Abstract
IFN-γ secreted from immune cells exerts pleiotropic effects on tumor cells, including induction of immune checkpoint and antigen presentation, growth inhibition, and apoptosis induction. We combined a dual promoter system with an IFN-γ signaling responsive promoter to generate a reporter named the interferon sensing probe (ISP), which quantitates the response to IFN-γ by means of fluorescence and bioluminescence. The integration site effect of the transgene is compensated for by the PGK promoter-driven expression of a fluorescent protein. Among five potential IFN-γ-responsive elements, we found that the interferon γ-activated sequence (GAS) exhibited the best performance. When ISP-GAS was introduced into four cell lines and subjected to IFN-γ stimulation, dose-dependency was observed with an EC50 ranging from 0.2 to 0.9 ng/mL, indicating that ISP-GAS can be generally used as a sensitive biosensor of IFN-γ response. In a syngeneic transplantation model, the ISP-GAS-expressing cancer cells exhibited bioluminescence and fluorescence signals in an IFN-γ receptor-dependent manner. Thus, ISP-GAS could be used to quantitatively monitor the IFN-γ response both in vitro and in vivo.
Key words: in vivo imaging, tumor microenvironment, interferon-gamma, dual promoter system
Introduction
IFN-γ is a cytokine secreted by immune cells and has pleiotropic effects on anti-tumor immune response (Dighe et al., 1994; Kaplan et al., 1998). IFN-γ suppresses tumor growth by inducing cell death or arresting the cell cycle (Bromberg et al., 1996; Chin et al., 1997; Chin et al., 1996; Fulda and Debatin, 2002). Besides IFN-γ increases MHC class I-dependent antigen presentation, and, thereby, susceptibility to CD8+ T cell-mediated killing (Meunier et al., 2005). Conversely, IFN-γ has been reported to be involved in tumor immune escape (Beatty and Paterson, 2000; Taniguchi et al., 1987). IFN-γ induces ligands such as PD-L1 and PD-L2 on cancer cells and contributes to their escape from T cells infiltrating the tumor (Dong et al., 2002; Freeman et al., 2000; Garcia-Diaz et al., 2017). IFN-γ also increases the instability of the tumor cell genome (Takeda et al., 2017), contributing to the ability of cancer cells to evade the immune system.
It is crucial to analyze the spatiotemporal spread of IFN-γ in the tumor microenvironment to understand the pleiotropic function of IFN-γ in tumors. Visualization of the IFN-γ production in vivo is helpful to understand its temporal changes and tissue specificity (Reynolds et al., 2019). For this purpose, JAK-STAT signaling pathway can be a versatile indicator to visualize the response of cancer cells to IFN-γ. Nuclear translocation of STAT1 can be used as a surrogate marker for the initial response to IFN-γ stimulation (Samsonov et al., 2013; Sanderson et al., 2012; Thibaut et al., 2020). When monitoring the expression of downstream transcription factors induced by STAT1, the bioluminescence system is also helpful for sensitive detection, and fluorescence can be used to monitor transcriptional activity at the cellular level (Hoekstra et al., 2020). However, pre-existing biosensors are subject to the integration site effect or the copy number of the transgene; therefore, it would be necessary to standardize the fluorescence/bioluminescence signal to quantitatively compare the IFN-γ activity.
In this study, we combined a dual promoter system and an IFN-γ signal-responsive elements to create a probe that could compensate for the integration site effect and the copy number of the biosensor gene. Using this probe, we visualized the IFN-γ signaling in transplanted cancer in vivo.
Material and Methods
Plasmids
To generate the ISP (IFN-γ sensing probe) constructs, cDNA encoding mCherry-Akaluc (Iwano et al., 2018) was inserted into the pPB piggyBac transposon vector (Yusa et al., 2009), and the 3-phosphoglycerate kinase (PGK) promoter-driven Turquoise-GL-NLS gene cassette was inserted in the opposite direction with a stuffer sequence upstream of it (Adra et al., 1987). Then, annealed oligoDNA containing a minimal promoter (MinP) and either four tandem repeats of the interferon γ-activated sequence (GAS) element (5'-AGTTTCATATTACTCTAAATC-3') (Hoekstra et al., 2020), five tandem repeats of the interferon-stimulated response element (ISRE) (5'-TAGTTTCACTTTCCC-3') (Weihua et al., 1997), five tandem repeats of the IRF1 elements (5'-TTTCCCCGAAA-3') (Pellegrini and Schindler, 1993), five tandem repeats of the Ly-6E elements (5'-ATTCCTGTAAG-3') (Pellegrini and Schindler, 1993) or five tandem repeats of the MIG elements (5'-CTTACTATAAA-3') (Pellegrini and Schindler, 1993) was cloned in front of the coding sequences of mCherry-Akaluc (Fig. S1). pCMV-mPBase was provided by Kosuke Yusa (Kyoto University, Kyoto, Japan).
