Special reviews
Evolutionary and phylogeographic views on Mc1r and Asip variation in mammals
2013 年 88 巻 3 号 p. 155-164
詳細
2013 年 88 巻 3 号 p. 155-164
The purpose of this article is to provide basic knowledge about the Mc1r-Asip system that promotes the evolution of coat color in mammals, and to stimulate genetic, ecological, and phylogeographic studies focusing on color variation in natural populations. The topics reviewed herein include: the genetic system of the Mc1r and Asip genes related to phenotypic variation; the evolutionary implications of the genetic features recorded in their nucleotide sequences; and the validity of surveys in the wild of genetic variations in coat color, which would facilitate a better understanding of the genetic system, ecological meaning, natural history, and taxonomic reevaluation of species and local populations.
Coloration is one of the most variable morphological traits in a variety of animals, and there are three hypotheses for its function: concealment, communication, and regulation of physiological processes (Caro, 2005; Takahashi, 2013, Miyagi and Terai, 2013). In mammals, intense study of genetic systems of pigmentation has resulted in the discovery of more than 100 genes related to coat color determination (Barsh, 1996). These findings have helped to advance evolutionary studies on the body color variation observed among wild animals (Nachman et al., 2003; Hoekstra, 2006), and have revealed that two genes, Mc1r and Asip, encoding melanocortin-1 receptor and agouti signaling protein, respectively, are essential to coat color changes in wildlife. These genes play a major role in red-yellow (pheomelanin) and black-brown (eumelanin) melanization (Klungland et al., 1995; MacDougall-Shackleton et al., 2003) and single-site mutations can sometimes lead to significant changes in phenotype, making genes particularly susceptible to rapid evolutionary change (Hoekstra and Price, 2004). Hence, variation in gene sequences can be used to assess the adaptive evolution of coat color, as well as to infer phylogenetic and phylogeographic relationships among organisms. This review addresses the evolutionary nature of the two enigmatic genes Mc1r and Asip, as a step toward using the variation in these genes to better understand the evolutionary history of a given set of species.
The melanocyte is the only cell that produces pigments in mammals and provides the two pigments, eumelanin and pheomelanin (Fig. 1, A and B). MC1R is a G-protein-coupled receptor that is expressed primarily in melanocytes and plays important roles in determining the pigment type, responding to two ligands of signaling molecules, the α-melanocyte stimulating hormone (α-MSH) and ASIP. α-MSH functions as an agonist, activating cells brought about by an increase in the intracellular level of cAMP, leading to the production of eumelanin by melanocytes. ASIP is normally released from dermal papilla cells at the base of hair follicles (see Bradley and Mundy, 2008 for further details). The interaction between ASIP and MC1R reduces the level of cAMP, causing the cells to become inactive and to produce pheomelanins as a default state (see Schiaffino, 2010 for further details).
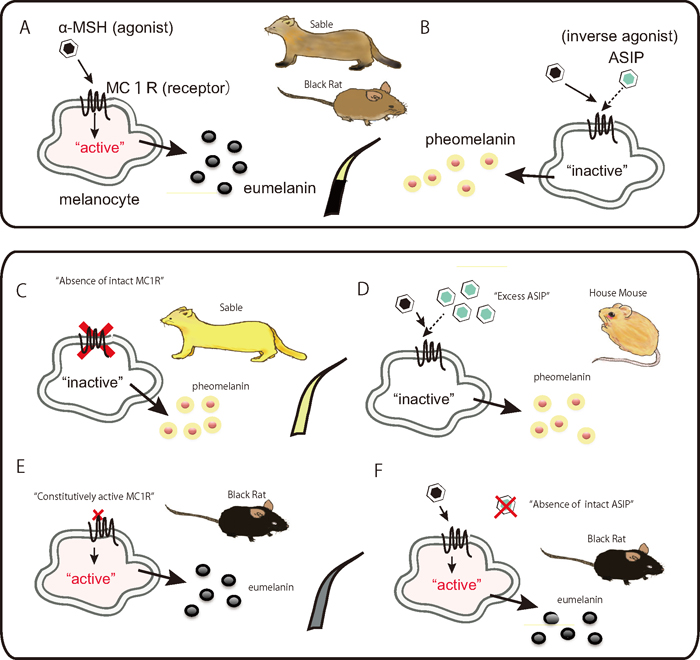
Schematic representation of the melanogenesis pathway in vertebrate melanocytes that produce eumelanins and pheomelanins (Schiaffino, 2010). The agonist, α-MSH activates melanocytes to generate eumelanins, brought by contraction to the cell-surface receptor MC1R, resulting in an elevated intracellular cAMP level (Bowers et al., 1997) (A). In contrast, the inverse agonist ASIP inactivates melanocytes to generate pheomelanins, leading to a reduction in cAMP levels (B). Typical wild-type hairs in rodents exhibit the agouti color pattern, in which single hairs have bands of light-colored pheomelanin and darker eumelanin. A loss-of-function mutation on the MC1R gene leads to complete loss of the regulatory system and brings melanocytes into the default state, pheomelanin production (C). An Asip allele that increases the level of ASIP expression is thought to counter the effect of the gain-of-function mutation of Mc1r, leading to pheomelanin production (D). A gain-of-function mutation on the MC1R gene constitutively activates melanocytes without contraction of the agonist α-MSH, causing the cells to produce eumelanin (E). A loss-of-function mutation in Asip causing depletion of intact inverse-agonist ligands makes the animal melanistic (F). ASIP: agouti signaling protein; α-MSH: alpha-melanocyte-stimulating hormone; MC1R: melanocortin 1 receptor; cAMP: cyclic adenosine monophosphate.
