Abstract
The nuclear volume component of equilibrium field shift isotope fractionations in europium and other lanthanide elements is estimated using Mössbauer spectroscopy and electronic structure calculations. This effect goes in the opposite direction from equilibrium mass-dependent fractionation, and in the case of europium is predicted to dominate over mass dependent fractionation for most materials. Including both effects, Eu2+-bearing species will have approximately 0.4–1‰ higher 153Eu/151Eu than Eu3+-bearing species at 298 K (25°C), and about 0.3‰ higher 153Eu/151Eu at 973 K (700°C). Field shift fractionation mainly depends on oxidation state; differences in coordination structure without changes in oxidation state appear to have much weaker associated fractionations. Nuclear volume isotope fractionation will become even more dominant over mass dependent fractionation at higher temperatures because nuclear volume effects scale with 1/T (K), vs. 1/T2 for mass-dependent fractionation. Fractionation favoring high 153Eu/151Eu in minerals that preferentially incorporate Eu2+, such as plagioclase, is consistent with recent measurements on igneous rocks showing low 153Eu/151Eu in samples with large negative europium anomalies (Lee and Tanaka, 2021). The present results agree with the recent conclusion that equilibrium fractionation cannot explain cosmochemical REE fractionations in primitive meteoritical materials (Hu et al., 2021), because the net fractionation is too small (~0.2‰ or less) at temperatures >1200 K where vapor-phase REE species are relevant.
The field shift effect will also tend to drive 142Ce/xxxCe ratios higher in reduced species (+2 > +3 > +4), for instance by 0.4‰ for 142Ce/140Ce in Ce3+ vs. Ce4+ at 298 K, approximately canceling the mass-dependent fractionation. The nuclear volume component of fractionation in cerium isotope pairs not involving 142Ce will be smaller (<0.1‰) because the other nuclei have very similar volumes. As a result, mass-dependent fractionation will drive 138Ce/140Ce and 136Ce/140Ce lower in Ce4+ species while 142Ce/140Ce hardly fractionates at equilibrium. This will yield an atypical mass vs. fractionation pattern in redox pairs.
Introduction
The geochemical and cosmochemical fractionation of the isotopes of rare earth elements is a topic of emerging interest, driven by the development of new analytical techniques and the recognition that these isotopic signatures may provide useful, complementary information to the well-established uses of rare-earth element abundance variations and radiogenic isotope geochronometers. Fractionations have been observed in meteoritic samples (Hu et al., 2021), as well as in experiments and terrestrial sample suites (e.g., Nakada et al., 2017; Lee and Tanaka, 2021). Meteoritic fractionations have been attributed mainly to either nucleosynthetic processes or disequilibrium mass-dependent effects (Hu et al., 2021). A field shift effect (Bigeleisen, 1996a; Nomura et al., 1996) in europium isotope fractionation in Eu(II)-Eu(III) partitioning on laboratory cation exchange columns has previously been proposed (Ismail et al., 1998), and explored with initial theoretical modeling (Abe et al., 2008), but the potential for this effect to drive measurable natural variations in isotopic abundances of rare earth elements has not been studied in depth. Field shift isotope fractionation effects are expected to scale in proportion to the reciprocal of absolute temperature (1/T), in contrast to the ~1/T2 scaling typical of equilibrium mass-dependent fractionation in elements heavier than hydrogen, so the relative importance of field-shift fractionation increases with increasing temperature (Bigeleisen, 1996b; Bigeleisen and Mayer, 1947). Here we estimate the nuclear volume component of equilibrium stable isotope fractionations in europium using ab initio electronic structure methods and isomer shift data from the Mössbauer spectroscopy literature. Reconnaissance calculations on cerium and other rare earth elements are also presented and briefly discussed.
Europium isotopes are particularly well suited to this type of study because 1) the 151Eu isotope has the largest, most diverse, and best quality set of Mössbauer spectra available among REEs, 2) there is a very large increase in nuclear charge radius between 151Eu and 153Eu (Tanaka et al., 1984), which will tend to drive large nuclear volume fractionations, 3) the Eu2+/Eu3+ oxidation transition is of great interest in rare-earth element geochemistry, 4) the vibrational contribution to mass-dependent isotope fractionation in crystals can now be estimated via 151Eu-NRIXS techniques (Hu et al., 2023; Hu, 2021; Hu et al., 2021), and 5) the position of europium near the middle of the lanthanide elements makes Eu3+ well positioned to serve as an analogue for other rare-earth elements. Cerium is also of interest because of natural Ce3+/Ce4+ redox geochemistry, and this system will also be briefly investigated here. Although there is not a cerium isotope suitable for Mössbauer spectroscopic determination of isomer shifts, the 145.4 keV transition in 141Pr, adjacent to cerium in the lanthanide series, provides a useful Mössbauer probe of materials with light rare-earth elements in the +2, +3, and +4 oxidation states (e.g., Kapfhammer et al., 1971; Groves et al., 1973; Moolenaar et al., 1996). It is also worth noting that at least some other lanthanide elements may occur in the +2 oxidation state in certain environments, such as the solar nebula (e.g., Hu et al., 2021; Ireland and Fegley, 2000).
Mössbauer spectroscopy based on the 21.5 keV transition in 151Eu has been extensively used to characterize europium sites in a wide variety of crystals, glasses, and molecules (e.g., Brix et al., 1964; Grandjean and Long, 1989). Of particular interest for the present study are isomer shifts, which scale in proportion to changes in the electron density at the europium nucleus. This scaling with electron density is closely analogous to the field shift effect, originally discovered by observation of slight changes in the energies of spectral lines in gas-phase atoms and ions, and now known to drive chemical fractionation of isotopes at equilibrium (e.g., Bigeleisen, 1996a; Nomura et al., 1996; Schauble, 2007; Schauble, 2013). Overall, 151Eu isomer shifts show a strong dichotomy, with Eu3+-bearing species characterized by shifts near 0 mm/sec (relative to the EuF3 standard), and typical shifts near –13 mm/sec (–0.09 J/mol) typically found in Eu2+-bearing species. This dichotomy reflects higher electron densities at Eu3+ nuclei; in Eu2+-species the s-electrons that dominate near-nucleus electron density are more effectively screened away from the nucleus by the additional 4f-electron. This dichotomy suggests that equilibrium field shift fractionations will lead to preferential partitioning of the larger volume europium isotope (153Eu) into low-electron-density (Eu2+) sites. This is the opposite sense to that predicted for equilibrium mass-dependent fractionation (Hu et al., 2021; Hu, 2021; Hu et al., 2023).
Methods
The net equilibrium europium isotope fractionation factor α153–151 between two substances EuX and EuZ is defined for present purposes as:
|
α153–151=(Eu153/Eu151)EuX(Eu153/Eu151)EuZ
|
In this study, α153–151 is estimated by combining mass dependent and nuclear volume components:
|
ln α153–151=ln α153–151NV+ln α153–151MD
| (1) |
(Schauble, 2007). This is a simplified approximation of an earlier formulation that also included non-spherical nuclear shape effects, anharmonicity, and non-Born Oppenheimer effects on fractionation (Bigeleisen, 1996a). For rare-earth elements, it is expected that these additional effects are secondary or negligible in importance (e.g., Bigeleisen, 1996a; Zhang and Liu, 2018). The general fractionation factor α is used here rather than fractionation relative to atomic vapor (β) because the nuclear volume component of fractionation in atomic Eu vapor is not trivial to calculate and has significant uncertainty, indicating that it is not the best species to normalize to for the purpose of model comparisons.
Nuclear volume fractionation
The nuclear volume component αNV of the field shift isotope fractionation is determined by the electronic energy difference (per europium atom) between isotopically substituted species of two substances EuX and EuZ:
|
ln α153–151NV=EEuZ153-E[EuZ151]-(EEuX153-E[EuX151])kBT
| (2) |
Here kB is Boltzmann’s constant and T is the absolute temperature. In contrast to mass-dependent fractionation, where the vibrational energies of the more massive 153Eu-isotopologues are lower (i.e., they are more stable thermodynamically) relative to their 151Eu-bearing counterparts, the nuclear volume effect leads to higher energies in isotopologues containing the larger (more voluminous) 153Eu-nucleus. In this study the nuclear volume effect on energy is estimated primarily by scaling tabulated 151Eu Mössbauer isomer shifts (converted to energy units ΔEisomer = (hνγ/c)·ΔIS) by the ratio of the isotopic difference in ground-state nuclear mean-squared radii to the isomeric change in the mean-squared 151Eu radius:
|
EEuZ153-E[EuZ151]-EEuX153-EEuX151=ΔEisomer<r1532>-<r1512><r151*2>-<r1512>
| (3) |
Here 151* represents the excited 7/2+ nuclear state of 151Eu at 21.5 keV used in Mössbauer spectroscopy. ΔEisomer is the difference in the isomer shifts of the two substances in energy units, Eisomer(EuZ) – Eisomer(EuX) measured relative to a common standard material, typically anhydrous EuF3. Because EuF3 is an exotic material in geochemistry, we will use EuPO4 in the monazite structure as the standard of comparison for isotope fractionations, assuming a shift of +0.19 mm/sec in EuPO4 relative to EuF3 (Klobes et al., 2016; Golbs et al., 2013 report +0.16 ± 0.01 mm/sec at 77 K). EuPO4-monazite has greater geochemical relevance, and 151Eu-Mössbauer isomer shifts and the lattice dynamics of Eu in this structure have both been investigated (Klobes et al., 2016; Hu, 2021; Hu et al., 2023). The mean squared radius change associated with the 21.5 keV transition has been estimated by various methods in the literature (Table 1), with published values ranging from 0.018–0.030 fm2 (1.8–3.0 × 10–32 m2; Walter et al., 1972; Brix et al., 1964). The most recent estimates, based on Mössbauer spectroscopy, muonic x-ray spectroscopy, and electronic structure calculations, span a slightly narrower range from 0.019–0.025 fm2 (Kalvius and Shenoy, 1974; Tanaka et al., 1984). Davydov et al. (2022) recently calculated a somewhat larger radius change of 0.031 fm2, however they used a rough approximation of the ground state 151Eu charge radius that may affect the estimated radius change in the Mössbauer transition, which makes the accuracy of their estimate difficult to quantify. The mean squared radius difference between 151Eu (in the 5/2+ ground state) and 153Eu has been determined to be 0.57–0.61 fm2 (e.g., Tanaka et al., 1984; Ahmad et al., 1985). Taken together, these radii yield an energy scale factor of 23–32, for which we choose the mid-point value (28 ± 5). A scale factor of 28 ± 5 means that a typical isomer shift of ~13 mm/sec (0.09 J/mol) between Eu2+- and Eu3+-bearing crystals corresponds to a 153Eu–151Eu nuclear volume effect of 2.5 ± 0.5 J/mol, enough to drive 1.0 ± 0.2‰ fractionation at 298.15 K and favoring high 153Eu/151Eu in Eu2+-bearing crystals. This field shift/isomer shift scaling method is useful for determining relative energy differences between solids—including many geochemically interesting phases and/or close chemical analogues, but because it depends on Mössbauer spectroscopy the library of available data is limited. The limitation is particularly acute for natural materials and melts, including major and REE-bearing rock-forming minerals. A significant advantage of this method is that errors in the scale factor (i.e., due to uncertainties in nuclear charge radii determinations) uniformly magnify or diminish the estimated nuclear volume component of equilibrium fractionation for all substances. The relative order of fractionation is then only sensitive to errors in isomer shift measurements on the substances being compared.