Tumor cell lines and culture conditions
The B16F10 melanoma cell line was purchased from the Cell Resource Center for Biomedical Research, Institute of Development, Aging and Cancer, Tohoku University (Sendai, Japan). The Panc02 mouse pancreatic ductal adenocarcinoma cell line was obtained from the National Institutes of Health (Bethesda, MD). The MC-38 mouse colon adenocarcinoma cell line was provided by Takeshi Setoyama and Tsutomu Chiba (Kyoto University). The BrafV600E melanoma cell line (BrafV600E melanoma) was provided by Caetano Reis e Sousa (The Francis Crick Institute, London, UK). Panc02 and MC-38 cells were maintained in DMEM high glucose (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). B16F10 and BrafV600E melanoma cells were cultured in RPMI 1640 (Thermo Fisher Scientific, Waltham, MA). All culture media were supplemented with 10% heat-inactivated FBS (SIGMA, St. Louis, MO), 100 U/mL penicillin, and 100 μg/mL streptomycin (penicillin-streptomycin mixed solution; Nacalai Tesque, Kyoto, Japan). Cell cultures were maintained at 37°C with 5% CO2.
Stable cell lines
PiggyBac transposon plasmids encoding the ISP and pCMV-mPBase were cotransfected into Panc02 and MC-38 cells by using Lipofectamine 3000 (Thermo Fisher Scientific), and into B16F10 and BrafV600E melanoma cells by using an Amaxa nucleofector system (Lonza, Basel, Switzerland). In Amaxa nucleofector electroporation, 5×105 cells were pelleted and resuspended in 100 μl of homemade buffer (4 mM KCl, 10 mM MgCl2, 107 mM NaH2PO4, 13 mM Na2HPO4, 11 mM HEPES pH 7.75) with 5 μg of DNA, and then electroporated with program P-020 for B16F10 and T-023 for BrafV600E melanoma. Two days after transfection, the cells were selected with 10 μg/mL blasticidin S (Invitrogen, San Diego, CA) for at least 1 week. For the cell lines for injection into mice, the fluorescence intensity of Turquoise-GL was normalized by cell sorting.
Knockout cell lines
The Gnaq–/– BrafV600E melanoma was developed according to a previously reported method with modification (Konishi et al., 2021). The CRISPR/Cas9 system was used to disrupt the expression of the Ifngr1 genes. The targeted gRNA oligos (5'-CTGATGCTGTCTGCGAAGGT-3') were introduced into the pX459 vector (Addgene plasmid #48139; Cambridge, MA). Then, 5 μg of pX459 plasmid was transfected into 5×105 cells using an Amaxa nucleofector system. Cells were subjected to single cell dilution cloning and examined for knockout by nucleotide sequencing (Fig. S2).
In vitro characterization of reporter cells
Cells expressing ISPs were trypsinized and suspended in PBS containing 3% FBS, then filtrated through a 40 μm nylon cell strainer (Corning, Corning, NY) and analyzed with a FACS Aria IIIu cell sorter (Becton Dickinson, Franklin Lakes, NJ). The following combinations of lasers and emission filters were used for the detection of fluorescence: Turquoise-GL, a 407 nm laser and an ET470/24m filter (Chroma Technology Corporation, Bellows Falls, VT); mCherry, a 561 nm laser and a DF610/20 filter (Omega Optical, Brattleboro, VT). Cells were gated for size and granularity to exclude cell debris and aggregates. Data were analyzed using FlowJo software (Tree Star, Ashland, OR). The fluorescence intensity ratio of mCherry/Turquoise-GL was used to represent the promoter activity. To quantify the response to IFN-γ, the mean mCherry/Turquoise-GL value in the absence of IFN-γ was used for the normalization. To compensate for the actual expression level of the fluorescent protein and the acquired fluorescence intensity, the sensitivity of the detector was adjusted so that the average fluorescence intensity of mCherry and Turquoise-GL was about 100 times higher than that in cells not expressing the fluorescent protein.
Cell proliferation assay
Cell proliferation was assessed by counting the number of Turquoise-GL-labeled nuclei by time-lapse imaging with widefield microscopy. Cells cultured on glass-base 96-well plates were observed under an IX83 inverted microscope (Olympus, Tokyo, Japan) equipped with a UPlanSApo 40x/0.95 objective lens (Olympus), a PRIME scientific CMOS camera (Photometrics, Tucson, AZ), a Spectra-X light engine (Lumencor, Beaverton, OR), an IX2-ZDC laser-based autofocusing system (Olympus), and an incubation chamber (Tokai Hit, Fujinomiya, Japan). The filters and dichroic mirrors used for time-lapse imaging were as follows: 430/24 (Olympus) excitation filters, XF2034 (455DRLP) (Omega Optical) dichroic mirrors, and FF01-483/32 (Semrock, Rochester, NY) emission filters. Image acquisition and analysis were carried out with MetaMorph (Molecular Devices Japan, Tokyo, Japan) and ImageJ (National Institute of Health), respectively.
