ABSTRACT
We developed an insertion sequence transposition detection system called the “jumping cat assay” and applied it to the Bacillus subtilis chromosome using IS256Bsu1 derived from B. subtilis natto. The high frequency of transposition enabled us to explore host factors; combining the assay and genetic analyses revealed that recA is essential for the transposition of IS256Bsu1. Detailed analyses using various domain mutants of recA demonstrated that this essentiality is not related to the function of recA in homologous recombination. Instead, the ATP binding and hydrolysis function seemed to be crucial for IS transposition. To elucidate the role of recA, we focused on the muB gene of the enterobacteriophage Mu. Based on information from the NCBI Conserved Domain Database, both MuB and RecA belong to the P-loop dNTPase superfamily. Further experiments revealed that muB complements the transposition-defective phenotype of a recA deletant, although it could not rescue UV sensitivity. These results suggest that recA shares a common function with muB that helps the transposition of IS256Bsu1 in B. subtilis.
INTRODUCTION
Insertion sequences (ISs) are small and simple transposons in bacteria, and many IS elements have been identified. They are classified on the basis of transposase homology, inverted repeat sequences and the length of the target sequences. Bacillus subtilis Marburg 168, however, is unique in that it lacks typical ISs (Kunst et al., 1997; Barbe et al., 2009). In contrast, the closely related B. subtilis natto strains, the starter strains for a traditional Japanese fermented soybean food called natto, harbor various IS copies (Nishito et al., 2010; Kamada et al., 2014, 2015) including IS4Bsu1 (Nagai et al., 2000) and IS256Bsu1 (Kimura and Itoh, 2007). To explore the genetic and evolutionary background of the absence of ISs in B. subtilis 168, we artificially introduced modified IS4Bsu1 into the 168 strain and developed several new assay systems for transposition frequency (Takahashi et al., 2007a, 2007b). An intermolecular transposition assay system revealed an increase in transposition frequency under high-temperature and competence-inducing conditions (Takahashi et al., 2007b). A green fluorescent protein (GFP) hop-on assay system facilitated quantitative detection of the transposition of the fluorescence-activated cell sorting (FACS)-optimized GFP mutant gene (Takahashi et al., 2007a), and was used to measure the frequency of Escherichia coli IS1 transposition (Saito et al., 2010). Although these assay systems improved our understanding of the cellular conditions under which transposition occurs, the low frequency of transposition restricted the efficient search for host factor(s).
In the original natto strain, transposition of IS4Bsu1 into the comP gene was frequently observed (Nagai et al., 2000). The ComP-ComA two-component regulatory system is responsible for the regulation of cell density-dependent phenotypes including genetic competence, flagellation and degradative enzyme production (Liu et al., 1998; Dubnau, 1999; Lazazzera, 2000; Tran et al., 2000). The synthesis of gamma-polyglutamic acid (γ-PGA), which confers viscous and sticky properties on natto products, is also controlled by this system and, therefore, IS transposition into comP is critical for natto starters. A previous study by Kimura and Itoh (2007) examined γ-PGA-negative mutants and found that 85% of IS elements transposed into a relevant plasmid were IS4Bsu1 and 15% were IS256Bsu1. This result indicates that both IS elements are active in the natto strain but that IS4Bsu1 appears to be more mobile than IS256Bsu1. However, the results we present here indicate that the 168 strain is distinct in terms of IS transposition and the function of the host factor involved.
The host transposition mechanism has been studied extensively in E. coli. The transposition activity of transposons is mediated by various host factors. Histone-like proteins such as HU and integration host factor function in the transposition reaction of some bacterial transposons and bacteriophage Mu (Kleckner et al., 1996; Lavoie and Chaconas, 1996), and a nucleoid-associated protein, H-NS, is required for IS1 transposition (Shiga et al., 2001). Although it should be noted that RecA is not necessary for the transposition of E. coli transposons (Johnson and Reznikoff, 1984; Sekine and Ohtsubo, 1989; Polard et al., 1992; Jain and Kleckner, 1993), the host factors in B. subtilis have not been investigated, even in natto strains.