Table 1.
Differences in root mean squared charge radii of
151Eu in the ground state relative to the excited (21.5 keV) state, and to
153Eu
The estimated field shift/isomer shift scale factor can be tested by comparing measured isomer shifts for molecules and molecular ions with first principles estimates of nuclear volume effects on electronic energy. This is not a fully independent test, because literature estimates of Mössbauer and 151Eu–153Eu nuclear radii are partly based on (typically atomic) electronic structure calculations. This molecular calibration method depends on the availability of isomer shift measurements for Eu0 atoms (–6.1 or –4.8 mm/sec relative to EuF3; Montano, 1982; Litterst et al., 1976) and EuCl2 molecules (–12.36 mm/sec relative to EuF3; Baggio-Saitovitch et al., 1976) trapped in inert-gas matrices, and for the Eu(H2O)93+ complex found in crystalline Eu(H2O)9.I3 (isomer shift of +0.75 mm/sec relative to EuF3; Jenden and Lyle, 1982). Eu(H2O)93+ is also a likely structure for Eu3+ in dilute, acidic aqueous solutions. Ground state relativistic electronic structures for each of these molecules are calculated at the Dirac Hartree-Fock level using the exact two component (X2C) Hamiltonian, as implemented in the 2019 version of the DIRAC software package (Gomes et al., 2019). For open-shell atoms and molecules, the energies of individual spin-orbit states are resolved from the average-of-configuration open-shell Hartree-Fock solution via a subsequent configuration interaction calculation (Visser et al., 1992). The calculations use all-electron double-zeta quality basis sets for Eu and the def2-TZVP basis set family for other elements (Gomes et al., 2010; Dolg et al., 1989; Gulde et al., 2012; Weigend and Ahlrichs, 2005). The results appear to be only slightly sensitive to the choice of basis sets. Nuclear charge radii tabulated by Angeli and Marinova (2013) are used for 151Eu and 153Eu, with a mean squared radius difference of 0.602 fm2. Relative to atomic Eu0, the calculations indicate a 1.86 J/mol smaller 151Eu–153Eu nuclear volume shift in EuCl2(v), and a 0.98 J/mol larger 151Eu–153Eu shift in Eu(H2O)93+. Using the Montano (1982) datum for Eu0, a best-fit linear regression through these three points (r2 = 0.96) gives a scale factor of 31 ± 6 (±1 std. error), consistent with the literature-based estimate above. The scale factor changes only slightly if the earlier Litterst et al. (1976) shift of –4.76 mm/sec is used for Eu0, and the fit is somewhat tighter (32 ± 3, r2 = 0.99).
Mössbauer data
Measured 151Eu-Mössbauer isomer shifts used in the present work are tabulated in Table 2. These data draw largely from the compilation of Barton and Greenwood (1973), which overlaps and is consistent with the more recent compilation of Grandjean and Long (1989). A datum for EuPO4 in the monazite structure is taken from the recent Klobes et al. (2016) study, and approximate isomer shifts for Eu2+ and Eu3+ in BaAl2Si2O8 (celsian – feldspar) and β-Ca2SiO4 (larnite) are taken from Clabau et al. (2008) and Nakanishi et al. (2009), respectively. The isomer shift in metallic europium is from Taylor and Farrell (1987). Isomer shifts are somewhat sensitive to temperature (especially for EuO, which has a temperature-dependent electronic structure), so room-temperature measurements are chosen when possible. Data for aqueous Eu3+ and Eu2+ in water ice, and [Eu(H2O)x]3+ aquo ions in crystalline chloride and ethyl sulfate hydrates are from the thesis of Nozik (1967; the isomer shift for each substance is averaged over all concentrations and temperatures measured). Data for crystalline Eu(H2O)9.I3 are from Jenden and Lyle (1982). Some species that are geochemically interesting require additional discussion, including Eu0-vapor and natural Eu-bearing silicate melts.
Table 2.
151Eu-Mössbauer isomer shifts
Atomic Eu0 vapor is of interest because it is thought to be the dominant gas-phase europium species present in the solar nebula (e.g., Lodders and Fegley, 1993; Ireland and Fegley, 2000; Hu et al., 2021), with europium being one of the most volatile rare earth elements in this environment. Two 151Eu-Mössbauer isomer shift measurements have been reported for dilute Eu0 frozen into a noble gas matrix. The earliest measured shift is –5.80 ± 0.05 mm/sec relative to the 151Sm2O3 source (Litterst et al., 1976). Assuming a correction of +1.04 (e.g., Klencsár et al., 2021), this would be equivalent to a shift of –4.76 ± 0.05 mm/sec relative to the modern EuF3 standard. Montano (1982) reports a shift of –6.1 ± 0.1 mm/sec relative to their 151SmF3 source (which is considered equivalent to the EuF3 standard). The difference of ~1.3 mm/sec between the two studies is much larger than the reported measurement uncertainties. Montano (1982) argue that this is due to the asymmetric peak shapes observed in both studies. For purposes of the present study we will primarily use the more recent Montano (1982) datum.
To the best of our knowledge, isomer shift measurements have not yet been reported for natural igneous melts or close synthetic analogues. However, a rough estimate of 0.6 ± 0.3 mm/sec can be proposed for Eu3+, and –13.5 ± 0.5 for Eu2+ in natural silicate melts, respectively, based on a variety of literature data for synthetic glasses. These ranges are also consistent with isomer shifts measured in the silicate crystals BaAl2Si2O8 celsian, a feldspar, and β-Ca2SiO4 larnite, an olivine-like structure. Overall fractionation in melts with both Eu3+ and Eu2+ can be approximated via the abundance-weighted average of these components. Virgo et al. (1981) report isomer shifts for melts of diopside-monticellite and diopside-silica mixtures with 1–10% added europium oxide. They detect an Eu3+ component at a characteristic shift of +0.6 mm/sec in a variety of compositions and an Eu2+ component with a more variable shift of –13.6 to –15.4 mm/sec. The Eu2+ spectral component is broader and possibly less precisely resolvable than the Eu3+ component in these data. It is not clear whether EuF3 is the isomer shift standard for these measurements, and it is worth cautioning that isomer shifts <–14 mm/sec relative to EuF3 appear to be very rare in general data compilations. Nemov et al. (2007) report measured isomer shifts in Al2O3 + SiO2 + MnO + Eu2O3 glasses, detecting an Eu3+-peak at 0.39 ± 0.07 mm/sec and an Eu2+-peak at –12.06 ± 0.07 mm/sec (both adjusted by 1.04 mm/sec to re-scale relative to the EuF3 standard from Eu2O3). Tanabe et al. (1989) investigated Eu3+ in synthetic SiO2 + Eu2O3, Al2O3 + Eu2O3, and SiO2 + Al2O3 + Eu2O3 glasses, measuring shifts ranging from +0.3 to +0.7 mm/sec. They noted a positive correlation between the Eu3+ isomer shift and optical basicity, with lower shifts of +0.3 to +0.4 mm/sec at geochemically relevant basicities of 0.55–0.65. These are similar to the calculated basicities of the basaltic (0.60) and rhyolitic (0.54) compositions measured in vibrational 151Eu-NRIXS studies (Hu, 2021; Hu et al., 2023). Tanabe et al. (1993) and Todoroki et al. (1993) found higher shifts of ~0.7–0.9 for Eu3+ in highly alkali-rich and alkaline earth-rich silicate glasses, whereas Conchas et al. (1996) report a shift of +0.55 in a multicomponent SiO2-Al2O3-SrO-MgO-Eu2O3-Li2O-Cs2O-ZnO composition formulated as an analogue to a Ce-bearing scintillating glass. In addition to the Eu2+ shifts measured by Virgo et al. (1981), Tanaka et al. (1998) report shifts of –13.1 to –13.5 mm/sec in a range of alkali oxide + EuO + SiO2 glass compositions, identical to the range found in sodium-europium-borate (non-silicate) glasses by Fujita et al. (1998). The latter study finds the most positive values of –13.0 to –13.2 mm/sec at the highest optical basicities sampled (0.54–0.56). The assumed shifts of 0.6 ± 0.3 mm/sec for Eu3+ and –13.5 ± 0.5 for Eu2+ for the present study are chosen to roughly span the range of shifts measured in all silicate glasses, representing widely varying compositions, and it may be that common igneous melts are more uniform than this range suggests. However, the range assumed for Eu2+ is narrower than Virgo et al. (1981) and Nemov et al. (2007) report and this could be a source of error in estimated nuclear volume fractionations.
Similarly, the Mössbauer properties of 151Eu substituted into major rock-forming minerals are not known. Perhaps the most critical mineral unknown is plagioclase feldspar, which plays a key role in the generation of europium abundance anomalies in igneous melts, minerals, and rocks. For the purposes of this study, it is assumed that the isomer shifts of Eu2+ and Eu3+ in Eu:BaAl2Si2O8 celsian, a non-plagioclase feldspar structure that occurs rarely in nature, are a reasonable analogue for more common feldspar compositions.
Mass dependent fractionation
The mass dependent component αMD can be determined approximately from the effects of isotope substitution on vibrational frequencies in the harmonic approximation, using a well-established formula (Urey, 1947) and its extension for phonons in crystals (Elcombe and Hulston, 1975). In the present study, this frequency-based method is mainly used as a test of the force constant method, described below, applied to EuO-lime and EuPO4-monazite.
Rather than using vibrational frequencies directly, in the present study the mass dependent component is estimated via a force constant method, allowing the use of Nuclear Resonance Inelastic Xray Scattering (NRIXS) measurements (Hu, 2021; Hu et al., 2023). Tabulated force constants derived from NRIXS data are supplemented with ab initio calculations using Density Functional Theory (DFT). Estimated mass dependent fractionations are calculated from force constants using the expression proposed by Bigeleisen and Mayer (1947), and subsequently adapted for use in calculating equilibrium 88Sr/86Sr fractionation (Widanagamage et al., 2014):
|
β153–151MD≈1+h296π2kB2T2m153Eu-m151Eum153Eu×m151EuA
| (4) |
Here βMD153–151 is the mass dependent fractionation between a substance and atomic vapor, h is Planck’s constant, kB is the Boltzmann constant, T is the absolute temperature, m153Eu and m151Eu are the isotopic masses in kg/atom, and A is the sum of force constants opposing displacement of a europium atom in three perpendicular directions. A, the force constant sum, is three times the average force constant typically reported in NRIXS studies (Table 3). Hu (2021) and Hu et al. (2023) propose a refinement of this method for estimating mass dependent 153Eu/151Eu fractionation that accounts for the deviation of mass-dependent fractionation from 1/T2 proportionality at low temperatures. They find that higher-order terms have little impact on calculated europium isotope fractionations at geochemically-relevant temperatures. In this study the simpler equation above is used. In order to be consistent with the way nuclear volume component of fractionation is reported, βMD153–151 is converted to a fractionation relative to EuPO4 in the monazite structure using:
Table 3.