Mice
B6 albino mice were obtained from Charles River Laboratories Japan (Yokohama, Japan). Mice were housed in a specific pathogen-free facility and received a routine chow diet and water ad libitum. The animal protocols were reviewed and approved by the Animal Care and Use Committee of Kyoto University Graduate School of Medicine (MedKyo20081).
Tumor cell injections
Cells were harvested by trypsinization, washed three times with PBS, and injected subcutaneously into the flank of recipient mice at 2×105 cells in 100 μl of 50% Matrigel (Corning) in PBS. Tumor size was quantified by multiplying the longest diameter, its perpendicular, and its thickness.
Processing of tumor tissue and flow cytometry
Tumors were dissected into pieces and digested with Collagenase Type IV (200 U/mL) (Worthington Biochemical Corporation, Lakewood, NJ) and DNase I (10 U/mL) (Roche Diagnostics, Indianapolis, IN) for 30 minutes at 37°C. After washing of cells with PBS, the fluorescence intensity of individual cells was determined with a FACS Aria IIIu.
Bioluminescence imaging
Mice bearing tumors were anesthetized with 2% isoflurane (FUJIFILM Wako Pure Chemical Corporation) inhalation (the O2 to air gas ratio was over 95%) and placed on a custom-made heating plate in the supine position. Bioluminescent images were acquired using a MIIS system (Molecular Devices Japan) equipped with an iXon Ultra EMCCD camera (Oxford Instruments, Belfast, UK) and a lens (MDJ-G25F095, φ16 mm, F: 0.95; Tokyo Parts Center, Saitama, Japan). Akalumine-HCl, also called TokeOni, was obtained from Kurogane Kasei (Nagoya, Japan) and used as the substrate of Akaluc. Five minutes after the start of image acquisition, 100 μL of 5 mM AkaLumine-HCl was administered (i.p.). Images were acquired under the following conditions: binning, 4; EM gain, 0. Acquisition of bioluminescent images was repeated every 30 seconds, and the maximum bioluminescent intensity during the imaging was adopted in each mouse. Image acquisition and analysis were carried out with MetaMorph and ImageJ, respectively.
Intravital imaging by two-photon excitation microscopy
Intravital imaging was performed as previously described with some modifications (Konishi et al., 2021; Yamauchi et al., 2016). In brief, mice were anesthetized with 2% isoflurane inhalation (the O2 to air gas ratio was 80:20) and placed in the prone position on an electric heating pad. The body temperature was maintained at 36.5°C. The skin flap was then placed on a coverglass.
Mice were observed with an FV1000MPE-IX-83 inverted microscope (Olympus) equipped with a UPLSAPO 30XS/1.05 numerical aperture (NA) silicon-immersion objective lens (Olympus) and an InSight DeepSee Ultrafast laser (Spectra Physics, Mountain View, CA). The excitation wavelength for Turquoise-GL was 840 nm, and that for mCherry was 1040 nm. Fluorescent images were acquired with two different detector channels using the following filters and mirrors: an infrared (IR)-cut filter, BA685RIF-3 (Olympus), two dichroic mirrors, DM505 and DM570 (Olympus), and three emission filters, FF01-425/30 (Semrock) for the second harmonic generation channel (SHG), BA460-500 (Olympus) for Turquoise-GL, and BA575-630 (Olympus) for mCherry. The microscope was equipped with a two-channel GaAsP detector unit and two multialkali detectors. FLUOVIEW software version 4.1a (Olympus) was used to control the microscope and to acquire images, which were saved in the multilayer 16-bit tagged image file format.
For the measurement of ISP-GAS activity in vivo, regions of interest (ROIs) were set at nuclei in a single plane by Turquoise-GL signal with Cellpose (Stringer et al., 2021). The integrated mCherry signal in the ROI was divided by the integrated Turquoise-GL intensity in the ROI and shown as ISP-GAS activity.
Statistical analysis
The cell EC50 was calculated by undertaking a two-parameter logistic curve fitting of the normalized dose-response curve using Python (version 3.7). All statistical analyses were performed using Python. No statistical analysis was used to predetermine the sample size. Welch’s t-test was used to evaluate statistically significant differences. The p-values less than 0.05 were considered statistically significant.
For all the box plots, the edges on the box plots indicate the first and third quartiles, with the line in the middle being the mean. The whiskers on the box plots extend another 1.5 x the interquartile range from the edges of the boxes, respectively.