MC1R has seven transmembrane domains and a standard amino acid length of 317, although deleted versions exist in some species: 315 (genus Mus: Shimada et al., 2009) and 302 (Holarctic martens; Hosoda et al., 2005, Ogawa et al., unpublished; Fig. 2A). Mc1r is a single-exon gene and mutations that change its amino acids have the potential to change the affinity of the receptor for interacting with its ligands and G-protein. Asip consists of a peptide of 131 amino acids with a conserved cysteine-rich region near the C-terminus and putative signal peptide at the N-terminus (Bultman et al., 1992; Fig. 2). The cysteine-rich domain containing 10 cysteine residues is conserved across taxa (Dinulescu and Cone, 2000; Klovins and Schiöth, 2005) and is responsible for its biological activity as an inverse agonist of MC1R (Lu et al., 1994; Willard et al., 1995; Jackson et al., 2006). The ASIP C-terminal loop, a six amino acid segment closed by a final disulfide bond, is essential for high-affinity MC1R binding and inverse agonism and may interact with the first extracellular loop (EC1) of MC1R (Patel et al., 2010). Asip has several promoters with various functions. Promoters of transcription during the hair growth cycle produce a sub-apical yellow band on each black hair shaft, which is called the “agouti pattern” and a sort of wild type in rodents (Fig. 2B). A ventral-specific promoter is responsible for yellow pelage (Nadeau et al., 2007; Hubbard et al., 2010).
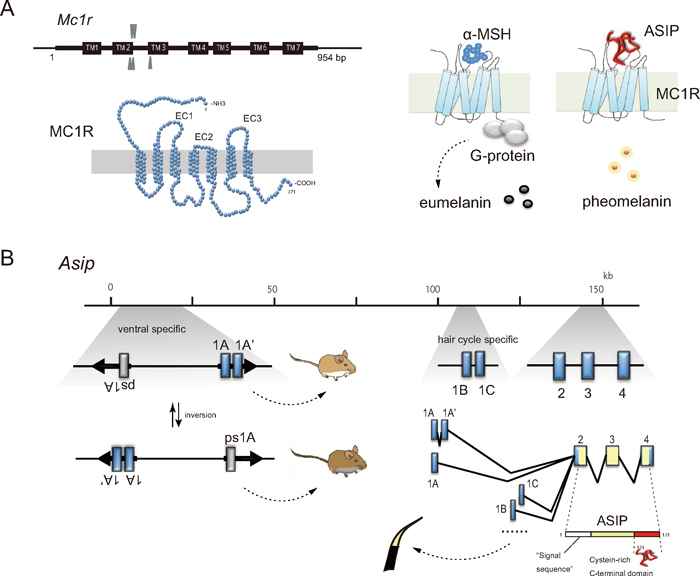
The mammalian Mc1r consists of a single exon with the most common length of 954 bp (A). The gene product MC1R comprises 317 amino acids with three extracellular loops (EC1, EC2, EC3). A conceptual presentation of the G-protein-coupled receptor MC1R in the plasma membrane with the characteristic seven α-helical transmembrane segments, being docked with the C-terminal functional domain of ASIP (Chai et al., 2003; Patel et al., 2010). Genomic organization of the ASIP gene represented by that of the house mouse M. musculus (Vrieling et al., 1994) is presented schematically with exons (blue boxes; 1A, 1A’, 1B, 1C, 2, 3 and 4) and alternative splicing patterns, in which the coding regions are marked with yellow color (B). Two regions of exon 1A (and/or 1A’) and exon 1B (or 1C) are known to be involved in the ventral specific and hair-cycle specific gene expression, respectively. Inverted repeats partly interrupted by unique sequences including exon 1A, exon 1A’, and pseudoexon 1A (ps1A) are indicated with arrows. The structure creates an interrupted palindrome of 22 kb in length and the repeats mediate intrachromosomal homologous recombination, which give rise to the inversed orientation of the 22-kb segment and generate allelic exchanges that are responsible for white and gray ventral colors in mice responding the position of active promoter (Chen et al., 1996).