Force constant sums
|
αMD153–151[X]=βMD153–151[X]/βMD153–151[EuPO4]
|
NRIXS-based force constants are available for crystalline EuO, EuS, EuSe, EuTe, Eu2(CO3)3, Eu2O3, EuTiO3, and EuPO4 in the monazite structure, metallic Eu, and rhyolite and basalt analogue glasses synthesized with high and low fO2 (Hu, 2021; Hu et al., 2023; Bessas et al., 2013; Klobes et al., 2016; Pradip et al., 2019; Lazewski et al., 2021; Stankov et al., 2022; Stankov et al., 2008). The NRIXS studies of Hu (2021) and Hu et al. (2023) are particularly comprehensive, and their results will be a primary basis for modeling the mass dependent component of fractionation. For EuTiO3 (a perovskite structure), a range of average force constants from 70–78 N/m has been reported over temperatures from 110–360 K (Bessas et al., 2013) without identifying the measured force constants at any particular temperature. A value of 74 ± 4 N/m (A = 222 ± 12 N/m) is therefore assumed for this material.
For other substances, and for purposes of comparison with NRIXS, Raman, and infrared and inelastic neutron scattering data, DFT calculations are performed using the PBE functional (Perdew et al., 1996). Projector Augmented Wave (PAW) datasets from the REE library generated by Topsakal and Wentzcovitch (2014) are used for calculating mass-dependent fractionation of europium in Eu2+-bearing species. In order to counter the systematic underestimation of electronic band gaps in conventional density functional theory, a Hubbard U correction of 7.5 eV is applied to the 4f orbitals. The value of 3.0 eV recommended by the creators of the Eu-pseudopotential used in the present study (Topsakal and Wentzcovitch, 2014) yields a near-metallic electronic structure for crystalline EuO (calculated band gap <0.1 eV), in contrast to the observed band gap of ~1.1 eV. Setting U = 7.5 eV is consistent with the range of ~6–8 eV used in previous work on Eu(II)- and Eu(III)-oxides (e.g., Davydov et al., 2022; Zhang et al., 2014; An et al., 2011; Tong et al., 2014), and yields a more reasonable band gap of ~0.8 eV for EuO. It was not possible to reliably obtain self-consistent electronic structure solutions for Eu3+-bearing species with this PAW dataset, so a 4f-in-core pseudopotential from the PSLibrary collection was used instead. The use of different PAW data sets for Eu2+ and Eu3+ likely adds somewhat to the uncertainty of estimated fractionation factors when species with different oxidation states are compared. A combination of PAW and ultrasoft pseudopotentials was used to represent other elements, roughly corresponding to recommendations from the Standard Solid State Pseudopotential project (https://www.materialscloud.org/discover/sssp). Plane wave basis sets are generated with a kinetic energy cutoff of 60 Rydberg (816 eV) for calculations with the Topsakal and Wentzcovitch (2014) PAW dataset, and 80 Rydberg (1088 eV) for calculations with the PSLibrary dataset. DFT force constant and phonon frequency calculations are made with the Quantum Espresso software package (Giannozzi et al., 2009; https://www.quantum-espresso.org/).
Eu2+-substituted Ca2SiO4-larnite and Ca2Al2Si2O8-anorthite are of particular interest as analogues for natural Eu2+-bearing silicates and silicate melts. However, NRIXS-based force constants are not available for either structure. In order to obtain initial estimates, DFT models were generated in which an Eu2+ is substituted for one Ca2+ in each crystal’s unit cell, and the structure is then allowed to relax until interatomic forces and unit-cell stresses are minimized. In the case of larnite, a single-atom substitution corresponds to a formula of Eu0.25Ca1.75SiO4 (a 1:7 Eu:Ca ratio; Yamnova et al., 2011). There are two distinct calcium sites in the larnite structure (Ca1 and Ca2), and it is found that the 0 K enthalpies are very similar when Eu2+ is substituted at either site, with the Ca1 site ~2 kJ/mol lower in energy. This small difference may not be significant, given the limitations of the DFT method, and even taken at face value it would imply substantial mixing across sites at moderate to high temperatures. The calculated total force constants at both sites are also very similar (400 N/m vs. 405 N/m). Therefore, we report an overall fractionation for this structure simply by taking the arithmetic mean of the fractionation at both sites. In the case of anorthite, the substitution yields a formula of Eu0.125Ca0.875Al2Si2O8, also a 1:7 Eu:Ca ratio. There are four distinct Ca-sites (Foit and Peacor, 1973), and in the present work it was found that Eu-substitution at the Ca(zi0) site is most favorable, with an enthalpy ~5 kJ/mol lower than the Ca(000) site, ~9 kJ/mol lower than the Ca(z00) site, and ~7 kJ/mol lower than the Ca(0i0) site. The 7-fold coordination and average Eu-O bond length (2.66 Å) in the Ca(zi0)-substituted structure are similar to the coordination environment of Eu2+ in synthetic EuAl2Si2O8-anorthite (2.70 Å; Kimata, 1988). For these reasons, the force constant for an Eu-atom substitution at the Ca(zi0) site is assumed to be representative for the whole structure.
Results
Estimated nuclear volume and mass dependent components of equilibrium 153Eu/151Eu fractionation are shown in Table 4 and Fig. 1.
Table 4.
Equilibrium
153Eu/
151Eu fractionations. Nuclear volume fractionation in anorthite is assumed equal to celsian, and Eu(H
2O)
8(OH)
2 is assumed equal to Eu(H
2O)
xCl
2
| Substance |
103ln αNV153–151 vs. EuPO4-monazite |
103ln αMD153–151 vs. EuPO4-monazite |
Total 103ln α153–151 vs. EuPO4-monazite |
| 298.15 K |
1000 K |
1500 K |
298.15 K |
1000 K |
1500 K |
298.15 K |
1000 K |
1500 K |
| Eu3+ species |
| EuPO4 - monazite |
0.00 |
0.00 |
0.00 |
0.00 |
0.000 |
0.000 |
0.00 |
0.00 |
0.00 |
| Eu2O3 |
–0.07 |
–0.02 |
–0.01 |
0.13 |
0.009 |
0.004 |
0.06 |
–0.01 |
–0.01 |
| Eu2(CO3)3 |
0.02 |
0.01 |
0.00 |
0.14 |
0.011 |
0.005 |
0.16 |
0.02 |
0.01 |
| Eu3+:BaAl2Si2O8 - celsian |
0.01 |
0.00 |
0.00 |
|
|
|
|
|
|
| Eu3+:CaAl2Si2O8 - anorthite |
0.01* |
0.00* |
0.00* |
|
|
|
|
|
|
| Eu3+:Ca2SiO4 - larnite |
–0.02 |
–0.01 |
0.00 |
|
|
|
|
|
|
| Eu(H2O)9I3 |
–0.04 |
–0.01 |
–0.01 |
0.17 |
0.013 |
0.006 |
0.13 |
0.00 |
0.00 |
| Eu3+ silicate melt |
–0.03 |
–0.01 |
–0.01 |
0.19 |
0.015 |
0.007 |
0.16 |
0.01 |
0.00 |
| Eu2+ species |
| EuO - lime |
0.97 |
0.29 |
0.19 |
–0.24 |
–0.023 |
–0.010 |
0.73 |
0.27 |
0.18 |
| EuS - oldhamite |
0.95 |
0.28 |
0.19 |
–0.31 |
–0.029 |
–0.013 |
0.64 |
0.25 |
0.18 |
| EuTiO3 - perovskite |
0.99 |
0.29 |
0.20 |
–0.41 |
–0.038 |
–0.017 |
0.58 |
0.26 |
0.18 |
| Eu2+:BaAl2Si2O8 - celsian |
1.11 |
0.33 |
0.22 |
|
|
|
|
|
|
| Eu2+:CaAl2Si2O8 - anorthite |
1.11* |
0.33* |
0.22* |
–0.26 |
–0.025 |
–0.011 |
0.85 |
0.31 |
0.21 |
| Eu2+:Ca2SiO4 - larnite |
1.07 |
0.32 |
0.21 |
–0.15 |
–0.015 |
–0.007 |
0.92 |
0.30 |
0.21 |
| Eu(H2O)xCl2 |
1.02 |
0.31 |
0.20 |
|
|
|
|
|
|
| Eu(H2O)8(OH)2 |
1.02* |
0.31* |
0.20* |
–0.30 |
–0.029 |
–0.013 |
0.72 |
0.28 |
0.19 |
| Eu2+ silicate melt |
1.07 |
0.32 |
0.21 |
–0.37 |
–0.035 |
–0.016 |
0.70 |
0.28 |
0.20 |
| Eu0 species |
| Eu-metal |
0.59 |
0.17 |
0.12 |
–0.58 |
–0.054 |
–0.024 |
0.00 |
0.12 |
0.09 |
| Eu0 vapor |
0.49 |
0.15 |
0.10 |
–0.72 |
–0.066 |
–0.030 |
–0.23 |
0.08 |
0.07 |
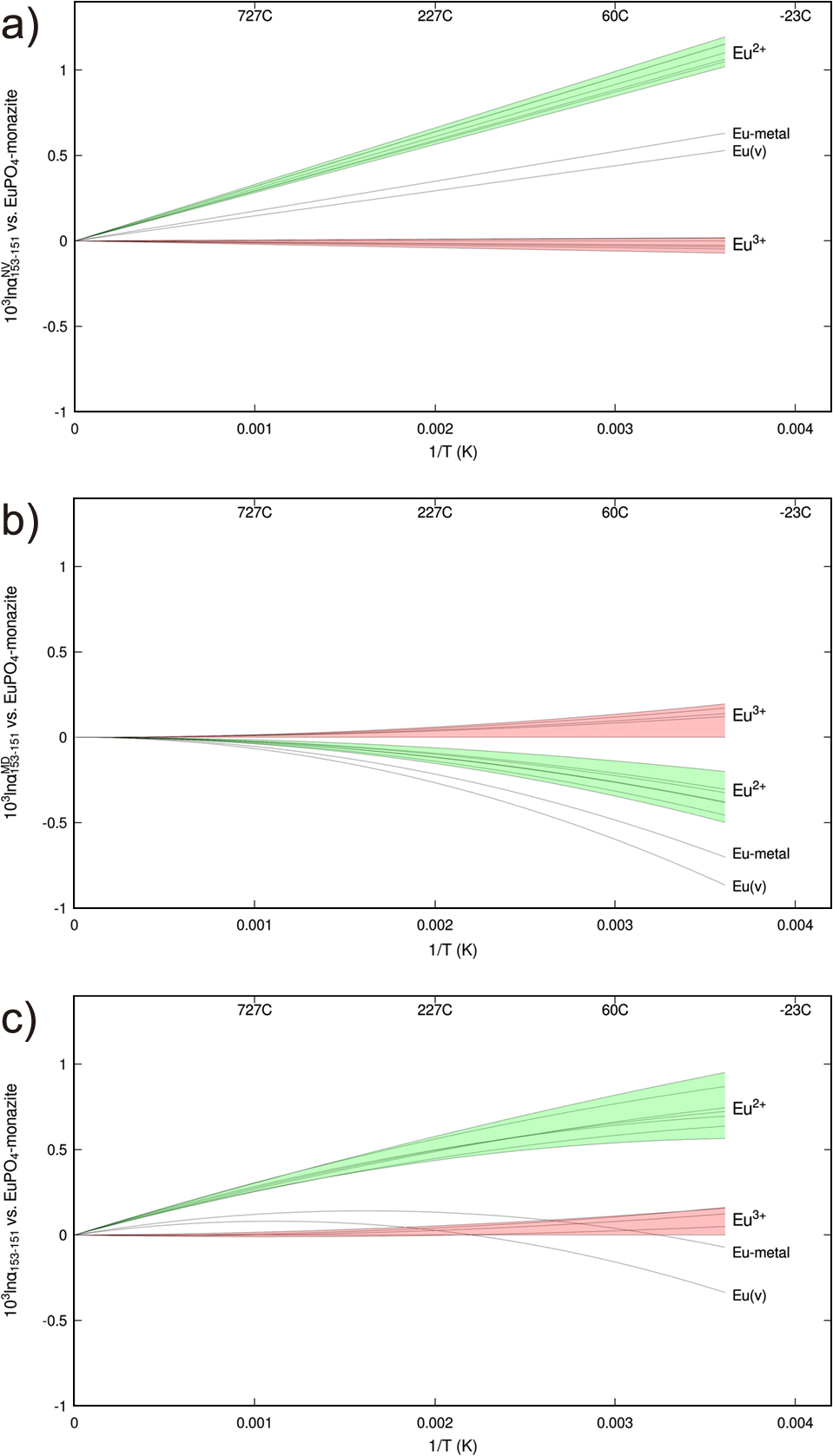
As is typical in exchanges involving different oxidation states, the mass-dependent component favors high 153Eu/151Eu in more oxidized Eu3+-bearing species, and lower 153Eu/151Eu in more reduced Eu2+-bearing species, by as much as 0.5‰ at 298 K (25°C). Because mass-dependent fractionation scales in approximate proportion to 1/T2, it becomes much more subtle at elevated temperatures, shrinking to ~0.05‰ or less at 1000 K (727°C). This result agrees well with previous estimates (Hu, 2021; Hu et al., 2021; Hu et al., 2023), which is not surprising given that many of the same force constant data have been used all these studies. Eu-metal, with a very low measured average force constant, is predicted to have the lowest 153Eu/151Eu among the compounds studied, only slightly higher than Eu0-vapor (by 0.14‰ at 298 K and 0.01‰ at 1000 K). Basaltic and rhyolitic melts compositions are also intermediate between Eu2+-bearing and Eu3+-bearing crystalline species, with 153Eu/151Eu decreasing in proportion to the percentage of europium in the +2 oxidation state (Hu, 2021; Hu et al., 2023).
