parental and Hrd1-KO cells expressed a similar level of PIGK. This result indicates that Hrd1 does not regulate the protein level of PIGK in cells expressing PIGT.
Discussion
This study revealed that excess PIGK was degraded in a proteasome-dependent manner (Fig. 2B). In PIGT-KO cells, endogenous PIGK was degraded in a Hrd1-dependent manner (Fig. 3A, C). KO of Hrd1 did not increase the mRNA level of PIGK (Fig. 3D). Moreover, KO of Hrd1 did not increase the level of PIGK in cells expressing PIGT (Fig. 3E). Taken together, we conclude that the Hrd1-dependent ERAD pathway is required for degradation of unassembled PIGK, but not of PIGK incorporated into the GPI-TA.
Hrd1 forms a complex by interacting with accessory factors. Specifically, Hrd1 interacts with OS-9 and XTP3-B via SEL1L. OS-9 and XTP3-B recognize the mannose-trimmed N-glycan of misfolded proteins and recruit these proteins to the Hrd1-SEL1L complex for degradation (Hosokawa et al., 2009, 2008). PIGK does not harbor the N-X-S/T consensus sequence that is required for N-glycosylation. Therefore, Hrd1 may recognize unassembled PIGK in an OS-9/XTP3-B-independent manner.
T cell receptor alpha (Tα) is a well-characterized ERAD substrate. Tα harbors a single transmembrane domain containing a charged residue that functions as a degradation signal. Normally, the transmembrane domain is masked by a binding partner. However, the transmembrane domain of unassembled Tα is exposed and functions as a degradation signal for the ERAD pathway (Bonifacino et al., 1990). Therefore, the exposed transmembrane domain of PIGK may function as a degradation signal for the Hrd1-dependent ERAD pathway.
KO of Hrd1 did not completely restore the protein level of PIGK in PIGT-KO cells (Fig. 3A). Therefore, we cannot exclude the possibility that a factor other than Hrd1 is also involved in degradation of unassembled PIGK. A recent study revealed that the Asi1/Asi2/Asi3 E3 ligase complex is required for degradation of unassembled Gpi8 (ortholog of PIGK) and Gpi16 (ortholog of PIGT), whereas the contribution of Hrd1 and Doa10 is minor in yeast(Natarajan et al., 2020). Mammalian homologs of Asi1, Asi2, and Asi3 have not been identified. The functional homologs of these proteins may be involved in degradation of unassembled PIGK.
A recent study showed that the intramembrane protease RHBDL4 cleaves the unassembled STT3A subunit of the oligosaccharyltransferase complex. This cleavage is required for ERAD-dependent degradation of unassembled STT3A (Knopf et al., 2020). We showed that RHBDL4 was not required for degradation of unassembled PIGK (Fig. 3B). A further study is required to investigate the involvement of a factor other than Hrd1 in degradation of unassembled PIGK.
The amount of PIG-K was not increased in Hrd1-KO cells (Fig. 3E), indicating that PIG-K is not unassembled in normally growing cells and that the Hrd1-ERAD system does not contribute to the elimination of unassembled PIG-K from cells in such situations. Aneuploidy is a hallmark of cancer cells (Kolodner et al., Science, 2011). An increased number of PIGK loci may require the elimination of unassembled PIGK. The Hrd1-ERAD system may degrade unassembled PIGK to relieve ER stress in cancer cells. If ERAD does not degrade unassembled PIG-K in such a situation, the exposed transmembrane domain may harm other proteins, resulting in cell death because of ER stress.
Unbalanced stoichiometry of the GPI-TA is believed to be involved in the onset and progression of cancer. PIGT, PIGU, GPAA1, and PIGS are overexpressed in many cancers, whereas PIGK is downregulated (Nagpal et al., 2008). It is unknown how the stoichiometry of the GPI-TA is regulated. This study showed that the ERAD machinery is involved in regulation of this stoichiometry. The ERAD machinery may prevent the onset and progression of cancer by regulating the stoichiometry of the GPI-TA.
Acknowledgments
This study was supported by grants-in-aid from the Ministry of Education, Culture, Sports, Science and Technology in Japan (KAKENHI grants JP17H0641301, JP17H06421, 19K06445 to S.G., JP20K06629 to M.Y.-H., and 19K16132 to K.K.).
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Bonifacino, J.S., Cosson, P., and Klausner, R.D. 1990. Colocalized transmembrane determinants for ER degradation and subunit assembly explain the intracellular fate of TCR chains. Cell, 63: 503–513.
- Eisenhaber, B., Eisenhaber, S., Kwang, T.Y., Gruber, G., and Eisenhaber, F. 2014. Transamidase subunit GAA1/GPAA1 is a M28 family metallo-peptide-synthetase that catalyzes the peptide bond formation between the substrate protein’s omega-site and the GPI lipid anchor’s phosphoethanolamine. Cell Cycle, 13: 1912–1917.