Results and Discussion
Generation of the IFN-γ sensing probe, ISP
To monitor the response to IFN-γ in tumor cells, the probe must satisfy two requirements. First, the cDNA of the probe must be stably expressed in tumor cells. Second, the integration site effect on the IFN-γ-dependent transcriptional activity must be compensated. For this, we generated a dual promoter plasmid named ISP. This reporter transcribes an mCherry-Akaluc fusion protein under an IFN-γ signal-responsive promoter (Fig. 1A). In addition, a Turquoise-GL protein with a nuclear localization signal (NLS) is expressed under the ubiquitous promoter, PGK, to compensate for the locus effect. With this, the promoter activity can be measured by the mCherry/Turquoise-GL fluorescence intensity ratio. We screened five promoter elements that may respond to IFN-γ in B16F10 melanoma cells. Among them, GAS (interferon γ-activated sequence) and ISRE (interferon-stimulated response element) responded to the INF-γ stimulation (Fig. 1B). As anticipated, cells expressing ISP-GAS or ISP-ISRE showed linear correlation between the mCherry and Turquoise-GL intensities (Fig. 1C), suggesting effective compensation of the integration site effect in the population. We next validated the specificity of the ISP-GAS and ISP-ISRE by using IFNGR1, the IFN-γ receptor, knockout cell line (Fig. 1D). Collectively, the ISPs can monitor the IFN-γ response with little or no integration site effect.

To validate the versatility of ISP, we expressed ISP in four C57BL/6-derived cell lines, B16F10 melanoma cells, Panc02 pancreatic adenocarcinoma cells, BrafV600E melanoma cells, and MC-38 colon adenocarcinoma cells, and examined the dose response to IFN-γ (Fig. 2A). In all cell lines, clear dose-response curves were obtained by both ISP-GAS and ISP-ISRE. The EC50 values were between 0.2 and 0.9 ng/mL in ISP-GAS-expressing cell lines and between 0.1 and 0.3 ng/mL in ISP-ISRE-expressing cell lines (Fig. 2B). The fold increase of the mCherry/Turquoise-GL fluorescence intensity ratio was always smaller in the cell line harboring ISP-GAS than in that harboring ISP-ISRE (Fig. 2C), demonstrating that the sensitivity of ISP-ISRE, specifically in vitro situation, is higher than that of ISP-GAS.

IFN-γ directly inhibits tumor cell growth and promotes tumor cell apoptosis (Trubiani et al., 1994) via the JAK-STAT pathway (Chin et al., 1997; Detjen et al., 2001). We asked whether the response detected by ISP correlates with the biological susceptibility to IFN-γ. We found that B16F10 and Panc02, but not BrafV600E or MC-38, showed growth retardation after 10 hours of IFN-γ treatment (Fig. 2D), as reported previously (Gerber et al., 2013; Mazzolini et al., 2003; Zoller, 1988). What was responsible for this difference? We found that the mCherry/Turquoise-GL fluorescence intensity ratios of ISP-GAS and ISP-ISRE were higher in IFN-γ-stimulated B16F10 and Panc02 than in BrafV600E or MC-38 cells (Fig. 2E), when the Turquoise-GL signals were similar among cell lines (Fig. 2F). These results demonstrate that the absolute transcriptional activity of B16F10 and Panc02 with IFN-γ stimulation is higher than those of BrafV600E or MC-38 cells. The difference of the susceptibility to IFN-γ, which is regulated by intracellular signaling pathway, may explain the difference in this growth retardation. Although the difference was marginal, the response detected by ISP-GAS may have been suitable for the prediction of the biological response.
ISP-GAS, an IFN-γ-specific reporter in vivo
We next examined whether ISP-GAS could be used to monitor the IFN-γ response in vivo. In ISPs, mCherry is fused to the ultrasensitive bioluminescent protein Akaluc (Fig. 1A). Therefore, GAS-dependent transcription was monitored by Akaluc bioluminescence in B6 albino mice implanted with ISP-GAS-expressing B16F10 cells (Fig. 3A). As a control, Ifngr1–/– B16F10 cells were also challenged. Seven days after inoculation, the bioluminescence signal was higher in the parental B16F10 cells than in the Ifngr1–/– cells (Fig. 3B, left panel). Meanwhile, the difference was not reproducible when ISP-ISRE was used (Fig. 3B, right panel). The difference in the tumor size was not significant between the parent and Ifngr1–/– cells (Fig. 3C). Thus, we concluded that ISP-GAS specifically monitors the IFN-γ signaling pathway in vivo.