βMD153–151 is assumed to be proportional to the force constant sum, and thus uncertainties in βMD153–151 are also expected to scale with uncertainties in the NRIXS-based force constants. These force constant uncertainties range considerably, from <10% to >30% in the data of Hu (2021) and Hu et al. (2023), though other authors quote somewhat smaller error estimates of 5–7% (Bessas et al., 2013; Stankov et al., 2008; Klobes et al., 2016; Pradip et al., 2019) for individual crystal structures. This suggests uncertainties on the order of ±0.1‰ up to as much as ±0.2‰ in βMD153–151 for Eu3+ species at 298 K, and somewhat smaller uncertainties for Eu2+ species and Eu0-metal. It is not clear whether errors in force constant determinations are systematic across species, or random; a conservative assumption that they are random suggests typical errors of ~0.1–0.3‰ at 298 K (0.01–0.03‰ at 1000 K) when βMD153–151 for different species are compared to generate αMD153–151. An independent check comes from the phonon-based DFT models of βMD153–151 in crystalline EuO and EuPO4-monazite, which agree within 0.14‰ and 0.01‰, respectively, with NRIXS-based determinations at 298 K. These deviations are consistent with reported uncertainties in NRIXS force constants, as was previously found by Hu (2021) and Hu et al. (2023) for a broader test suite of DFT models.
Nuclear volume fractionation strongly favors high 153Eu/151Eu in Eu2+-bearing species, with Eu0-bearing atomic vapor and metal intermediate. The total range is approximately 1.2‰ at 298 K (Eu2O3 vs. Eu2+:BaAl2Si2O8), shrinking to ~0.4‰ at 1000 K. For most redox pairs studied, the nuclear volume effect dominates mass dependent fractionation at all temperatures above 0°C, and particularly at higher temperatures. In contrast, different crystals with the same oxidation state are very similar, spanning only ~0.1‰ at 298 K and ~0.04‰ at 1000 K. Relativistic electronic structure models also suggest that vapor-phase molecular EuO is very similar to crystalline EuO.
The main source of error in estimated nuclear volume fractionations is likely in the scale factor used to convert isomer shifts to 153Eu–151Eu energy shifts. This translates into an approximate uncertainty of ±5/28 × αNV153–151 ≈ ±0.18 × αNV153–151. This error will be systematic across all Mössbauer-based αNV153–151 estimates. Other sources of error include uncertainties in the measurement of isomer shifts, the neglect of nuclear shape effects, quadrupole moments, & other secondary contributors to the field shift effect, and temperature dependencies in electronic structure.
The data in Table 1 can be extrapolated to other temperatures by scaling the nuclear volume component as 1/T, and the mass dependent component as 1/T2.
Discussion
The present results indicate that equilibrium 153Eu/151Eu fractionation will typically be dominated by the nuclear volume effect, especially at the elevated temperatures of interest in cosmochemical, igneous, and metamorphic contexts. The results also indicate that measurable stable europium isotope fractionation is likely to be found in any reducing geochemical environments where Eu2+ and Eu3+ coexist—even at igneous temperatures. As an example, the Eu2+ component of celsian (as an analogue to plagioclase) is expected to be 0.3‰ heavier than an “oxidized” endmember of Eu3+ in rhyolite at 1000 K, and 0.2‰ heavier than an oxidized basalt end member at 1500 K. This finding significantly extends the range of elements with large (≥1‰ at 298 K) predicted field shift fractionations, which was previously limited to elements of atomic number 80 (Hg) or greater (Tl, Pb, U) (e.g., Nomura et al., 1996; Bigeleisen, 1996a; Abe et al., 2008; Schauble, 2007; Yang and Liu, 2015). Rhenium, a 5d-block transition element with atomic number intermediate between Hg and Eu, has previously been predicted to show dominantly mass-dependent equilibrium fractionation, particularly at typical Earth surface temperatures (Miller et al., 2015). The present results also suggest that nuclear volume effects may be significant for other lanthanide elements. Particular interest arises in situations where oxidation state changes are possible, including Ce3+ vs Ce4+ in oxidizing terrestrial environments, and vapor/liquid or vapor/solid interactions involving REE-monoxide molecules (or neutral atomic vapor) in the nebular environment of the early solar system. Other lanthanide elements will be discussed in a later section.
Comparison with previous measurements and predictions
Abe et al. (2008) also estimated the nuclear volume fractionation between Eu2+ and Eu3+, based on ab initio modeling of vapor-phase atomic ions, predicting an effect of only ~0.4‰ at 308 K, which is substantially smaller than estimated in the present study. Although there are several differences in the methods used in the two studies, the main cause of the discrepancy seems to be a much smaller assumed difference in the nuclear charge radii (5.0413 vs. 5.0218 fm, Δr2 ≈ 0.2 fm2) in the earlier study, relative to the current one (Δr2 ≈ 0.6 fm2). There does not appear to be a direct citation of the reference used for nuclear radii in the Abe et al. (2008) study, and the small quoted difference in radii is inconsistent with estimates going back to at least the 1970’s (e.g., the tabulation in Ahmad et al., 1985). Scaling the Abe et al. (2008) result to the same radius difference used in our calculation of the Mössbauer scale factor yields an adjusted Eu2+(v)/Eu3+(v) fractionation of 1.2‰ at 308 K, in good agreement with the present study.
Ismail et al. (1998) analyzed 153Eu/151Eu fractionation between Eu2+ and Eu3+ in aqueous EuCl2 solutions in contact with a cation exchange resin pre-loaded with Fe3+, in which the Eu3+ had a slight preference for binding to the resin and Eu2+ had a slight preference for remaining in solution. After accounting for the incomplete separation of the oxidation states, they found effective fractionation factors of ~0.2‰ between Eu2+ and Eu3+ at 308 K, increasing to ~0.6‰ at 343 K. If we take the present estimates for isomer shifts in Eu(H2O)x2+ in water ice (Nozik, 1967) and the force constant in crystalline Eu(H2O)8(OH)2 to be representative of Eu2+-species in the Ismail et al. (1998) experiments, and the isomer shift and force constant for Eu(H2O)9.I3 to be representative of the Eu3+-species, the present study estimates a total fractionation factor of 0.5–0.6‰ that is invariant within 0.02‰ over the 308 K–343 K temperature range. This estimate agrees well with the highest-temperature data of Ismail et al. (1998), but not with their lower temperature results. The reason for this discrepancy is not clear.
There are as yet only a few published high-precision measurements examining possible geochemical fractionation of europium isotopes. These include a recent study of isotopic compositions of multiple rare earth elements in group II calcium-aluminum–rich inclusions (CAIs), among the oldest solar system materials (Hu et al., 2021). This study found a range of more than 3‰ in 153Eu/151Eu in what was likely a high-temperature fractionating environment. Such large fractionations are difficult to reconcile with the relatively modest (<0.4‰ at T ≥1000 K) equilibrium fractionations predicted by the present study. From 1000–1500 K, a temperature range spanning the expected condensation/vaporization of europium in the solar nebula (e.g., Lodders, 2003), the predicted total fractionation between atomic Eu0 vapor and Eu2+ in a silicate melt (the total fractionation at these temperatures is dominated by the field shift effect) is 0.1–0.2‰, an order of magnitude smaller than the measured range of variation. Vapor-phase EuO, calculated using the same method described for molecules above, appears to be similar to other Eu2+ species, with an estimated equilibrium fractionation of <0.1‰ relative to Eu2+-bearing silicate melt at the same temperatures. The present results therefore support the conclusion of Hu et al. (2021) that equilibrium fractionation is unlikely to play a significant role in generating the observed variations in REE isotope composition in CAIs.
Perhaps the most interesting recent study, in light of the results presented here, presents measurements of igneous rocks and minerals (Lee and Tanaka, 2021). In highly fractionated granites with large negative Eu-anomalies, it is found that the Eu-anomaly is correlated with low whole-rock 153Eu/151Eu. This correlation is suggestive of crystal fractionation of high 153Eu/151Eu, high Eu2+/Eu3+ feldspar during the magmatic evolution of these rocks. The total range spanned by the highly fractionated granites is ~0.4‰ (considering only 147Sm/149Sm-normalized data), in qualitative agreement with the predicted Eu2+-anorthite vs. Eu3+-melt fractionation at ~1000 K (ignoring e.g., Rayleigh distillation effects that could magnify the observed fractionation). Although a detailed geochemical model of the evolution of natural magmas and cumulates is beyond the scope of the present study, it is worth making a more quantitative predictive assessment of feldspar-melt fractionation, which follows in the next section.