Sables from Hokkaido, Japan, with a bright yellow winter coat (Photo taken by Yoshinobu Nishimura, February, 2012).
The coat color in mammals is determined by the density of melanin and the distribution of melanin types on individual hairs (Hoekstra, 2006). Extreme phenotypic changes in the hair color patterns associated with single mutations of Mc1r and Asip have been reported in a variety of animals. Such allelic variation in Mc1r and Asip is classified as either a loss-of-function or gain-of-function mutation, which results in solid hair color patterns with either red-yellow or black-brown (melanistic) pigments, respectively (Robbins et al., 1993).
Extreme yellow coat colors are caused by destructive Mc1r mutations (e.g., nonsense and frameshift mutations) that prevent the production of functional receptor molecules. The removal of intact MC1R from melanocytes is known to situate melanocytes in a default state, i.e., inactive state, resulting in biased generation of pheomelanin. Examples of this are observed in some bright-yellow sables in Hokkaido, Japan (Ogawa et al., unpublished; Figs. 1C and 3) and American black bear in North America (Ritland et al., 2001). The totally yellowish or white coat color is also known to be generated by the ubiquitous expression of the antagonist ASIP (Fig. 1D), and typical examples can be found among laboratory mice (Miller et al., 1993), domestic sheep (Norris and Whan, 2008), and domestic quail (Nadeau et al., 2008). It is worth to note that in combination with the Asip-Mc1r system, other coat color related gene, such as the tyrosinase gene (Ollmann et al., 1998) could be involved in determining the yellow or white hair color.
Mc1r and Asip are also the major genes responsible for the opposite peculiar phenotype, melanism. Modulated MC1R proteins that become constitutively active irrespective of the presence or absence of the signaling ligand molecules continue to produce eumelanin (Fig. 1E). The state of depletion of the antagonistic molecule ASIP results in the continuous activation of MC1R (Fig. 1F). Mc1r-included melanism has been described in laboratory and wild animals. The most well-known mutation is that from glutamate to lysine at position 94 (nucleotide position 280G > A) or its equivalent, which has been documented in a number of animals including mice (Robbins et al., 1993) and black rats (Kambe et al., 2011; Fig. 1E). The comparative mutation is associated with black plumage in domestic chickens (Takeuchi et al., 1996) and a passerine bird, the bananaquit (Coereba flaveola; Theron et al., 2001). In black rats, the commonly observed melanistic form is Mc1r-dependent (280G > A; Kambe et al., 2011), whereas the relatively rare mutation observed in melanistic individuals of Okinawa Island, Japan, is caused by a recessive mutation in Asip that causes a point mutation at its C-terminal region where the conserved five pairs of S-S bonds are located (Sasamori et al., unpublished; Fig. 2B).
Whether the genetic systems of melanism are dominant or recessive is a matter of discussion. The factors that determine the colors are likely to depend on the efficiency of alleles of Mc1r and Asip. Certain alleles of Mc1r are responsible for melanism, overwhelming the allelic power of Asip, in which we refer to Mc1r as being epistatic to Asip. In contrast, Asip can sometimes affect the resultant hair color markedly, as is observed in red foxes (Våge et al., 1997), in which Asip alleles counteract a constitutively active MC1R. Another unique interaction has been documented in beach mice. When a mutation leading to increased Asip expression is present, Mc1r mutations that lead to a lighter coloration become appreciable (Steiner et al., 2007). The dominant inheritance of the melanistic allele in chicken (Glu92Lys) is reported to be inherited as an incomplete dominant trait in Japanese quail (Nadeau et al., 2006). A deletion in the MC1R gene causing the loss of one amino acid (Phe256del) is reported to be perfectly associated with melanism in the guinea fowl (Numida meleagris), being a case of “recessive” melanism linked to Mc1r (Vidal et al., 2010). These cases are possibly explained by the allelic variation in other genes, such as Asip. These considerations are consistent with our recent experimental work on black rats from the Ogasawara Islands, Japan, where the Mc1r allele responsible for melanism exists in a polymorphic state. The specific allele for melanism in Mc1r, 280A (94Lys), which is known to yield “constitutively active MC1R” as mentioned above, is affected by alleles of Asip, implying that both genes are involved in determining the extent of eumelanin (Fig. 4, Suzuki et al., unpublished). These studies lead us to note that the phenotypic effects of both MC1R and ASIP depend on their allelic status and are thereby likely to vary among populations and species (Hubbard et al., 2010). The cooperative link between the two genes in turn portrays a specific strategy for the evolutionary tuning of coat color, in which high genetic diversity is created by combinations of polymorphic alleles of Mc1r and Asip to cope with rapid environmental changes.