In the present study, using our newly developed “jumping cat assay” system, we serendipitously discovered that IS256Bsu1, one of the IS256 family of transposons (Kimura and Itoh, 2007), which encodes DDE recombinase-like MuA (Eisen et al., 1994; Yuan and Wessler, 2011; Montaño et al., 2012; Guérillot et al., 2014), transposed very frequently in B. subtilis 168. We then explored the host factors associated with the transposition of IS256Bsu1, and found that recA is genetically essential for transposition, but that this function is probably not related to its capacity for homologous recombination. Moreover, the recA function in transposition could be complemented by the muB gene derived from bacteriophage Mu.
MATERIALS AND METHODS
Bacterial strains, plasmids and media
The bacterial strains and plasmids used in this study are listed in Table 1. A jumping cat assay system (see below) was constructed in B. subtilis Marburg 168; the resulting strain was named NBS801, and is referred to below as wild type (WT). Gene disruptions or point mutations within the recA gene were effected using a recombinant polymerase chain reaction (PCR) method (Higuchi, 1989), with the set of primers listed in Supplementary Table S1, and introduced into NBS801. All gene disruptants are written with the suffix “d” attached to the gene name (e.g., recAd). For NBS2665, the recA gene in NBS801 was replaced with the muB gene, which was amplified from E. coli ATCC 9637 harboring the lysogenized Mu phage genome (CP002967.1, bps 325221–326159, inserted reversely) in combination with a tetracycline resistance marker. These constructs were verified by sequencing. All bacterial strains derived from B. subtilis 168 were grown in Luria-Bertani (LB), competence induction (CI) (Anagnostopoulos and Spizizen, 1961) or 2xSG medium (Leighton and Doi, 1971) at 37 ℃. Escherichia coli DH10B (Grant et al., 1990) was grown in LB medium at 37 ℃. The E. coli-B. subtilis shuttle vector pDR111a (ampicillin-resistant for E. coli and spectinomycin-resistant for B. subtilis) (Maamar and Dubnau, 2005) was used to construct the jumping cat assay system. Antibiotics were used at the following concentrations: chloramphenicol (Cm), 5 μg/ml; ampicillin, 100 μg/ml; spectinomycin, 50 μg/ml; erythromycin, 1 μg/ml; and tetracycline, 15 μg/ml.
Table 1. Strains and plasmids used in this study
| Strain or plasmid | Genotype | Source |
|---|
| B. subtilis strains | | |
| BEST195 | a natto-fermenting strain | Itaya and Matsui, 1999 |
| 168 | trpC2 | Laboratory stock |
| NBS801 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE | This study |
| NBS2648 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE comK::tet | This study |
| NBS2649 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE comP::tet | This study |
| NBS2650 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE comA::tet | This study |
| NBS2651 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE rok::tet | This study |
| NBS2652 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA::tet | This study |
| NBS2653 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recO::erm | This study |
| NBS2654 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recU::erm | This study |
| NBS2655 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA K70R-erm | This study |
| NBS2656 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA R58C-erm | This study |
| NBS2657 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA E154R-erm | This study |
| NBS2658 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA E154V-erm | This study |
| NBS2659 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA G155P-erm | This study |
| NBS2660 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA G155R-erm | This study |
| NBS2661 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA D159A-erm | This study |
| NBS2662 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA G202I-erm | This study |