Plagioclase/melt fractionation
Although the isomer shifts of europium in major silicate minerals have not been measured, the strongly systematic pattern of isomer shifts in other crystals suggests that reasonable estimates of equilibrium fractionation factors are possible by referencing analogous structures with the appropriate Eu-oxidation state. Plagioclase is of particular interest, because of the importance of bulk europium partitioning and redox segregation into feldspars relative to silicate melts. Here, plagioclase-melt 153Eu/151Eu fractionation is estimated as a function of the temperature, oxygen fugacity, and the optical basicity of the silicate melt. Nuclear volume and mass-dependent fractionation in the melt are assumed to be determined by the abundance-weighted average of the Eu2+-melt and Eu3+-melt components (see Methods, above, as well as Hu, 2021 and Hu et al., 2023). It is assumed that isomer shifts for Eu2+ and Eu3+ in plagioclase are –14.0 mm/sec and 0 mm/sec, respectively, as observed in the analogous BaAl2Si2O8-celsian structure (Clabau et al., 2008). Total force constants driving mass-dependent fractionation in plagioclase are assumed to be the same as in Eu2+-substituted CaAl2Si2O8-anorthite (327 N/m) and the Eu3+-bearing silicate melt end member (642 N/m; Hu, 2021 and Hu et al., 2023). These analogies are crude, but it should be noted that the mass-dependent component of equilibrium fractionation is so small at magmatic temperatures that even large uncertainties in total force constants barely matter to the final result. For instance, using the smaller force constant for Eu3+PO4 (524 N/m) instead of the larger Eu3+-melt value of Hu (2021) and Hu et al. (2023) changes the Eu3+-plagioclase—melt total fractionation by less than 0.02‰ at 900 K, and by less than 0.01‰ at 1500 K. An error of 1 mm/sec in the assumed isomer shift likewise translates to errors of <0.03‰ and <0.02‰ at 900 K and 1500 K in the nuclear volume component of the total fractionation, respectively.
The fraction of Eu3+ vs. Eutot in melt is estimated using the equation proposed by Burnham et al. (2015):
|
Eu3+ΣEu=11+10-0.25logfO2-6410/T-14.2Λ+10.1
|
Here fO2 is the oxygen fugacity, T is absolute temperature, and Λ is the optical basicity of the melt.
Plagioclase/melt partition coefficients for Eu2+ and Eu3+ have been determined by Wilke and Behrens (1999), based on data from experiments on tonalitic and basaltic compositions. These data and subsequent studies (e.g., Sun et al., 2017) indicate that mineral-melt partitioning has significant temperature and compositional dependence, but it is beyond the scope of the present reconnaissance treatment to attempt a full treatment of this complexity. For present purposes, the 750°C partitioning data of Wilke and Behrens (1999), with DEu2+ = 1.85 ± 0.17 and DEu3+ = 0.055, are assumed to be valid for all temperatures, melt compositions, and plagioclase compositions. The most important parameter is the ratio of partition coefficients DEu2+/DEu3+, because
|
(Eu2+Eu3+)Plag=(Eu2+Eu3+)Melt×DEu2+DEu3+
| (5) |
Calculated equilibrium plagioclase-melt fractionation factors are plotted against log fO2 in Fig. 2a. The parameters chosen for this calculation yield maximal fractionation factors that decline from 0.27‰ at 900 K (627°C) to 0.15‰ at 1600 K (1327°C). At each temperature, the fractionation is smallest at extreme fO2 conditions, because these are conditions where both plagioclase and the melt are dominated by the same oxidation state of europium. Peak fractionation occurs at fO2 very close to the Fayalite-Magnetite-Quartz buffer (FMQ, Fig. 2b), with shoulders extending roughly from the Iron-Wüstite buffer (IW) on the reducing side to the Magnetite-Hematite buffer (MH) on the oxidizing side. However, the position of the peak depends on the choice of plag-melt partition coefficients and optical basicity. Figs. 2c and 2d show the sensitivity of the fractionation to DEu3+/DEu2+ and optical basicity (Λ), respectively. Increasing contrast in partitioning of Eu2+ vs. Eu3+ increases the fractionation factor, and displaces the maximum fractionation to higher fO2. Increasing basicity does not directly affect the size of the maximum fractionation, but does shift the position of the peak to lower fO2, whereas decreasing basicity increases the fO2 of the peak fractionation.

Given the strong control of the oxidation state of europium on the plagioclase-melt fractionation, and the weak influence of other structural factors, a reasonable approximation is that the fractionation is simply proportional to the difference in the fraction of total europium as Eu2+ in each phase:
|
ln α153–151(Plag-Melt)≈KT[Eu2+Eu2++Eu3+Plag-(Eu2+Eu2++Eu3+)Melt]
| (6) |
Here T is absolute temperature and K ≈ 0.33 ± 0.06 K is a constant of proportionality such that K/T is nearly identical to the nuclear volume component of fractionation between Eu2+ in plagioclase and Eu3+ in melt.
The overall trend vs. fO2, in which the maximum isotopic fractionation occurs while Eu3+ is still the most abundant europium oxidation state in the melt, is distinct from bulk europium partitioning into plagioclase. For elemental partitioning, the fractionation is expected to reach a plateau as fO2 decreases, allowing formation of europium abundance anomalies even when Eu2+ is the major component of the melt’s europium reservoir. This suggests that igneous rocks that evolve europium anomalies through plagioclase fractionation will display a spectrum of correlations between Eu/Eu* and 153Eu/151Eu. Under relatively oxidizing conditions (defined here as those where Eu3+ is much more abundant in the melt than Eu2+), elemental and isotopic fractionation should track each other closely. At moderately lower fO2 (where Eu3+ and Eu2+ are subequal in the melt), the correlation will persist but 153Eu/151Eu will vary less as Eu/Eu* changes. At extremely low fO2, where Eu2+ dominates the melt, 153Eu/151Eu may be nearly invariant (at least with respect to closed-system fractionation) and decoupled from Eu/Eu*. This may partly explain the scatter in 153Eu/151Eu relative to Eu/Eu* observed by Lee and Tanaka (2021).
Equilibrium isotopic fractionation in cerium and other lanthanide elements
Two factors combine to make 153Eu/151Eu the most promising lanthanide system for finding significant equilibrium isotope fractionation effects in natural materials: 1) the importance of different oxidation states (Eu2+ and Eu3+), and 2) the large difference in nuclear charge radii between the two stable isotopes. Factor (1) is not unique to europium, as cerium takes on 3+ and 4+ charges in terrestrial environments and other REE’s can occur as +2-bearing molecules or neutral atomic species in the reducing, low-pressure conditions of the solar nebula (e.g., Ireland and Fegley, 2000; Hu et al., 2021). While the contrast in charge radii between 151Eu and 153Eu is uniquely large (on a per neutron basis), significant variation is common for lanthanides with multiple naturally occurring isotopes. Collectively, these suggest that measurable, potentially useful (if smaller) fractionations may be found in other REE elements in appropriate geochemical and cosmochemical contexts. After europium, cerium would appear to be the next most promising isotopic system, and high precision measurements of cerium stable isotope abundance variation have recently begun to be published (e.g., Nakada et al., 2013; Nakada et al., 2017; Hu et al., 2021).
One interesting aspect of the cerium isotope system is that one (142Ce) of the four naturally occurring cerium isotopes has a nuclear charge radius that contrasts substantially with the others (136Ce, 138Ce, 140Ce), with δ<r2> ≈ –0.031, –0.033, and +0.281 fm2 for the 136Ce–140Ce, 138Ce–140Ce, and 142Ce–140Ce pairs, respectively (Angeli and Marinova, 2013). This indicates that the nuclear volume effect will be distinguishable from both equilibrium and disequilibrium mass-dependent fractionation by the magnitude of e.g. 142Ce/140Ce fractionation relative to any pairs without the 142Ce isotope. This may be complicated somewhat by daughter product accumulation in the radiogenic 138La-138Ce system in natural materials, possibly requiring precise measurements of the rarer 136Ce isotope. Modern, high precision studies reporting both 142Ce/140Ce and 136Ce/140Ce variations do not yet seem to be available in the literature.
Equilibrium 142Ce/140Ce/138Ce/136Ce fractionations can be estimated using similar techniques to those used here for 153Eu/151Eu. Unfortunately, none of the cerium isotopes has a useful database of measured Mössbauer isomer shifts. However, the 145.4 keV Mössbauer transition in 141Pr has been studied (Groves et al., 1973; Bent et al., 1971), with results showing a reasonably consistent offset of ~0.7–1 mm/sec (0.03–0.05 J/mol) between Pr4+-bearing crystals such as PrO2 and Pr3+-bearing crystals such as PrPO4 and Pr2O3. δ<r2> for the 141Pr-Mössbauer transition is estimated to be 0.012–0.014 fm2 (Kapfhammer et al., 1971; Groves et al., 1973; Kalvius and Shenoy, 1974). Taking 0.013 ± 0.001 fm2 as an average value, and assuming identical electron densities for cerium-bearing species relative to their praseodymium-bearing analogues, the (142Ce–140Ce)/141Pr-isomer shift scale factor is approximately 0.281/0.013 = 21 ± 2. With this scale factor, a 1 mm/sec Pr3+/Pr4+ isomer shift implies a 142Ce vs. 140Ce nuclear volume effect of 1 ± 0.1 J/mol, enough to drive 0.3–0.4‰ fractionation at 298.15 K and 0.1‰ at 1000 K. Relativistic all-electron quantum mechanical calculations on Ce-ions and small molecules also indicate a 142Ce–140Ce field shift of ~1 J/mol between Ce3+ and Ce4+ species. As with 153Eu/151Eu, nuclear volume fractionation favors low 142Ce/140Ce in more oxidized (Ce4+-bearing) species. The corresponding fractionations for the 138Ce/140Ce and 136Ce/140Ce pairs are only 0.05‰ at 298.15 K and 0.01‰ at 1000 K, in the opposite direction. Nuclear volume fractionation of 142Ce/140Ce is thus expected to be less pronounced than the nuclear volume effect in europium, but still large enough to be potentially measurable.
Mass dependent cerium-isotope fractionation can be estimated using phonons calculated with DFT-based methods as described above. The resulting force constant sum for CePO4-monazite is 502 N/m, and for CeO2-ceria (cerianite-Ce) it is 664 N/m. Previous theoretical estimates of 142Ce/140Ce fractionations in solvated Ce3+-bearing aqueous complexes (Nakada et al., 2017) yield results similar to the present CePO4 model. For the CeO2-ceria – CePO4-monazite pair, mass-dependent 142Ce/140Ce fractionation is predicted to be roughly +0.3‰ at 298.5 K and +0.02‰ at 1000 K (Table 5, Fig. 3), favoring high 142Ce/140Ce in CeO2-ceria and approximately cancelling the nuclear volume effect at 298.15 K.
Table 5.