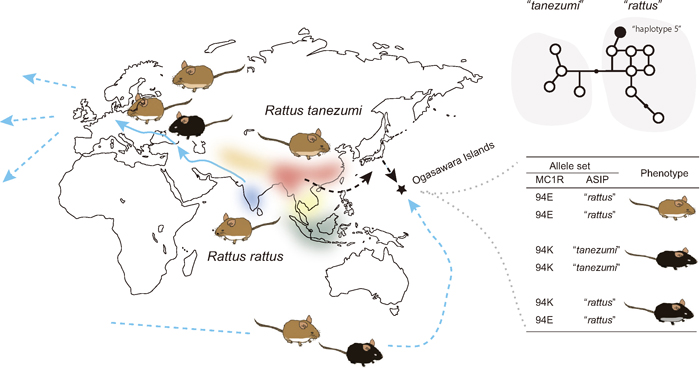
A schematic representation of the homeland of the R. rattus species complex, which includes six distinct mitochondrial phylogroups, including R. rattus and R. tanezumi (Pagès et al., 2010; Aplin et al., 2011). The two major members of this complex expanded their lineages across the world. In the case of R. rattus, it originated from the Indian Subcontinent and spread to Europe through the movements of prehistoric humans. The two lineages experienced intensive human-mediated dispersal (Aplin et al., 2011). Melanistic individuals are rare or altogether absent in India (Kambe et al., 2011), possibly with the exception of major ports and towns where rats of secondary European origin are very likely present (Marshall, 1976). Melanistic rats, in contrast, occur throughout the Middle East and Europe (Schwarz and Schwarz, 1967). European populations of the rats exhibit three main color variants: an ‘alexandrinus’ variant with agouti dorsum and gray to slate gray venter, a ‘frugivorous’ variant with agouti dorsum and pure white venter, and an essentially melanistic ‘rattus’ variant that varies from grayish black upperparts merging into slate-gray underparts through to all-black coats. Such variability in the coat color in Europe could be explained in part by the emergence of the 94E variation that is responsible for the melanism, during the dispersal from India to East Asia or East Asia to Europe (Kambe et al., 2011). Human activity in the ancient and recent past brought the rats to almost the entire world, including remote islands. An example is the Ogasawara Islands, Japan, where two lineages of R. rattus and R. tanezumi are found and the coat colors are variable, including melanistic and whitish individuals. Recent work on Mc1r and Asip variants has indicated that these coat color variants are shaped by allelic combinations of these two genes (Suzuki et al., unpublished).
Mc1r and Asip have had primary roles in the evolution of coat color in mammals. But why these two genes?
Overall, mutations in Mc1r and Asip are likely to be less harmful for survival than those in other genes related to coat color and color pattern (e.g., c-KIT, EDNRB genes), which can be rather destructive or even lethal (Maeda et al., 1992; Hosoda et al., 1994). The less pleiotropic effects of the two genes may explain why many evolutionary changes in coat color are related to these two genes. Vertebrate ancestors had a single set of melanocortin receptors (MCRs) for energy balance and coloration, and these experienced gene duplication several hundred million years ago, resulting in several MCR subtypes (e.g., MC3R, MC4R); the same occurred to agouti-related protein genes (AgRP) (see Kaelin et al., 2008 for review). This led to the development of the MC1R-ASIP system, which played a limited role in the evolution of pigmentation (Jackson et al., 2006; Patel et al., 2010). MC1R is expressed primarily by melanocytes in the skin, and its major ligand, ASIP, by nearby dermal papilla cells. In this way, deleterious pleiotropic effects are minimized, which may have allowed the two genes to evolve rather rapidly toward playing a role in adaptive changes related to coat color (Kingsley et al., 2009: Patel et al., 2010).