| NBS2663 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA F215Q-erm | This study |
| NBS2664 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA K241Q K243N-erm | This study |
| NBS2665 | trpC2 amyE′::(Phyper-spank IS256Bsu1 tnp IRL-cat-IRR spc)::′amyE recA::muB-tet | This study |
| E. coli strains | | |
| DH10B | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 deoR recA1 araD139Δ(ara leu)7697 galU galK λ−rpsL endA1 nupG | Grant et al., 1990 |
| Plasmids | | |
| pDR111a | bla spc; an integration vector of B. subtilis amyE locus | Masaya Fujita;
Britton et al., 2002 |
| pDR111a-T2 | pDR111a containing glyQ/StRNA attenuator sequence and the terminator sequence of the xkd operon | This study |
| pJMPcat101 | pDR111a-T2 containing the transposase gene (tnp) of IS256Bsu1, the “mini-IS” gene cassette harboring a chloramphenicol resistance gene (cat) from pBEST4C between the two inverted repeat sequences of IS256Bsu1, and the lacI gene | This study |
| pBEST4C | bla cat | Itaya et al., 1990 |
We first constructed a plasmid carrying strong terminators to prevent read-through upstream of the IS element containing the reporter gene (Fig. 1). According to information from the database of transcriptional regulation in B. subtilis (http://dbtbs.hgc.jp/, Sierro et al., 2008), we selected the glyQ/StRNA attenuator sequence and the terminator sequence of the xkd operon (the rod and circle in Fig. 1). These sequences were amplified by PCR from B. subtilis 168 genomic DNA using the specific primer pairs A1-F and A1-R, and A2-F and A2-R, respectively. Both fragments were then concatenated by recombinant PCR (Higuchi, 1989) with the primers A1-F and A2-R. The resulting fragment was digested with NheI/SphI and cloned into the same sites of pDR111a, an integration vector for the B. subtilis amyE locus; the resulting plasmid was named pDR111a-T2. We next constructed a “mini-IS” gene cassette as a transposition-monitoring element, which harbored a Cm resistance (Cmr) gene, cat, between the two inverted repeat sequences of IS256Bsu1. cat, encoding chloramphenicol acetyltransferase derived from plasmid pC194 (Horinouchi and Weisblum, 1982), with the Shine-Dalgarno sequence but without a promoter, was amplified from pBEST4C by PCR (Itaya et al., 1990) using the primers B1-F and B1-R. The resulting fragment was further amplified with the partially overlapping primers B2-F and B2-R. This second PCR resulted in the addition of 40-bp left- and right-inverted repeat sequences (abbreviated to IRL and IRR, respectively) of IS256Bsu1 as well as an NheI site at one end and a SalI site at the other end. Alternatively, the transposase gene (tnp) of IS256Bsu1 was amplified by PCR from the B. subtilis BEST195 genome using the primers C-F and C-R. The first codon of the transposase was changed from GTG to ATG so that it would function reliably in the jumping cat assay. The mini-IS and tnp gene fragments were digested with NheI/SalI and HindIII/SalI, respectively, and cloned into the NheI/HindIII sites of pDR111a-T2. The resulting plasmid, named pJMPcat101 and now containing terminators, the mini-IS fragment and the tnp gene, was introduced by transformation into the amyE locus of various B. subtilis 168 strains. Integration was verified by an amylase-negative phenotype. The constructed strains harbored the tnp gene under the control of the LacI-repressed/isopropyl-β-D-thiogalactopyranoside (IPTG)-induced promoter Phyper-spank (a gift from M. Fujita, University of Houston; Britton et al., 2002), and the mini-IS containing cat was placed in its immediate vicinity.
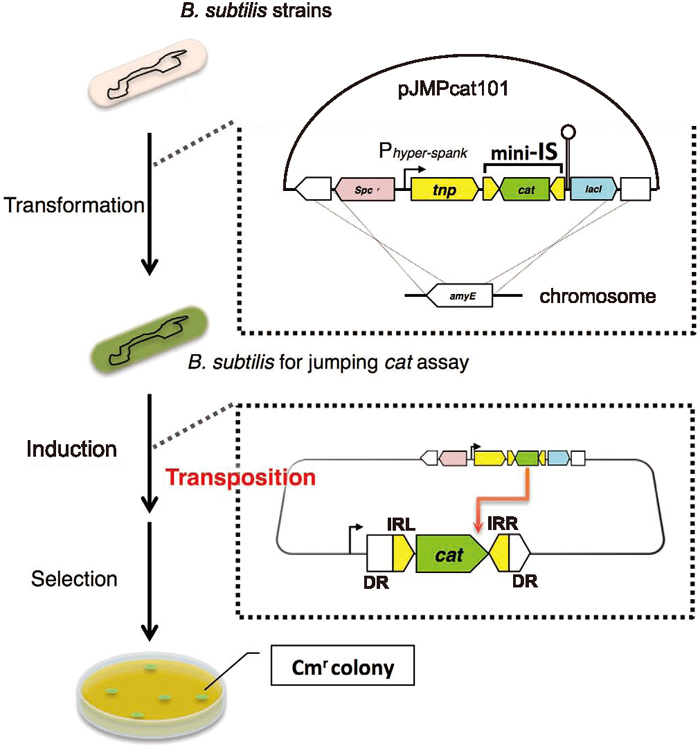