Equilibrium
xxxCe/
140Ce fractionation factors in CeO
2-ceria relative to CePO
4-monazite
| Isotope ratio |
103ln αNV |
103ln αMD |
Total 103ln α |
| 298.15 K |
1000 K |
1500 K |
298.15 K |
1000 K |
1500 K |
298.15 K |
1000 K |
1500 K |
| 142Ce/140Ce |
–0.29 |
–0.09 |
–0.06 |
0.27 |
0.024 |
0.011 |
–0.02 |
–0.06 |
–0.05 |
| 138Ce/140Ce |
0.03 |
0.01 |
0.01 |
–0.28 |
–0.024 |
–0.011 |
–0.24 |
–0.01 |
0.00 |
| 136Ce/140Ce |
0.03 |
0.01 |
0.01 |
–0.56 |
–0.050 |
–0.022 |
–0.53 |
–0.04 |
–0.02 |
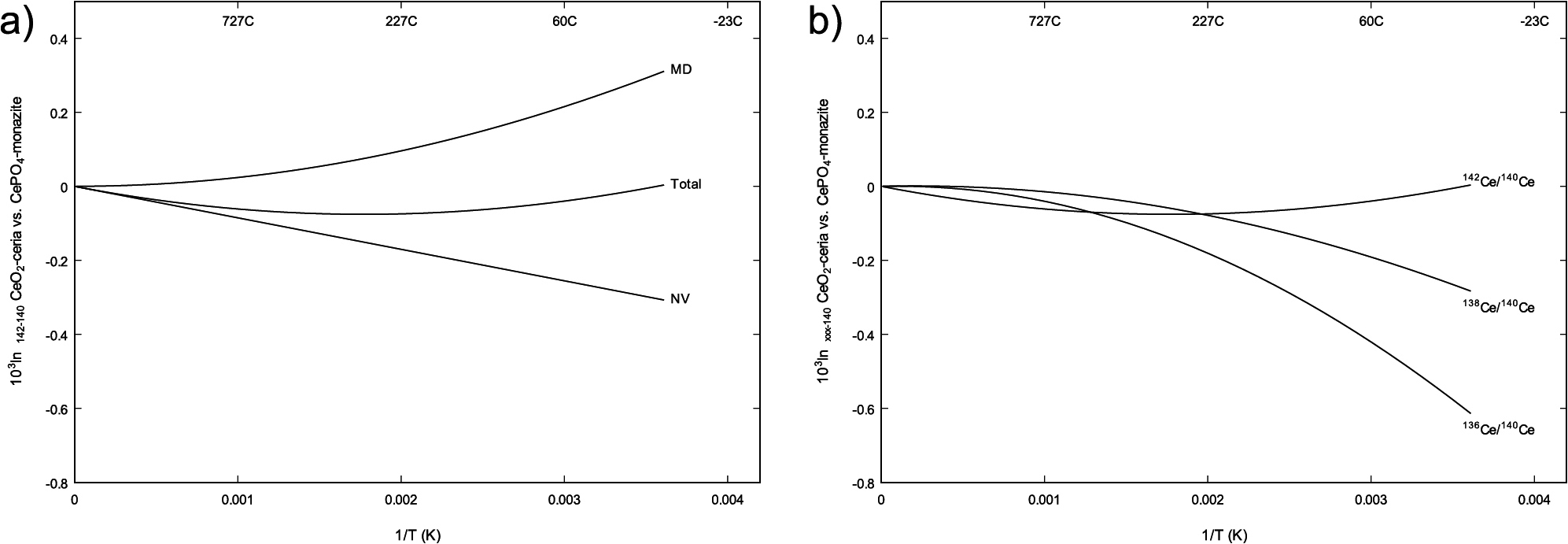
The total 142Ce/140Ce fractionation is predicted be subtle and have unusual temperature dependence, reaching a minimum of about –0.08‰ at hydrothermal temperatures (~550 K), and grading towards no fractionation at both higher and lower temperatures. 136Ce/140Ce and 138Ce/140Ce, in contrast, show the familiar approximate 1/T2 proportionality. In the case of 136Ce/140Ce, the more massive isotope is favored in Ce4+-bearing CeO2 by 0.6‰ at 298.15 K and 0.04‰ at 1000 K. Although these estimates are crude, they suggest that combined 142Ce/140Ce and 136Ce/140Ce measurements will yield more information than a single stable isotope ratio.
The remaining lanthanide group elements with at least two naturally occurring long-lived isotopes (La, Nd, Sm, Gd, Dy, Er, Yb, and Lu) are all dominantly found in the +3 oxidation state in Earth and planetary materials. For europium isotopes, the predicted equilibrium fractionations among 3+-bearing species are much smaller than fractionations between 2+ and 3+ species. This suggests small fractionations for the non-redox rare earths as a group. Some variation in measured force constants is permitted by measured force constants among Eu3+-bearing species, and among Dy3+-bearing species, though measurement uncertainties are similar in magnitude to the apparent variability (Hu, 2021; Hu et al., 2023). There is also a modest range of reported 151Eu-Mössbauer isomer shifts in Eu3+-bearing species, i.e. the roughly 1 mm/sec difference between Eu2O3 and Eu2(CO3)3, which scales to 0.2 J/mol for the 153Eu/151Eu nuclear volume shift. Using this range in isomer shifts as a guideline, and (very crudely) assuming fixed electronic structures across the entire lanthanide series, it is possible to obtain a ballpark estimate of plausible nuclear volume fractionations. In this case the isotope shift/151Eu isomer shift scale factor calculation is modified by simply inserting the most disparate nuclear radii for naturally occurring isotopes of each element (Table 6). 156Dy and 158Dy, two very rare light isotopes of dysprosium, are omitted from this treatment out of concern that they are difficult to measure to high precision, relative to the much more common 160Dy–164Dy isotopes. The estimated maximal nuclear volume component of non-redox fractionation is 0.1–0.2‰ at 298.15 K for Nd, Sm, and Gd, which have the largest range of nuclear charge radii (e.g., Angeli and Marinova, 2013); the calculated range is smaller than 0.1‰ for the other elements. Hu et al. (2021), Hu (2021), and Hu et al., (2023) also predict little mass-dependent fractionation for these elements.
Table 6.
Estimated maximal nuclear volume fractionation among REE
3+-bearing crystals for lanthanide elements, based on
151Eu-Mossubauer isomer shifts and published nuclear charge radii (
Angeli and Marinova, 2013)
| Element |
Smallest nucleus |
Largest nucleus |
δ<r2> fm2 |
Scaled REE3+ 103ln αNV |
| 298.15 K |
1000 K |
| La |
138 |
139 |
0.067 |
0.01 |
0.00 |
| Ce |
136 |
142 |
0.317 |
0.04 |
0.01 |
| Nd |
142 |
150 |
1.282 |
0.17 |
0.05 |
| Sm |
144 |
154 |
1.481 |
0.20 |
0.06 |
| Eu |
151 |
153 |
0.602 |
0.08 |
0.02 |
| Gd |
152 |
160 |
1.01 |
0.13 |
0.04 |
| Dy |
160† |
164 |
0.285 |
0.04 |
0.01 |
| Er |
162 |
170 |
0.551 |
0.07 |
0.02 |
| Yb |
168 |
176 |
0.541 |
0.07 |
0.02 |
| Lu |
175 |
176 |
0.042 |
0.01 |
0.00 |
†156Dy and 158Dy are very rare.
Conclusions
Theoretical estimates based on 151Eu-Mössbauer isomer shifts and force constants predict measurable netequilibrium 153Eu/151Eu fractionations of ~0.5–1‰ at 298.15 K and ~0.3‰ at 1000 K whenever Eu3+- and Eu2+-bearing phases equilibrate, favoring high 153Eu/151Eu in Eu2+-bearing phases. The fractionation is driven primarily by the nuclear volume component of the field shift effect, enhanced in europium by the large difference in charge radius between 151Eu and 153Eu. Mass dependent fractionation is usually secondary or subequal, opposing the dominant nuclear volume component. The predicted fractionations are qualitatively consistent with laboratory column-exchange experiments, and with measurements showing low 153Eu/151Eu in igneous rocks with negative europium anomalies. Simplified modeling of redox partitioning of europium in plagioclase relative to silicate melts indicates that 153Eu/151Eu fractionation will be less pronounced at extreme high and low fO2, and will reach a maximum of ~0.2–0.3‰ (at typical magmatic temperatures) at intermediate fO2 where Eu2+/(Eu2+ + Eu3+) in the melt is much lower than coexisting plagioclase. Because 136Ce, 138Ce, and 140Ce have very similar nuclear charge radii, their fractionation from each other will typically be dominantly mass dependent. In contrast, the larger radius of 142Ce means that nuclear volume fractionation can be significant. In the case of 142Ce/140Ce in the CeO2 (Ce4+) vs. CePO4 (Ce3+) equilibrium, nuclear volume and mass dependent fractionations nearly cancel each other out. Nuclear volume fractionation in other lanthanide elements is expected to be more subtle than in the redox-active Eu and Ce systems, but may still be measurable, especially for Nd, Sm, and Gd, which have isotopes spanning a large range of nuclear charge radii.
Acknowledgments
The author is grateful for helpful comments from Minori Abe, an additional anonymous reviewer, and associate editor Takeshi Ohno. This research is supported by US National Science Foundation grants EAR1530306 and EAR1524811, as well as funding from the UCLA Division of Physical Sciences.
References
- Abe, M., Suzuki, T., Fujii, Y. and Hada, M. (2008) An ab initio study based on a finite nucleus model for isotope fractionation in the U(III)-U(IV) exchange reaction system. J. Chem. Phys. 128, 144309. https://doi.org/10.1063/1.2898541
- Ahmad, S. A., Klempt, W., Ekström, C., Neugart, R. and Wendt, K. (1985) Nuclear spins, moments, and changes of the mean square charge radii of 140–153Eu. Z. Phys. A – At. Nucl. 321, 35–45. https://doi.org/10.1007/BF01411941
- An, J. M., Barabash, S. V., Ozolins, V., van Schilfgaarde, M. and Belashchenko, K. D. (2011) First-principles study of phase stability of Gd-doped EuO and EuS. Phys. Rev. B 83, 064105. https://doi.org/10.1103/PhysRevB.83.064105
- Angeli, I. and Marinova, K. P. (2013) Table of experimental nuclear ground state charge radii: An update. At. Data Nucl. Data Tables 99, 69–95. https://doi.org/10.1016/j.adt.2011.12.006
- Baggio-Saitovitch, E., Litterst, F. J. and Micklitz, H. (1976) Mössbauer absorption spectrum of 151EuCl2 molecules isolatedin an argon matrix. J. Phys. Colloques 37, C6-529–C6-530. https://doi.org/10.1051/jphyscol:19766111
- Barton, C. M. P. and Greenwood, N. N. (1973) Europium-151 Mössbauer spectroscopy. Mössbauer Effect Data Index (Stevens, J. G. and Stevens, V. E., eds.), 395–446, Springer.