- Eisenhaber, B., Sinha, S., Wong, W.-C., and Eisenhaber, F. 2018. Function of a membrane-embedded domain evolutionarily multiplied in the GPI lipid anchor pathway proteins PIG-B, PIG-M, PIG-U, PIG-W, PIG-V, and PIG-Z. Cell Cycle, 17: 874–880.
- Hong, Y., Ohishi, K., Kang, J.Y., Tanaka, S., Inoue, N., Nishimura, J., Maeda, Y., and Kinoshita, T. 2003. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Mol. Biol. Cell, 14: 1780–1789.
- Hosokawa, N., Wada, I., Nagasawa, K., Moriyama, T., Okawa, K., and Nagata, K. 2008. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J. Biol. Chem., 283: 20914–20924.
- Hosokawa, N., Kamiya, Y., Kamiya, D., Kato, K., and Nagata, K. 2009. Human OS-9, a lectin required for glycoprotein endoplasmic reticulum-associated degradation, recognizes mannose-trimmed N-glycans. J. Biol. Chem., 284: 17061–17068.
- Juszkiewicz, S. and Hegde, R.S. 2018. Quality Control of Orphaned Proteins. Molecular Cell, 71: 443–457.
- Kawaguchi, K., Sato, T., Kondo, S., Yamamoto-Hino, M., and Goto, S. 2019. Stability of the transamidase complex catalyzing GPI anchoring of proteins. Biochem. Biophys. Res. Commun., 512: 584–590.
- Kinoshita, T. 2020. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol., 10: 190290.
- Knopf, J.D., Landscheidt, N., Pegg, C.L., Schulz, B.L., Kühnle, N., Chao, C.-W., Huck, S., and Lemberg, M.K. 2020. Intramembrane protease RHBDL4 cleaves oligosaccharyltransferase subunits to target them for ER-associated degradation. J. Cell Sci., 133: jcs243790.
- Kolodner, R.D., Cleveland, D.W., and Putnam, C.D. 2011. Cancer. Aneuploidy drives a mutator phenotype in cancer. Science, 333: 942–943.
- Leto, D.E., Morgens, D.W., Zhang, L., Walczak, C.P., Elias, J.E., Bassik, M.C., and Kopito, R.R. 2019. Genome-wide CRISPR Analysis Identifies Substrate-Specific Conjugation Modules in ER-Associated Degradation. Mol. Cell, 73: 377–389.e311.
- Nagpal, J.K., Dasgupta, S., Jadallah, S., Chae, Y.K., Ratovitski, E.A., Toubaji, A., Netto, G.J., Eagle, T., Nissan, A., Sidransky, D., and Trink, B. 2008. Profiling the expression pattern of GPI transamidase complex subunits in human cancer. Mod. Pathol., 21: 979–991.
- Natarajan, N., Foresti, O., Wendrich, K., Stein, A., and Carvalho, P. 2020. Quality Control of Protein Complex Assembly by a Transmembrane Recognition Factor. Mol. Cell, 77: 108–119.e109.
- Ohishi, K., Inoue, N., Maeda, Y., Takeda, J., Riezman, H., and Kinoshita, T. 2000. Gaa1p and gpi8p are components of a glycosylphosphatidylinositol (GPI) transamidase that mediates attachment of GPI to proteins. Mol. Biol. Cell, 11: 1523–1533.
- Ohishi, K., Inoue, N., and Kinoshita, T. 2001. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. EMBO J., 20: 4088–4098.
- Ohishi, K., Nagamune, K., Maeda, Y., and Kinoshita, T. 2003. Two subunits of glycosylphosphatidylinositol transamidase, GPI8 and PIG-T, form a functionally important intermolecular disulfide bridge. J. Biol. Chem., 278: 13959–13967.
- Stefanovic-Barrett, S., Dickson, A.S., Burr, S.P., Williamson, J.C., Lobb, I.T., van den Boomen, D.J., Lehner, P.J., and Nathan, J.A. 2018. MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep., 19: e45603.
- Vainauskas, S. and Menon, A.K. 2004. A conserved proline in the last transmembrane segment of Gaa1 is required for glycosylphosphatidylinositol (GPI) recognition by GPI transamidase. J. Biol. Chem., 279: 6540–6545.
Abbreviations
DKOdouble knockout
ERendoplasmic reticulum
ERADendoplasmic reticulum-associated degradation
FLAERfluorescent-labeled inactive toxin aerolysin
GPIglycosylphosphatidylinositol
GPI-APglycosylphosphatidylinositol-anchored protein
GPI-TAglycosylphosphatidylinositol transamidase complex
KOknockout
PBSphosphate-buffered saline
RT-qPCRreverse-transcription quantitative PCR
TαT cell receptor alpha
WTwild-type