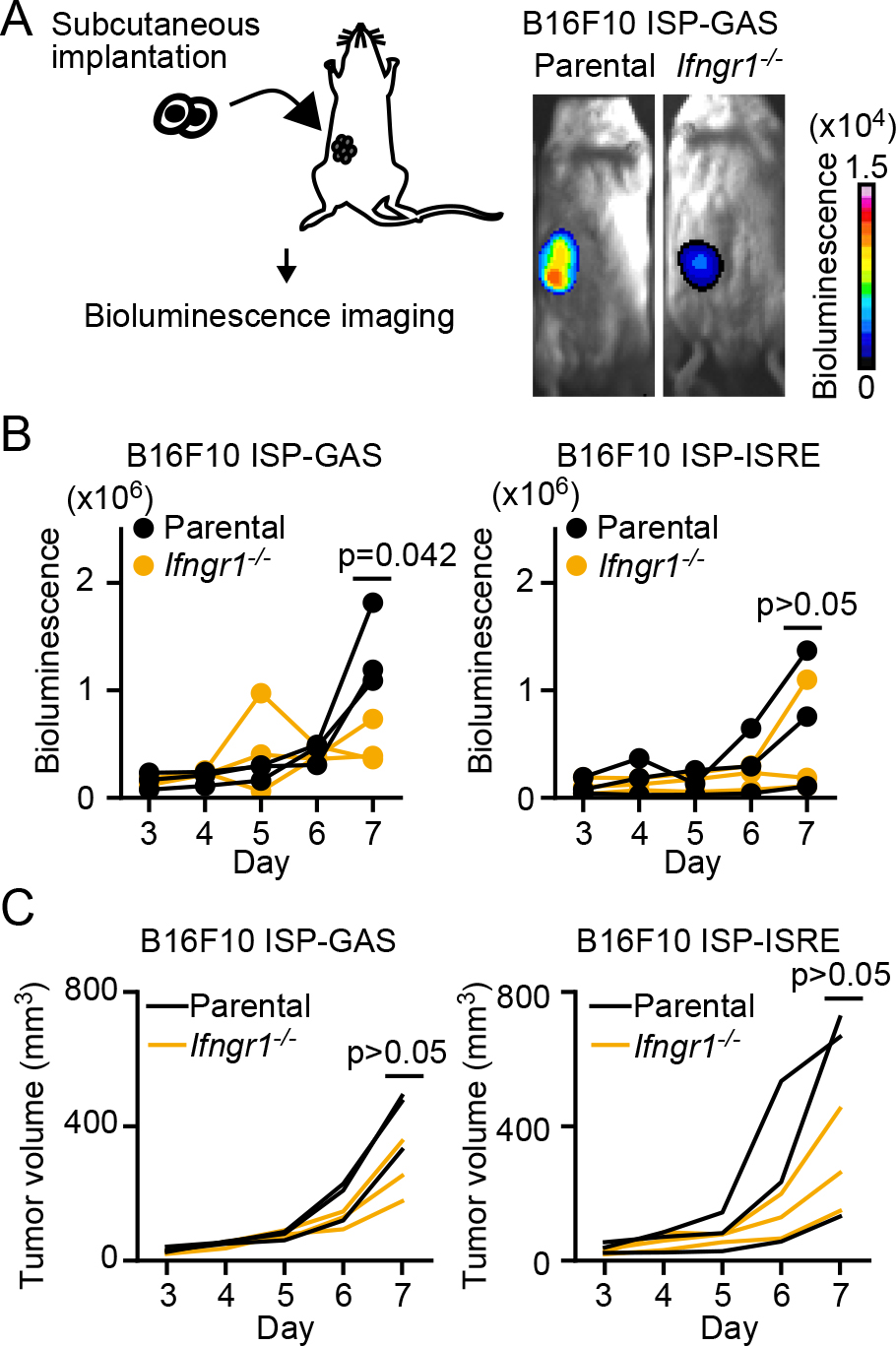
The reason that we failed to detect a significant difference in the ISP-ISRE response between the parent and Ifngr1–/– cells may be ascribable to the previous observation that ISRE is also activated by type-I IFNs including IFN-α, IFN-β, and IFN-ω (Khiar et al., 2017; Tanaka et al., 1993; Uccellini and Garcia-Sastre, 2018). If so, ISP-ISRE could be used as a pan-IFN reporter including the type-II IFN, IFN-γ, after further characterization.
Enhanced IFN-γ signaling in Gnaq–/– tumor cells in vivo
We recently reported that BrafV600E melanoma cells maintain an immunocompromised microenvironment by means of thromboxane A2-mediated activation of the GqPCR, Gq protein-coupled receptors, pathway (Konishi et al., 2021). Activated GqPCR triggers the secretion of prostaglandin E2 (PGE2) from tumor cells, which, in turn, suppress the recruitment of NK cells, a principal producer of IFN-γ in the tumor microenvironment (Zelenay et al., 2015). We therefore examined the effect of the suppressed GqPCR signaling pathway on the IFN-γ response by the knockout of Gnaq, the Gq protein alpha subunit. In the parental BrafV600E melanoma cells, the bioluminescence signal of ISP-GAS was higher than in the Ifngr1–/– cells, although the difference was not statistically significant (Fig. 4A, left). On the other hand, Gnaq–/– cells exhibited a markedly higher bioluminescence signal of ISP-GAS than Gnaq–/– Ifngr1–/– double-knockout cells (Fig. 4A, right), supporting the notion that IFN-γ is abundant in the tumor microenvironment. However, the difference in tumor volume would affect the bioluminescence signal (Fig. 4B), which motivated us to analyze the IFN-γ signaling at single-cell resolution.