- Bent, M. F., Cook, D. D. and Persson, B. I. (1971) Nuclear hyperfine interaction in 141Pr. Phys. Rev. C 3, 1419–1429. https://doi.org/10.1103/PhysRevC.3.1419
- Bessas, D., Rushchanskii, K. Z., Kachlik, M., Disch, S., Gourdon, O., Bednarcik, J., Maca, K., Sergueev, I., Kamba, S., Lezaic, M. and Hermann, R. P. (2013) Lattice instabilities in bulk EuTiO3. Phys. Rev. B 88, 144308. https://doi.org/10.1103/PhysRevB.88.144308
- Bigeleisen, J. (1996a) Nuclear size and shape effects in chemical reactions. Isotope chemistry of heavy elements. J. Am. Chem. Soc. 118, 3676–3680. https://doi.org/10.1021/ja954076k
- Bigeleisen, J. (1996b) Temperature dependence of the isotope chemistry of the heavy elements. Proc. Natl. Acad. Sci. USA 93, 9393–9396. https://doi.org/10.1073/pnas.93.18.9393
- Bigeleisen, J. and Mayer, M. G. (1947) Calculation of equilibrium constants for isotopic exchange reactions. J. Chem. Phys. 15, 261–267. https://doi.org/10.1063/1.1746492
- Brix, P., Hüfner, S., Kienle, P. and Quitman, D. (1964) Isomer shift on Eu151. Phys. Lett. 13, 140–142. https://doi.org/10.1016/0031-9163(64)90698-5
- Burnham, A. D., Berry, A. J., Halse, H. R., Schofield, P. F., Cibin, G. and Mosselmans, J. F. W. (2015) The oxidation state of europium in silicate melts as a function of oxygen fugacity, composition and temperature. Chem. Geol. 411, 248–259. https://doi.org/10.1016/j.chemgeo.2015.07.002
- Clabau, F., Garcia, A., Bonville, P., Gonbeau, D., Le Mercier, T., Deniard, P. and Jobic, S. (2008) Fluorescence and phosphorescence properties of the low temperature forms of the MAl2Si2O8:Eu2+ (M = Ca, Sr, Ba) compounds. J. Sol. State Chem. 181, 1456–1461. https://doi.org/10.1016/j.jssc.2008.03.011
- Conchas, G., Congiu, F., Muntoni, C., Bettinelli, M. and Speghini, A. (1996) Hyperfine interactions at europium sites in oxide glasses. Phys. Rev. B 53, 6197–6202. https://doi.org/10.1103/PhysRevB.53.6197
- Davydov, A., Sanna, A. and Concas, G. (2022) Y2O3: Eu and the Mössbauer isomer shift coefficient of Eu compounds from ab-initio simulations. J. Phys. Condens. Matter 34, 075502. https://doi.org/10.1088/1361-648X/ac37d8
- de Vries, J. W. C., Thiel, R. C. and Buschow, K. H. J. (1985) On the interpretation of Mössbauer isomer shifts. J. Phys. F: Met. Phys. 15, 2403–2408. https://doi.org/10.1088/0305-4608/15/11/021
- Dolg, M., Stoll, H. and Preuss, H. (1989) Energy-adjusted ab initio pseudopotentials for the rare earth elements. J. Chem. Phys. 90, 1730–1734. https://doi.org/10.1063/1.456066
- Elcombe, M. M. and Hulston, J. R. (1975) Calculation of sulphur isotope fractionation between sphalerite and galena using lattice dynamics. Earth Planet. Sci. Lett. 28, 172–180. https://doi.org/10.1016/0012-821X(75)90224-1
- Foit, F. F. and Peacor, D. R. (1973) The anorthite crystal structure at 410 and 830°C. Am. Min. 58, 665–675.
- Fricke, G. and Heilig, K. (2004) Nuclear charge radii·63-Eu Europium. Nuclear Charge Radii (Schopper, H., ed.), Landolt-Börnstein - Group I Elementary Particles, Nuclei and Atoms, 1–8, Springer-Verlag. https://doi.org/10.1007/10856314_65
- Frost, B. R. (1991) Chapter 1. Introduction to oxygen fugacity and its petrologic importance. Oxide Minerals: Petrologic and Magnetic Significance (Donald H. Lindsley, ed.), 1–10, De Gruyter. https://doi.org/10.1515/9781501508684-004
- Fujita, K., Tanaka, K., Hirao, K. and Soga, N. (1998) Mössbauer spectroscopy of borate glasses containing divalent europium ions. J. Am. Ceram. Soc. 81, 1845–1851. https://doi.org/10.1111/j.1151-2916.1998.tb02556.x
- Gerth, G., Kienle, P. and Luchner, L. (1968) Chemical effects on the isomer shift in 151Eu. Phys. Lett. A 27, 557–558. https://doi.org/10.1016/0375-9601(68)90919-5
- Giannozzi, P., Baroni, S., Bonini, N., Calandra, M., Car, R., Cavazzoni, C., Ceresoli, D., Chiarotti, G. L., Cococcioni, M., Dabo, I., Dal Corso, A., de Gironcoli, S., Fabris, S., Fratesi, G., Gebauer, R., Gerstmann, U., Gougoussis, C., Kokalj, A., Lazzeri, M., Martin-Samos, L., Marzari, N., Mauri, F., Mazzarello, R., Paolini, S., Pasquarello, A., Paulatto, L., Sbraccia, C., Scandolo, S., Sclauzero, G., Seitsonen, A. P., Smogunov, A., Umari, P. and Wentzcovitch, R. M. (2009) QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys.: Condens. Matter 21, 395502. https://doi.org/10.1088/0953-8984/21/39/395502
- Golbs, S., Schappacher, F. M., Pöttgen, R., Cardoso-Gil, R., Ormeci, A., Schwarz, U., Schnelle, W., Grin, Y. and Schmidt, M. (2013) Europium phosphate, europium arsenate, and europium antimonate – Correlation of crystal structure and physical properties. Z. Anorg. Allg. Chem. 639, 2139–2148. https://doi.org/10.1002/zaac.201300285
- Gomes, A. S. P., Saue, T., Visscher, L., Jensen, H. J. Aa., Bast, R., Aucar, I. A., Bakken, V., Dyall, K. G., Dubillard, S., Ekström, U., Eliav, E., Enevoldsen, T., Faßhauer, E., Fleig, T., Fossgaard, O., Halbert, L., Hedegård, E. D., Helgaker, T., Helmich-Paris, B., Henriksson, J., Iliaš, M., Jacob, Ch. R., Knecht, S., Komorovský, S., Kullie, O., Lærdahl, J. K., Larsen, C. V., Lee, Y. S., Nataraj, H. S., Nayak, M. K., Norman, P., Olejniczak, G., Olsen, J., Olsen, J. M. H., Park, Y. C., Pedersen, J. K., Pernpointner, M., Di Remigio, R., Ruud, K., Sałek, P., Schimmelpfennig, B., Senjean, B., Shee, A., Sikkema, J., Thorvaldsen, A. J., Thyssen, J., van Stralen, J., Vidal, M. L., Villaume, S., Visser, O., Winther, T. and Yamamoto, S. (2019) DIRAC, a relativistic ab initio electronic structure program, Release DIRAC19 (http://diracprogram.org). https://doi.org/10.5281/zenodo.3572669
- Gomes, A. S. P., Dyall, K. G. and Visscher, L. (2010) Relativistic double-zeta, triple-zeta, and quadruple-zeta basis sets for the lanthanides La–Lu. Theor. Chem. Acc. 127, 369–381. https://doi.org/10.1007/s00214-009-0725-7
- Grandjean, F. and Long, G. J. (1989) Mössbauer spectroscopy of europium-containing compounds. Mössbauer Spectroscopy Applied to Inorganic Chemistry (Long, G. J. and Grandjean, F., eds.) 513–598, Springer. https://doi.org/10.1007/978-1-4899-2289-2_11
- Groves, J. L., DePasquali, G. and Debrunner, P. G. (1973) Mössbauer scattering in 14159Pr. Phys. Rev. B 7, 1974–1984. https://doi.org/10.1103/PhysRevB.7.1974
- Gulde, R., Pollak, P. and Weigend, F. (2012) Error-balanced segmented contracted basis sets of double-z to quadruple-ζ valence quality for the lanthanides. J. Chem. Theory Comput. 8, 4062–4068. https://doi.org/10.1021/ct300302u
- Heilig, K. and Steudel, A. (1974) Changes in mean-square nuclear charge radii from optical isotope shifts. At. Data Nucl. Data Tables 14, 613–638. https://doi.org/10.1016/S0092-640X(74)80006-9
- Hu, J. (2021) Rare earth isotope variations and implications for the evolution of the early solar system. PhD Thesis, U. Chicago., 199 pp. https://doi.org/10.6082/uchicago.3391
- Hu, J. Y., Dauphas, N., Tissot, F. L. H., Yokochi, R., Ireland, T. J., Zhang, Z., Davis, A. M., Ciesla, F. J., Grossman, L., Charlier, B. L. A., Roskosz, M., Alp, E. E., Hu, M. Y. and Zhao, J. (2021) Heating events in the nascent solar system recorded by rare earth element isotopic fractionation in refractory inclusions. Sci. Adv. 7, eabc2962. https://doi.org/10.1126/sciadv.abc2962
- Hu, J. Y., Dauphas, N., Nie, N. X., Roskosz, M., Chen, X., Heard, A. W., Zhang, Z. J., Zeng, H., Alp, E. E., Hu, M. Y. and Zhao, J. (2023) Equilibrium fractionation of REE isotopes in nature: Insights from NRIXS and DFT + U studies of Eu and Dy phonon density of states. Geochim. Cosmochim. Acta 348, 323–339. https://doi.org/10.1016/j.gca.2023.03.002
- Ireland, T. R. and Fegley, Jr. B. (2000) The solar system’s earliest chemistry: Systematics of refractory inclusions. Int. Geol. Rev. 42, 865–894. https://doi.org/10.1080/00206810009465116
- Ismail, I., Nomura, M. and Fujii, Y. (1998) Isotope effects in Eu(II)/Eu(III) electron exchange system observed by using cation exchange chromatography. J. Nucl. Sci. Technol. 35, 801–807. https://doi.org/10.1080/18811248.1998.9733947
- Jenden, C. M. and Lyle, S. J. (1982) A Mössbauer spectroscopic study of the iodides of europium. J. Chem. Soc., Dalton Trans. 2409–2414. https://doi.org/10.1039/DT9820002409
- Kalvius, G. M. and Shenoy, G. K. (1974) Changes in mean-square nuclear charge radii from mössbauer isomer shifts. At. Data Nucl. Data Tables 14, 639–653. https://doi.org/10.1016/S0092-640X(74)80007-0
- Kapfhammer, W. H., Maurer, W., Wagner, F. E. and Kienle, P. (1971) Mössbauer isomer shifts and hyperfine splitting of the 145.4 keV γ-rays of 141Pr. Z. Naturforsch. A 26, 357–367. https://doi.org/10.1515/zna-1971-0305
- Kimata, M. (1988) The crystal structure of non-stoichiometric Eu- anorthite: An explanation of the Eu-positive anomaly. Mineral. Mag. 52, 257–265. https://doi.org/10.1180/minmag.1988.052.365.13
- Klencsár, Z., Wang, J., Ge, R., Zhou, W., Liu, D., Rykov, A. I. and Zhang, T. (2021) The new WEB-accessible online database of the Mössbauer effect data center. Hyperfine Interact. 242, 15. https://doi.org/10.1007/s10751-021-01733-7
- Klobes, B., Arinicheva, Y., Neumeier, S., Simon, R. E., Jafari, A., Bosbach, D. and Hermann R. P. (2016) Quadrupole splitting and Eu partial lattice dynamics in europium orthophosphate EuPO4. Hyperfine Interact. 237, 31. https://doi.org/10.1007/s10751-016-1211-y