Given that Mc1r has a primary role in the evolution of coat color, it may be possible to elucidate the long-term evolutionary trends in coat color in a specific lineage by assessing the rate of amino acid substitution in the coding region of this gene. Mc1r may have evolved differentially across taxa over evolutionary time (Nadeau et al., 2007), responding to environmental changes and intraspecies competition (sexual selection). Accelerated adaptive evolution can be measured by the ratio of nonsynonymous substitutions per nonsynonymous site (dN) to synonymous substitutions per synonymous site (dS). In lion tamarin, the Mc1r sequence is thought to have evolved rapidly with respect to changes in amino acids, with a high dN/dS ratio of 0.91 compared to other anthropoid lineages, implying a reduction of functional constraints or adaptive evolution (Mundy and Kelly, 2003). An evolutionary study on the Mus (Muridae, Rodentia) group leading to M. musculus is another example, in which dN/dS reflects specific evolutionary episodes related to coat color. This group has experienced speciation via a shift in niche from the subtropical ancestral species of forest dwellers to grassland dwellers (subgenus Mus), to that of commensalism (Suzuki et al., 2004; Suzuki and Aplin, 2012). Shimada et al. (2009) examined dN/dS in Mc1r sequences among species belonging to the genus Mus and found significantly higher ratios in the ancestral lineage of subgenus Mus (nearly 1; number of synonymous substitutions = 2, number of synonymous substitutions = 8), while the average ratio was 0.19 for all species examined (Fig. 5). Notably, a slight change in the data set, adding one species (M. lepidoides, a recently discovered taxon from Myanmar; Shimada et al., 2010), modified the outcome, showing no synonymous substitution along the ancestral branch (Suzuki and Aplin, 2012; Suzuki et al., unpublished), confirming that the unusual situation (dN/dS > 1.0) occurred specifically during the adaptation to grasslands. These findings suggest that examining such evolutionary patterns provides an opportunity to illuminate concealed evolutionary episodes of certain characteristics in certain lineages.
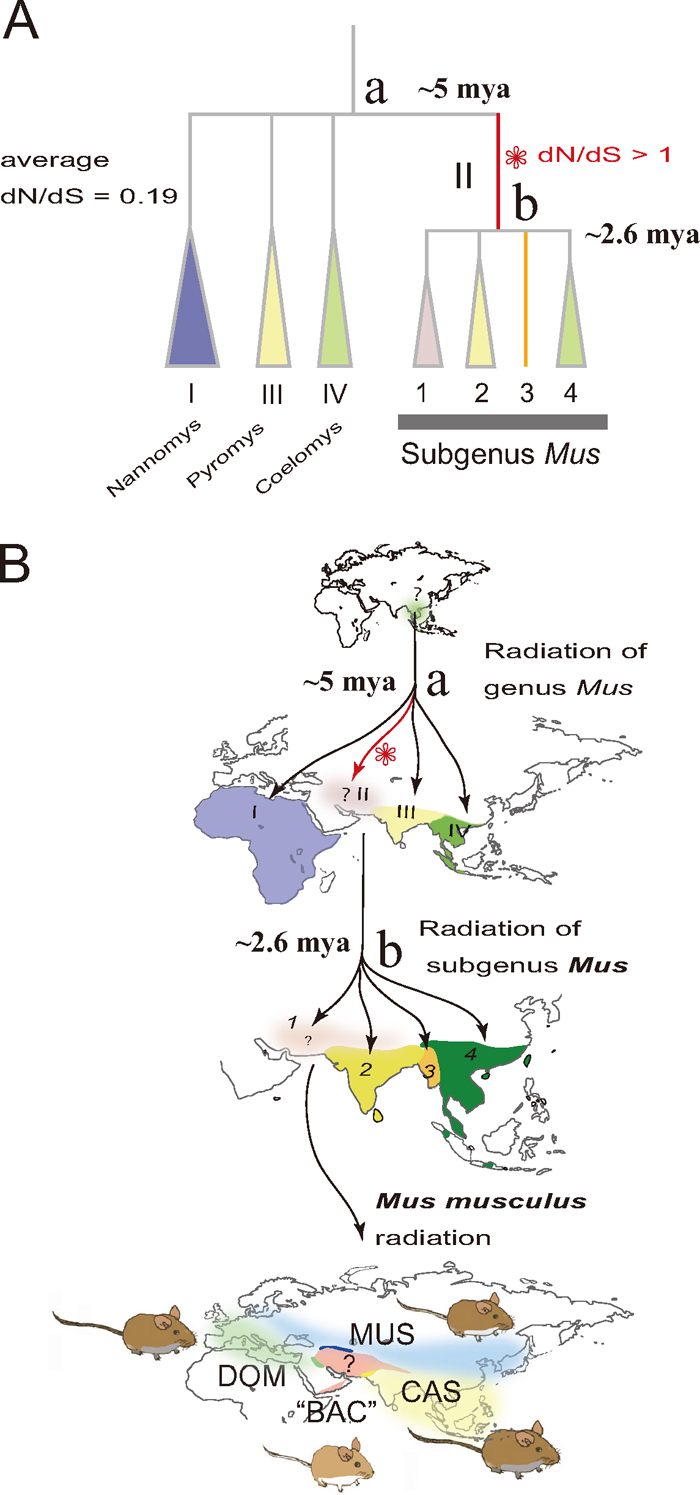
An evolutionary view of species diversity in the genus Mus (A), based on molecular phylogenetic studies (Suzuki et al., 2004; Shimada et al., 2007, 2010; Suzuki and Aplin, 2012). Species diversity leading to the house mouse M. musculus was achieved through three or four radiation processes (B): first, for the four subgenera Coelomys, Mus, Pyromys, and Nannomys around 5 million years ago (mya); second, for the four species groups represented by M. booduga, M. cervicolor, M. lepidoides, and M. musculus around 2.6 mya; and finally for four subspecies groups of M. musculus, three of which extended their geographic range by associating with humans over the last 100,000 years (M. m. castaneus, CAS; M. m. domesticus, DOM; M. m. musculus, MUS), leaving the fourth subspecies group “M. m. bactrianus” (“BAC”) in the central portion of the homeland. The step-wise diversification pattern would provide a good opportunity for understanding adaptive evolution. The evolution of amino acids in the Mc1r coding sequences (945 bp, without the stop codon) assessed by the ratio of non-synonymous substitutions to synonymous substitutions (dN/dS) in species of the genus Mus was significantly accelerated in the branch representing the ancestral lineage of the subgenus Mus (Shimada et al., 2009; Suzuki and Aplin, 2012).