Against our expectation, we did not observe a significant difference in bioluminescence between the parental and Gnaq–/– cells. Thus, we next analyzed tumor cells at single-cell resolution using two-photon microscopy (Fig. 4C, D) and flow cytometry (Fig. 4E). As anticipated, the GAS activity, mCherry/Turquoise-GL ratio, in Gnaq–/– cells was significantly higher than that in parental cells, supporting the abundance of IFN-γ in Gnaq–/– cells.
Why did the Ifngr1–/– cells show higher GAS activities than Gnaq–/– Ifngr1–/– double-knockout cells in flow cytometry analysis? We suppose that JAK-STAT pathway could be also activated via other cytokines rather than IFN-γ (O’Shea and Plenge, 2012; Salas et al., 2020), though it is not dominant for ISP-GAS. Since we have demonstrated that the removal of Gq protein transforms the microenvironment into tumor-killing immune microenvironment (Konishi et al., 2021), the microenvironment would be abundant in cytokines such as IL-12b and IL-10 (Zelenay et al., 2015).
What is the advantage of ISP-GAS in comparison to the preceding probes of the IFN-γ signaling pathway (Koster and Hauser, 1999; Reynolds et al., 2019; Samsonov et al., 2013; Sanderson et al., 2012; Stifter et al., 2019; Taniguchi et al., 1987; Thibaut et al., 2020; Uccellini and Garcia-Sastre, 2018)? First, because of the constitutive Turquoise-GL expression, the dual promoter system of ISP-GAS allows us to validate the IFN-γ signaling at single-cell resolution. The intensity of bioluminescence alone could be affected by the number of cells in the tumor mass, the depth of the tumor below the body surface, and the technical reproducibility of the substrate injection (Baba et al., 2007). These drawbacks would be minimized by using ISP-GAS with FACS analysis and intravital imaging, as shown in Fig. 4D and 4E. Second, we can compare the IFN-γ signaling in a variety of cell lines in vivo. In tumor microenvironments, several epigenetic modifiers have been reported, including IFNs (Ivashkiv, 2018; Ivashkiv and Donlin, 2014), which would alter the expression of luciferase and/or Turquoise-GL-NLS. Indeed, in our case, the intensity of Turquoise-GL-NLS was decreased in Gnaq–/– Ifngr1–/– double-knockout BrafV600E cells expressing ISP-GAS at 7 days after tumor implantation, while the mCherry/Turquoise-GL ratio was still low (Fig. 4D). Our dual promoter system successfully compensated for the epigenetic status and integration site effect. Third, ISP-GAS facilitates monitoring of the IFN-γ signaling in living mice. Fluorescent protein-tagged Stat1 is also widely used for detecting the IFN-γ signaling, because of its fast kinetics (Thibaut et al., 2020). However, it would be time-consuming to identify the appropriate spatio-temporal window under microscopy. A hybrid combining the advantages of bioluminescence and fluorescence will help to identify them.
Collectively, our results demonstrated that ISP-GAS allows us to monitor the activity of the IFN-γ signaling pathway at single-cell resolution and thereby monitor immune activity in the tumor microenvironment.
Acknowledgments
We are grateful to the members of the Matsuda Laboratory for their helpful input; to K. Hirano, K. Takakura, Y. Takeshita, and S. Hong for their technical assistance; and to the Medical Research Support Center of Kyoto University for in vivo imaging. Financial support was provided in the form of JSPS KAKENHI grants (nos. 21H02715 to K.T., 20H05898 to M.M.), and a Moonshot R&D grant (no. JPMJPS2022-11 to M.M.).
References
- Adra, C.N., Boer, P.H., and McBurney, M.W. 1987. Cloning and expression of the mouse pgk-1 gene and the nucleotide sequence of its promoter. Gene, 60: 65–74.
- Baba, S., Cho, S.Y., Ye, Z., Cheng, L., Engles, J.M., and Wahl, R.L. 2007. How reproducible is bioluminescent imaging of tumor cell growth? Single time point versus the dynamic measurement approach. Mol. Imaging, 6: 315–322.
- Beatty, G.L. and Paterson, Y. 2000. IFN-gamma can promote tumor evasion of the immune system in vivo by down-regulating cellular levels of an endogenous tumor antigen. J. Immunol., 165: 5502–5508.
- Bromberg, J.F., Horvath, C.M., Wen, Z.L., Schreiber, R.D., and Darnell, J.E. 1996. Transcriptionally active Stat1 is required for the antiproliferative effects of both interferon alpha and interferon gamma. Proc. Natl. Acad. Sci. USA, 93: 7673–7678.
- Chin, Y.E., Kitagawa, M., Su, W.C., You, Z.H., Iwamoto, Y., and Fu, X.Y. 1996. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science, 272: 719–722.
- Chin, Y.E., Kitagawa, M., Kuida, K., Flavell, R.A., and Fu, X.Y. 1997. Activation of the STAT signaling pathway can cause expression of caspase 1 and apoptosis. Mol. Cell. Biol., 17: 5328–5337.