- Lazewski, J., Sternik, M., Jochym, P. T., Kalt, J., Stankov, S., Chumakov, A. I., Göttlicher, J., Rüffer, R., Baumbach, T. and Piekarz, P. (2021) Lattice dynamics and structural phase transitions in Eu2O3. Inorg. Chem. 60, 9571–9579. https://doi.org/10.1021/acs.inorgchem.1c00708
- Lee, S.-G. and Tanaka, T. (2021) Europium isotope fractionation in highly fractionated igneous rocks with large Eu negative anomaly. Geochem. J. 55, e9–e17. https://doi.org/10.2343/geochemj.2.0631
- Litterst, F. J., Michlitz, H. and Schichl, A. (1976) Mössbauer absorption spectrum of 151Eu atoms isolated in an argon matrix. Phys. Lett. A 57, 70–72. https://doi.org/10.1016/0375-9601(76)90458-8
- Lodders, K. (2003) Solar system abundances and condensation temperatures of the elements. ApJ. 591, 1220–1247. https://doi.org/10.1086/375492
- Lodders, K. and Fegley, Jr. B. (1993) Lanthanide and actinide chemistry at high C/O ratios in the solar nebula. Earth Planet. Sci. Lett. 117, 125–145. https://doi.org/10.1016/0012-821X(93)90122-P
- Miller, C. A., Peucker-Ehrenbrink, B. and Schauble, E. A. (2015) Theoretical modelling of rhenium isotope fractionation, natural variations across a black shale weathering profile, and potential as a paleoredox proxy. Earth Planet. Sci. Lett. 430, 339–348. https://doi.org/10.1016/j.epsl.2015.08.008
- Montano, P. A. (1982) Matrix isolation study of 151Eu atoms and molecules. J. Phys. C: Solid State Phys. 15, 565–572. https://doi.org/10.1088/0022-3719/15/3/020
- Moolenaar, A. A., Gubbens, P. C. M., van Loef, J. J., Menken, M. J. V. and Menovsky, A. A. (1996) PrBa2Cu3O7 investigated by 141Pr Mössbauer spectroscopy. Physica C 267, 279–292. https://doi.org/10.1016/0921-4534(96)00343-7
- Nakada, R., Takahashi, Y. and Tanimizu, M. (2013) Isotopic and speciation study on cerium during its solid—Water distribution with implication for Ce stable isotope as a paleo-redox proxy. Geochim. Cosmochim. Acta 103, 49–62. https://doi.org/10.1016/j.gca.2012.10.045
- Nakada, R., Tanaka, M., Tanimizu, M. and Takahashi, Y. (2017) Aqueous speciation is likely to control the stable isotopic fractionation of cerium at varying pH. Geochim. Cosmochim. Acta 218, 273–290. https://doi.org/10.1016/j.gca.2017.09.019
- Nakanishi, T. and Tanabe, S. (2009) Quantitative analysis of Eu (II)/Eu (III) ratio in alkaline-earth silicate phosphors by 151Eu Mössbauer spectroscopy. IOP Conf. Ser.: Mater. Sci. Eng. 1, 012027. https://doi.org/10.1088/1757-8981/1/1/012027
- Nemov, S. A., Marchenko, A. V., Seregin, P. P. and Tomil’tsev, E. A. (2007) Europium(II) in Glasses of the Al2O3–SiO2–MnO–Eu2O3 System. Glass Phys. Chem. 33, 658–660. https://doi.org/10.1134/S1087659607060181
- Nomura, M., Higuchi, N. and Fujii, Y. (1996) Mass dependence of uranium isotope effects in the U(IV)-U(VI) exchange reaction. J. Am. Chem. Soc. 118, 9127–9130. https://doi.org/10.1021/ja954075s
- Nozik, A. J. (1967) Mössbauer resonance studies of ions in ice. PhD Thesis, Yale U., 216 pp.
- Perdew, J. P., Burke, K. and Ernzerhof, M. (1996) Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. https://doi.org/10.1103/PhysRevLett.77.3865
- Pradip, R., Piekarz, P., Merkel, D. G., Kalt, J., Waller, O., Chumakov, A. I., Rüffer, R., Oles, A. M., Parlinski, K., Baumbach, T. and Stankov, S. (2019) Phonon confinement and spin-phonon coupling in tensile-strained ultrathin EuO films. Nanoscale 11, 10968–10976. https://doi.org/10.1039/C9NR01931F
- Ryl’nikov, A. S., Egorov, A. I., Ivanov, G. A., Marushenko, V. I.., Mezentsev, A. F., Smirnov, A. I., Sumbaev, O. I. and Fedorov, V. V. (1973) Hyperfine broadening of the X-ray lines. Soviet Phys. JETP 36, 27–32 (translated from 1972, Zh. Eksp. Teor. Fiz. 63, 53–56, in Russian).
- Schauble, E. A. (2007) Role of nuclear volume in driving equilibrium stable isotope fractionation of mercury, thallium, and other very heavy elements. Geochim. Cosmochim. Acta 71, 2170–2189. https://doi.org/10.1016/j.gca.2007.02.004
- Schauble, E. A. (2013) Modeling nuclear volume isotope effects in crystals. Proc. Natl. Acad. Sci. USA 110, 17714–17719. https://doi.org/10.1073/pnas.1216216110
- Stankov, S., Piekarz, P., Oles, A. M., Parlinski, K. and Rüffer, R. (2008) Lattice dynamics of Eu from nuclear inelastic scattering and first principles calculations. Phys. Rev. B 78, 180301. https://doi.org/10.1103/PhysRevB.78.180301
- Stankov, S., Merkel, D. G., Kalt, J., Göttliher, J., Lazewski, J., Sternik, M., Jochym, P. T., Piekarz, P., Baumbach, T., Chumakov, A. I. and Rüffer, R. (2022) Phonon confinement and interface lattice dynamics of ultrathin high-k rare earth sesquioxide films: The case of Eu2O3 on YSZ(001). Nanoscale Adv. 4, 19–25. https://doi.org/10.1039/D1NA00728A
- Sun, C., Graff, M. and Liang, Y. (2017) Trace element partitioning between plagioclase and silicate melt: The importance of temperature and plagioclase composition, with implications for terrestrial and lunar magmatism. Geochim. Cosmochim. Acta 206, 273–295. https://doi.org/10.1016/j.gca.2017.03.003
- Tanabe, S., Hirao, K. and Soga, N. (1989) Mössbauer spectroscopy of 151Eu in oxide crystals and glasses. J. Non-Cryst. Solids 113, 178–184. https://doi.org/10.1016/0022-3093(89)90009-4
- Tanabe, S., Hanada, T., Ohyagi, T. and Soga, N. (1993) Correlation between 151Eu Mössbauer isomer shift and Judd-Ofelt Ω6 parameter of Nd3+ ions in phosphate and silicate laser glasses. Phys. Rev. B 48, 10591–10594. https://doi.org/10.1103/physrevb.48.10591
- Tanaka, K., Fujita, K., Matsuoka, N., Hirao, K. and Soga, N. (1998) Large Faraday effect and local structure of alkali silicate glasses containing divalent europium ions. J. Mater. Res. 13, 1989–1995. https://doi.org/10.1557/JMR.1998.0279
- Tanaka, Y., Steffen, R. M., Shera, E. B., Reuter, W., Hoehn, M. V. and Zumbro, J. D. (1984) Measurement and analysis of the muonic × rays of 151Eu and 153Eu. Phys. Rev. C 29, 1897–1904. https://doi.org/10.1103/PhysRevC.29.1897
- Taylor, R. D. and Farrell, J. N. (1987) Mössbauer effect of europium metal under pressure. J. Applied Phys. 61, 3669–3670. https://doi.org/10.1063/1.338683
- Todoroki, S., Hirao, K. and Soga, N. (1993) A study of local structure around Eu3+ ions in oxide glasses using Mössbauer spectroscopy. Nucl. Instrum. Methods. Phys. Res. B 76, 76–77. https://doi.org/10.1016/0168-583X(93)95138-U
- Tong, W.-Y., Ding, H.-C., Gao, Y.-C., Gong, S.-J., Wan, X. and Duan, C.-G. (2014) Spin-dependent optical response of multiferroic EuO: First-principles DFT calculations. Phys. Rev. B 89, 064404. https://doi.org/10.1103/PhysRevB.89.064404
- Topsakal, M. and Wentzcovitch, R. M. (2014) Accurate projected augmented wave (PAW) datasets for rare-earth elements (RE = La–Lu). Comput. Mater. Sci. 95, 263–270. https://doi.org/10.1016/j.commatsci.2014.07.030
- Urey, H. C. (1947) The thermodynamic properties of isotopic substances. J. Chem. Soc. 562–581. https://doi.org/10.1039/JR9470000562
- Virgo, D., Seifert, F. A. and Mysen, B. O. (1981) The oxidation state of europium in albite and alkali-earth silicate glasses. Carnegie Institution of Washington Year Book 80, 313–316.
- Visser, O., Visscher, L., Aerts, P. J. C. and Nieuwpoort, W. C. (1992) Molecular open shell configuration interaction calculations using the Dirac-Coulomb Hamiltonian: The f6-manifold of an embedded EuO69– cluster. J. Chem. Phys. 96, 2910–2919. http://dx.doi.org/10.1063/1.461987
- Walter, H. K., Backe, H., Engfer, R., Kankeleit, E., Petitjean, C., Schneuwly, H. and Schröder, W. U. (1972) Muonic isomer shifts in 153Eu: A re-evaluation of Mössbauer isomer shifts in rare earths. Phys. Lett. B 38, 64–66. https://doi.org/10.1016/0370-2693(72)90739-3
- Weigend, F. and Ahlrichs, R. (2005) Balanced basis sets of split valence, triple zeta valence and quadruple zeta valenc quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305. https://doi.org/10.1039/B508541A
- Widanagamage, I. H., Schauble, E. A., Scher, H. D. and Griffith, E. M. (2014) Stable strontium isotope fractionation in synthetic barite. Geochim. Cosmochim. Acta 147, 58–75. https://doi.org/10.1016/j.gca.2014.10.004
- Wilke, M. and Behrens, H. (1999) The dependence of the partitioning of iron and europium between plagioclase and hydrous tonalitic melt on oxygen fugacity. Contrib. Mineral. Petrol. 137, 102–114. https://doi.org/10.1007/s004100050585
- Yamnova, N. A., Zubkova, N. V., Eremin, N. N., Zadov, A. E. and Gazeev, V. M. (2011) Crystal structure of larnite β-Ca2SiO4 and specific features of polymorphic transitions in dicalcium orthosilicate. Crystallogr. Rep. 56, 210–220. https://doi.org/10.1134/S1063774511020209
- Yang, S. and Liu, Y. (2015) Nuclear volume effects in equilibrium stable isotope fractionations of mercury, thallium and lead. Sci. Rep. 5: 12626. https://doi.org/10.1038/srep12626
- Zhang, H., Wang, J., Xu, G., Xu, Y. and Zhang, S.-C. (2014) Topological states in ferromagnetic CdO/EuO superlattices and quantum wells. Phys. Rev. Lett. 112, 096804. https://doi.org/10.1103/PhysRevLett.112.096804
- Zhang, Y. and Liu, Y. (2018) The theory of equilibrium isotope fractionations for gaseous molecules under super-cold conditions. Geochim. Cosmochim. Acta 238, 123–149. https://doi.org/10.1016/j.gca.2018.07.001