When the mini-IS is transposed with the tnp gene product, cells become Cmr owing to cat gene expression resulting from the mini-IS having “jumped” into, and transcriptionally fused to, a certain gene locus, which should be a nonessential gene. This is the concept of the jumping cat assay. A frozen stock of each strain was precultured on LB plates, and then cultured in LB, CI or 2xSG liquid medium containing 1 mM IPTG at 37 ℃ for 24 h. Each culture was diluted and plated on LB medium, whereas the non-diluted cultures were plated on LB medium containing Cm. To determine the number of spore-forming Cmr cells, a portion of the 2xSG culture was heated at 80 ℃ for 10 min and plated on LB medium with Cm. Colony-forming units (CFUs) were calculated according to the number of colonies observed after a 24-h incubation. The transposition frequency (TPF) was defined as [CFU on LB plates with Cm] per [CFU on LB plates]. This test was performed at least three times for each strain. Statistical differences in TPF values between the WT and the mutants were estimated by a permutation test based on the Brunner–Munzel test (Neubert and Brunner, 2007).
Detection of direct repeat sequences (DRs) on both sides of the transposed mini-IS
To verify that the Cmr colonies obtained actually resulted from mini-IS transposition, we detected DRs that had been generated on both sides of the mini-IS sequence. The genome of the Cmr colonies was isolated and digested with HindIII, and then ligated with the HindIII cassette of the Takara LA-PCR in vitro Cloning Kit (Takara Bio, Otsu, Japan). The first PCR was performed with this ligation mixture using Cassette Primer C1 and either S1 or S3 (cat gene-specific primers). This reaction mixture served as a template for the second PCR using Cassette Primer C2 and either S2 or S4 to amplify the specific fragment containing the cat gene. C1 and C2 were manufacturer-supplied primers corresponding to the cassette sequence, and S1 to S4 comprised reverse direction (S1 and S2) and forward direction (S3 and S4) components of the cat gene. The primers for the second PCR (C2, S2 and S4) were based on the first PCR products. Sequence determination of these second PCR products revealed mini-IS-inserted regions, thereby indicating the flanking sequences of the IS insertion.
Molecular phylogenetic analysis
This analysis involved 406 nucleotide sequences that encode recombinases (recA, radA, rad51, uvsX and muB), obtained from the NCBI genome database. Sequences used in this analysis are shown in Supplementary Table S2. The sequences were initially categorized by gene, translated, and then aligned using a multiple alignment program for amino acids or nucleotide sequences, MAFFT (version 7.273), using the options “maxiterate1000” and “genafpair” to remove unique insertions (Katoh and Standley, 2013). The sequences were then aligned again by MAFFT using the “maxiterate1000” and “localpair” options (Katoh and Standley, 2013), and unreliable alignments were removed based on the transitive consistency score (TCS, filter 3), using back-translation to DNA sequences (Chang et al., 2014). Evolutionary history was inferred using the maximum likelihood method, based on the general time-reversible model (Nei and Kumar, 2000). The bootstrap consensus tree inferred from 200 replicates was taken to represent the evolutionary history of the taxa analyzed (Felsenstein, 1985). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (200 replicates) is shown adjacent to each branch (Felsenstein, 1985). Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the maximum composite likelihood approach, and then selecting the topology with the highest log-likelihood value. A discrete gamma distribution was used to model evolutionary rate differences among sites (five categories [+G, parameter = 0.9205]). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 0.764% sites). The final dataset comprised a total of 1,044 positions. Evolutionary analyses were conducted using the molecular evolutionary genetics analysis software MEGA7 (version 7.0.14) (Kumar et al., 2016). The phylogenetic tree was graphically edited in FigTree (version 1.4.2) (http://tree.bio.ed.ac.uk/software/figtree/).