- Detjen, K.M., Farwig, K., Welzel, M., Wiedenmann, B., and Rosewicz, S. 2001. Interferon gamma inhibits growth of human pancreatic carcinoma cells via caspase-1 dependent induction of apoptosis. Gut, 49: 251–262.
- Dighe, A.S., Richards, E., Old, L.J., and Schreiber, R.D. 1994. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity, 1: 447–456.
- Dong, H., Strome, S.E., Salomao, D.R., Tamura, H., Hirano, F., Flies, D.B., Roche, P.C., Lu, J., Zhu, G., Tamada, K., Lennon, V.A., Celis, E., and Chen, L. 2002. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med., 8: 793–800.
- Freeman, G.J., Long, A.J., Iwai, Y., Bourque, K., Chernova, T., Nishimura, H., Fitz, L.J., Malenkovich, N., Okazaki, T., Byrne, M.C., Horton, H.F., Fouser, L., Carter, L., Ling, V., Bowman, M.R., Carreno, B.M., Collins, M., Wood, C.R., and Honjo, T. 2000. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med., 192: 1027–1034.
- Fulda, S. and Debatin, K.M. 2002. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene, 21: 2295–2308.
- Garcia-Diaz, A., Shin, D.S., Moreno, B.H., Saco, J., Escuin-Ordinas, H., Rodriguez, G.A., Zaretsky, J.M., Sun, L., Hugo, W., Wang, X., Parisi, G., Saus, C.P., Torrejon, D.Y., Graeber, T.G., Comin-Anduix, B., Hu-Lieskovan, S., Damoiseaux, R., Lo, R.S., and Ribas, A. 2017. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep., 19: 1189–1201.
- Gerber, S.A., Sedlacek, A.L., Cron, K.R., Murphy, S.P., Frelinger, J.G., and Lord, E.M. 2013. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol., 182: 2345–2354.
- Hoekstra, M.E., Bornes, L., Dijkgraaf, F.E., Philips, D., Pardieck, I.N., Toebes, M., Thommen, D.S., van Rheenen, J., and Schumacher, T.N.M. 2020. Long-distance modulation of bystander tumor cells by CD8(+) T cell-secreted IFNgamma. Nat. Cancer, 1: 291–301.
- Ivashkiv, L.B. and Donlin, L.T. 2014. Regulation of type I interferon responses. Nat. Rev. Immunol., 14: 36–49.
- Ivashkiv, L.B. 2018. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat. Rev. Immunol., 18: 545–558.
- Iwano, S., Sugiyama, M., Hama, H., Watakabe, A., Hasegawa, N., Kuchimaru, T., Tanaka, K.Z., Takahashi, M., Ishida, Y., Hata, J., Shimozono, S., Namiki, K., Fukano, T., Kiyama, M., Okano, H., Kizaka-Kondoh, S., McHugh, T.J., Yamamori, T., Hioki, H., Maki, S., and Miyawaki, A. 2018. Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science, 359: 935–939.
- Kaplan, D.H., Shankaran, V., Dighe, A.S., Stockert, E., Aguet, M., Old, L.J., and Schreiber, R.D. 1998. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA, 95: 7556–7561.
- Khiar, S., Lucas-Hourani, M., Nisole, S., Smith, N., Helynck, O., Bourgine, M., Ruffie, C., Herbeuval, J.P., Munier-Lehmann, H., Tangy, F., and Vidalain, P.O. 2017. Identification of a small molecule that primes the type I interferon response to cytosolic DNA. Sci. Rep., 7: 017–02776.
- Konishi, Y., Ichise, H., Watabe, T., Oki, C., Tsukiji, S., Hamazaki, Y., Murakawa, Y., Takaori-Kondo, A., Terai, K., and Matsuda, M. 2021. Intravital Imaging Identifies the VEGF-TXA2 Axis as a Critical Promoter of PGE2 Secretion from Tumor Cells and Immune Evasion. Cancer Res., 81: 4124–4132.
- Koster, M. and Hauser, H. 1999. Dynamic redistribution of STAT1 protein in IFN signaling visualized by GFP fusion proteins. Eur. J. Biochem., 260: 137–144.
- Mazzolini, G., Narvaiza, I., Martinez-Cruz, L.A., Arina, A., Barajas, M., Galofre, J.C., Qian, C., Mato, J.M., Prieto, J., and Melero, I. 2003. Pancreatic cancer escape variants that evade immunogene therapy through loss of sensitivity to IFNgamma-induced apoptosis. Gene Ther., 10: 1067–1078.
- Meunier, M.C., Delisle, J.S., Bergeron, J., Rineau, V., Baron, C., and Perreault, C. 2005. T cells targeted against a single minor histocompatibility antigen can cure solid tumors. Nat. Med., 11: 1222–1229.