Considering that Mc1r and Asip continue to play important roles in the modulation of body color, after having done so over a long evolutionary history, it may be reasonable to assume that these genes have been built to accommodate evolutionary change. We assessed the evolutionary trends of nucleotide substitutions in Mc1r in carnivoran species with a special emphasis on martens, in which a relatively higher dN/dS was observed (nearly 1) in a previous study based on partial gene sequences. We measured the dN/dS ratio with longer coding sequences of Mc1r and the software codeml which carries out maximum likelihood analysis of protein-coding DNA sequences using codon substitution model implemented in Phylogenetic Analysis by Maximum Likelihood (PAML; Yang, 2007) with two different substitution models. The ratio was relatively high in martens (1.03, genera Martes plus Gulo), compared to that of weasels (0.35), dogs (0.37), and cats (0.21), under the “1/61 each” model in which codon usage bias is not taken into account (Hatanaka et al., unpublished). Estimation with the “F3 × 4” model resulted in a lower dN/dS, approximately half, suggesting possible constraints of codon usage bias on the sequence variation in the Mc1r of carnivorans. Accordingly, the relative level of dS in martens was substantially low, compared to those of the other taxon groups. These results suggest that the higher dN/dS ratio in martens is accompanied by certain genetic traits, depressed levels of synonymous substitutions, and higher levels of codon usage bias and GC content. This study illustrates the unstable features of the genetic traits being modulated in a lineage during the course of evolution (Hatanaka et al., unpublished).
In martens, the Mc1r coding region exhibits three independent length-modification events that resulted in a lack of a 15 bp, 18 bp, and 45 bp segment near TM2 and EC1 (Hosoda et al., 2005), whereas no such deletion or insertion events are visible in the other mustelid taxon, weasels. In-frame deletion events have been reported in other groups of mammals including domestic rabbits and jaguars (Fontanesi et al., 2006). Interestingly, in-frame deletions are attributed to the melanism of the jaguar (15 bp) and jaguarondis (24 bp; Eizirik et al., 2003). The golden-headed lion tamarin possesses a deletion of 24 bp at the same position as that seen in jaguarondis (Mundy and Kelly, 2003), serving as a good example of molecular parallel evolution. The in-frame deletion events in martens are concentrated in a particular portion of the Mc1r gene (i.e., EC1; Fig. 2A) and are thought to have arisen in association with specific nucleotide motifs, such as tandem repeats of two codons (6 bp) and an inverted repeat-like segment between the tandem repeats (Hosoda et al., 2005). It is plausible to presume that the emergence of such specific motifs could be the consequence of the above mentioned genomic constraints, and strong codon usage bias.
Rodent Asip has a specific intragenic architecture for evolution (Fig. 2B). In around the 150 kb region upstream of exons 2–4, there is a 22-kb fragment harboring inverted repeats in 3.3 kb, one of which holds an active promoter for exon 1A (and 1A’) and the other an inactive promoter for pseudo exon 1 (ps1A). The 22 kb fragment is predicted to change its direction by inversion in which the duplicated sequences mediate a palindrome structure that provides the means for intrachromosomal homologous recombination (Chen et al., 1996). This would provide ventral fur polymorphism rather instantly. The upstream region of Asip is conserved across muroid rodents including Mus and Pelomyscus (Kingsley et al., 2009), implying the long-term preservation of this system, perhaps as a tool for the evolution of coat color.
The molecular features embedded in the Mc1r and Asip genes suggest a degree of parallel or convergent evolution in coat color. It is worth noting that the Mc1r and Asip system would help us obtain a comprehensive view of the gene evolution afforded by both environmental and genomic constraints; how genetic architecture is affected by the environment through long-term adaptation and how genetic architecture affects the evolution of genes in adaptive evolution.
Variation within species provides a good opportunity for understanding the genetic systems underlying phenotypic changes and the evolutionary or ecological meanings of phenotypic characteristics (Nachman et al., 2003; Hoekstra and Nachman, 2003; Hoekstra et al., 2004, 2005; Mundy et al., 2004). In addition, intraspecies analyses of coat color would provide valuable clues to the natural history of the species examined (Bradley and Mundy, 2008), as well as resolution of taxonomic problems, if any, in which color variation is often used as a diagnostic character for subspecies discrimination. Several examples related to this issue are discussed below.
Camouflage appears to be the single most important evolutionary force explaining the overall coloration of mammals (Caro, 2005) and a typical example is the Japanese hare Lepus brachyurus. It shows dimorphic patterns in fur color during the winter season: white in the northern range and brown in the southern range, exhibiting clear geographic demarcation on the Japanese Sea coast and Pacific coast, respectively, with the predicted border being correlated with snowfall. For this reason, these two populations have long been considered different subspecies. However, their genetic structures are not correlated with the phenotypic differences (Nunome et al., 2010), suggesting that the coat color polymorphism is not linked to different phylogenetic origins but rather is an evolutionary consequence of long-standing natural selection on the phenotype responding to snowfall during the winter season. It is desirable to identify the gene responsible for the winter season pelage polymorphism to validate the factors shaping the variation and to settle the taxonomic status.
Studies on American black bear with a white hair phase found in northwest British Columbia, Canada (the so-called Kermode bear, Ursus americanus kermodei), have provided valuable insight into the biological factors maintaining the coat color polymorphism. The gene and variation responsible for the white hair have been identified: Tyr-Cys replacement at codon 298 (A893G) in Mc1r (Ritland et al., 2001). Analyses of population genetic structure with microsatellite markers have demonstrated that the genetic variation has been maintained in populations by a combination of gene flow with neighboring wild-type phase populations, genetic drift, selective pressure, and nonrandom assortative mating (Marshall and Ritland, 2002; Hedrick and Ritland, 2012). Interestingly, the Kermode bear’s white coat may give it an advantage for hunting salmon during the daytime (Klinka and Reimchen, 2009). The effect of positive-assortative mating on the maintenance of the unusual Mc1r polymorphism of a deficiency of heterozygotes is now being evaluated (Hedrick and Ritland, 2012). It is noteworthy that these studies on coat color gene variants are also valuable for a better understanding of the local ecosystem of British Columbia itself (Hedrick and Ritland, 2012).
A recent study on sables from Hokkaido, Martes zibellina brachyura, which have been considered a distinct subspecies from the continental sables, revealed the concealed evolutionary history of this local population and highlighted Hokkaido mammalian fauna with respect to a biogeographic view (Ishida et al., 2013). The sables show substantial variation in the color of winter pelage, ranging from dark brown to bright yellow (see Fig. 3 for example). Recent work has revealed that the bright yellow is caused by a recessive mutation, Cys35Phe, on Mc1r (Ogawa et al., unpublished). It is reasonable to think that this causes radical functional changes by the disruption of disulfide (S-S) bonds. Seven cysteine residues of human MC1R are conserved in all five members of the MCR subfamily (e.g., Mc3r, Mc5r), four of which (C35 on N-terminus; C267, C273, and C289 on EC3) are critical for maintaining receptor structure (Frändberg et al., 2001; Sánchez-Más et al., 2005). The replacement of cysteine at codon 35 in the sable Mc1r is predicted to cause a complete loss of function in the activation of melanocytes for eumelanin synthesis (Fig. 1C). Notably, in Hokkaido sables, analyses of the allelic sequences of Mc1r have shown that there are two clusters; one is distributed widely throughout the Russian continent and Hokkaido, and the other is endemic to Hokkaido (Ishida et al., 2013). Interestingly, the Hokkaido-specific alleles tend to be similar to those of the closely related species, Martes americana and Martes melampus. Together with inferences from mtDNA (e.g., Sato et al., 2011), these data suggest that Hokkaido hosted ancient marten lineages that experienced subsequent intensive backcrossing with M. zibellina (Ishida et al., 2013). Surveys of the causative mutations of coat color-related variation have thus uncovered the hidden natural history of Hokkaido sables as a valuable byproduct. On the other hand, the allele responsible for bright yellow pelage belongs to the continental cluster, although the specific allele is likely unique to Hokkaido; the frequency of the variant is roughly 20% on Hokkaido, while no observation has been made on the continent (Ishida et al., 2013; Ogawa et al., unpublished). This suggests that the allele responsible for bright yellow occurred on the way to Hokkaido or after arriving on Hokkaido. This may imply that the environments on Hokkaido or nearby regions assisted with the emergence and development of the variant. The study of Mc1r thus highlights the specific features of the local ecosystem as well. In future studies, it would be desirable to investigate the footprint of selective sweep along with specific alleles to assess the presence or absence of the adaptive trait in the Hokkaido sables.