- O’Shea, J.J. and Plenge, R. 2012. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity, 36: 542–550.
- Pellegrini, S. and Schindler, C. 1993. Early events in signalling by interferons. Trends Biochem. Sci., 18: 338–342.
- Reynolds, C.J., Chong, D.L.W., Li, Y., Black, S.L., Cutler, A., Webster, Z., Manji, J., Altmann, D.M., and Boyton, R.J. 2019. Bioluminescent Reporting of In Vivo IFN-gamma Immune Responses during Infection and Autoimmunity. J. Immunol., 202: 2502–2510.
- Salas, A., Hernandez-Rocha, C., Duijvestein, M., Faubion, W., McGovern, D., Vermeire, S., Vetrano, S., and Vande Casteele, N. 2020. JAK-STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol., 17: 323–337.
- Samsonov, A., Zenser, N., Zhang, F., Zhang, H., Fetter, J., and Malkov, D. 2013. Tagging of genomic STAT3 and STAT1 with fluorescent proteins and insertion of a luciferase reporter in the cyclin D1 gene provides a modified A549 cell line to screen for selective STAT3 inhibitors. PLoS One, 8: e68391.
- Sanderson, N.S., Puntel, M., Kroeger, K.M., Bondale, N.S., Swerdlow, M., Iranmanesh, N., Yagita, H., Ibrahim, A., Castro, M.G., and Lowenstein, P.R. 2012. Cytotoxic immunological synapses do not restrict the action of interferon-gamma to antigenic target cells. Proc. Natl. Acad. Sci. USA, 109: 7835–7840.
- Stifter, S.A., Bhattacharyya, N., Sawyer, A.J., Cootes, T.A., Stambas, J., Doyle, S.E., Feigenbaum, L., Paul, W.E., Britton, W.J., Sher, A., and Feng, C.G. 2019. Visualizing the Selectivity and Dynamics of Interferon Signaling In Vivo. Cell Rep., 29: 3539–3550 e3534.
- Stringer, C., Wang, T., Michaelos, M., and Pachitariu, M. 2021. Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods, 18: 100–106.
- Takeda, K., Nakayama, M., Hayakawa, Y., Kojima, Y., Ikeda, H., Imai, N., Ogasawara, K., Okumura, K., Thomas, D.M., and Smyth, M.J. 2017. IFN-γ is required for cytotoxic T cell-dependent cancer genome immunoediting. Nat. Commun., 8.
- Tanaka, N., Kawakami, T., and Taniguchi, T. 1993. Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol. Cell. Biol., 13: 4531–4538.
- Taniguchi, K., Petersson, M., Hoglund, P., Kiessling, R., Klein, G., and Karre, K. 1987. Interferon gamma induces lung colonization by intravenously inoculated B16 melanoma cells in parallel with enhanced expression of class I major histocompatibility complex antigens. Proc. Natl. Acad. Sci. USA, 84: 3405–3409.
- Thibaut, R., Bost, P., Milo, I., Cazaux, M., Lemaitre, F., Garcia, Z., Amit, I., Breart, B., Cornuot, C., Schwikowski, B., and Bousso, P. 2020. Bystander IFN-gamma activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat. Cancer, 1: 302–314.
- Trubiani, O., Bosco, D., and Di Primio, R. 1994. Interferon-gamma (IFN-gamma) induces programmed cell death in differentiated human leukemic B cell lines. Exp. Cell Res., 215: 23–27.
- Uccellini, M.B. and Garcia-Sastre, A. 2018. ISRE-Reporter Mouse Reveals High Basal and Induced Type I IFN Responses in Inflammatory Monocytes. Cell Rep., 25: 2784–2796 e2783.
- Weihua, X., Kolla, V., and Kalvakolanu, D.V. 1997. Interferon gamma-induced transcription of the murine ISGF3gamma (p48) gene is mediated by novel factors. Proc. Natl. Acad. Sci. USA, 94: 103–108.
- Yamauchi, F., Kamioka, Y., Yano, T., and Matsuda, M. 2016. In Vivo FRET Imaging of Tumor Endothelial Cells Highlights a Role of Low PKA Activity in Vascular Hyperpermeability. Cancer Res., 76: 5266–5276.
- Yusa, K., Rad, R., Takeda, J., and Bradley, A. 2009. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat. Methods, 6: 363–369.
- Zelenay, S., van der Veen, A.G., Bottcher, J.P., Snelgrove, K.J., Rogers, N., Acton, S.E., Chakravarty, P., Girotti, M.R., Marais, R., Quezada, S.A., Sahai, E., and Reis e Sousa, C. 2015. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell, 162: 1257–1270.
- Zoller, M. 1988. IFN-treatment of B16-F1 versus B16-F10: relative impact on non-adaptive and T-cell-mediated immune defense in metastatic spread. Clin. Exp. Metastasis, 6: 411–429.
Abbreviations
IFN-γinterferon-gamma
ISPinterferon sensing probe