Another interesting example of the phylogeographic study of coat color-related genes is that of black rats. The taxon is now often designated the Rattus rattus species complex, as its taxonomic status is still debated and much remains to be understood, in particular about their nuclear genomic background. Notably, the rats, in which two major members (R. rattus with 2n = 38 and R. tanezumi with 2n = 42) have been relatively well investigated, exhibit substantial variation in coat color. R. rattus is generally believed to have originated in western India (Yosida, 1980) and this view has been strengthened by a recent mtDNA phylogeographic study of R. rattus (sensu lato; Robins et al., 2007; Pagès et al., 2010; Aplin et al., 2011). In most areas, melanistic individuals are more abundant in heavily populated and industrialized contexts than in rural settings (e.g., Ondrias, 1966) and they are generally rare in wild populations (Kambe et al., 2011). It has been predicted that this mutation originated after dispersal from India to the Middle East or perhaps even to Europe (Fig. 4). This may have occurred only after the establishment of rat populations in more densely settled urban contexts where dark coloration may have conferred a selective advantage. The predicted history of the causative mutation of melanism can be tested by typing SNP 280 in the nearby chromosome region and looking for signals of selective sweep, indicative of natural selection.
The details of the spatial genetic structure of M. musculus remain unknown, especially for the genetic background of mice found in the predicted homeland encompassing Iran, Afghanistan, Pakistan, and north India (tentatively designated M. m. bactrianus, or BAC; Fig. 5B). The coat colors of the subspecies groups are variable and were traditionally used for taxonomic considerations (Marshall, 1998). It is desirable to clarify the genetic and phylogeographic backgrounds of the dorsal and ventral coat colors to better understand the evolutionary episodes of M. musculus and resolve the taxonomic debate. A sequence determination in the entire coding region of Mc1r (948 bp) confirmed the low variability within species and substantial similarity between mice representing DOM and a number of CAS mice, whereas subspecies-specific divergence was evident in the adjacent spacer region. Negative values of Tajima’s D were detected in the Mc1r coding region in CAS and DOM (Kodama et al., unpublished). These results indicate that CAS and DOM mice share a rapidly expanded unique allele in their current genomes, suggesting that they experienced ancient intersubspecies introgression targeting the Mc1r coding region at an ancient time such as before the human-mediated expansion event (Kodama et al., unpublished). To fully understand the evolutionary episodes, it is necessary to conduct a wild survey of the variation in Mc1r and Asip. It is desirable to carry out intensive phylogeographic studies on M. musculus to address the fur color polymorphism for a better understanding of the evolutionary meaning of the genetic variation in Mc1r and Asip.
The examination of genetic variation in the Mc1r and Asip genes has provided useful information for assessing the population genetic structure. In particular, geographic groups with different coat colors are of interest to assess two possible scenarios: that only coat color-related genes show spatial differentiation governed by certain selective forces, or that it reflects the state of lineage differentiation (e.g., subspecies or local races). These analyses in turn provide us with the opportunity to better understand the natural history of a given species, and provide useful clues for taxonomic reconsideration of local populations.
The Mc1r-Asip system can provide useful clues to better understand the nature of adaptation by linking molecular changes (e.g., changes in amino acids and gene expression) to functional traits during the course of evolution. It also helps us to understand gene evolution or genetic architecture as a consequence of long-term selection. Understanding the ecological meaning of variation in coat color is of great interest, and it offers valuable insight into the taxonomic status of geographic groups. The comparison of spatially different populations provides an important perspective on the biogeographic view of a given geographic area. Comparative studies on the two primary coat color-related genes, Mc1r and Asip, within and between species are thus highly desirable as an initial investigation into the phylogeny and phylogeography of a given taxonomic group, as are those typically conducted using mitochondrial DNA markers.
I thank Hiroaki Yamamoto, Kazuo Moriwaki, Ken Aplin, Alexey P. Kryukov, Jun J. Sato, Tomofumi Shimada, Mitsuo Nunome, Morihiko Tomozawa, Gohta Kinoshita, Sayaka Kodama, Kohei Ogawa, Kotaro Ishida, Kota Anjo, Takashi Kuwayama, Syoichi Sasamori, and Miyu Isobe for their valuable comments. I also thank Yoshinobu Nishimura for providing the photo of golden colored sables. This study was supported by Grant-in-Aid for Scientific Research (B) from Japan Society for the Promotion of Science (JSPS) to H. S. (2440513).