UV survival assay
Semi-quantitative measurements of UV sensitivity were carried out as described previously (Mustard and Little, 2000). Wild type and recAd (NBS2652) or recA::muB mutant (NBS2665) bacteria were grown overnight on LB plates. Precultures were made and grown in LB liquid medium until the OD600 reached 1.0. Bacterial cultures were streaked onto LB plates using disposable loops. The plates were exposed to increasing doses (J/m2) of UV light and then incubated at 37 ℃ for an additional 16 h in the dark. The growth of cultures was then examined.
RESULTS
recA gene is essential for the transposition of IS256Bsu1
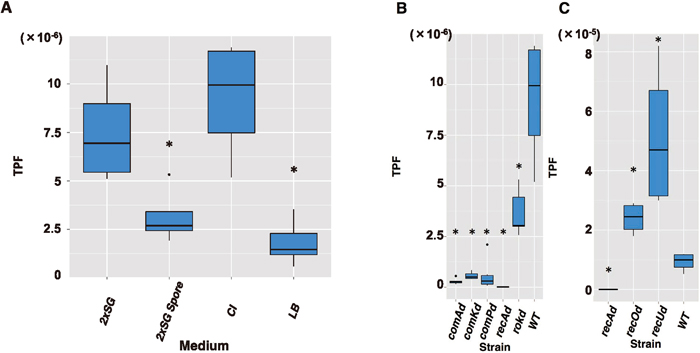
To identify host factors associated with the transposition of IS256Bsu1, we constructed the TPF measurement system for B. subtilis 168, which is known to be a transposon-free strain. An overview of this system is given in Fig. 1 and the system is described in detail in Materials and Methods. As a mutator-like element, the IS256Bsu1 sequence was acquired from B. subtilis BEST195, a natto-fermenting strain. In this experimental system, the cat gene was sandwiched between the IR sequences from IS256Bsu1; we refer to this as the mini-IS. The transposase gene was contiguous with but outside the IR left–right sandwich to prevent the mini-IS from undergoing a second transposition. Because the cat gene transposes on the genome replicatively, we named this system the jumping cat assay. If the mini-IS transposes, the cat gene is expressed via transcriptional fusion, and the cell consequently becomes Cmr. The strain harboring this system was cultivated in three different media (CI, LB and 2xSG), and the CI sample exhibited the highest TPF (Fig. 2A). Furthermore, the TPF of spore-forming cells was approximately 42% that of cells from the 2xSG medium (Fig. 2A). Therefore, transposition of the mini-IS was more frequent under nutrient starvation conditions, except in spore-forming cells. Although both CI medium and 2xSG medium contain smaller amounts of nutrient than LB medium, CI medium was originally designed as a transformation medium (Anagnostopoulos and Spizizen, 1961), whereas 2xSG medium was optimized for spore formation (Leighton and Doi, 1971). For this reason, we carried out the jumping cat assay in CI medium.
Insertion sequences that belong to the IS256 family, as well as those belonging to other IS families (Mahillon and Chandler, 1998; Jang et al., 2012), generate short DRs of the target DNA flanking the IS (Mahillon and Chandler, 1998). If Cmr colonies are derived from mini-IS transposition, DRs must be detectable. We randomly selected Cmr colonies from the assays that had been cultivated in CI and LB media, and investigated the generation of DRs. Consequently, six DRs consisting of non-conserved 8-bp sequences were detected (Table 2); this was the same length of IS256 as previously observed in Staphylococcus (Loessner et al., 2002). Whereas the inserted regions were located randomly in the genome, the cat gene on the mini-IS was oriented in the same direction as the open reading frame (ORF) of the inserted genes; this indicated that the Cmr phenotype was derived from transcriptional fusions of the cat gene, as expected. Thus, the jumping cat assay proved to be an efficient system for evaluating TPFs.
Table 2. Direct repeat sequences found at IS-inserted loci
| Direct repeat (5′ > 3′) | Positiona | Directionb | Insertedgenec | Function |
|---|
| TTTGAAGC | 44859–44866 | + | yabC | 16S rRNA methyltransferase |
| TGTTTATT | 1441408–1441415 | − | queF | nitrile reductase |
| ACTCCTCT | 2553019–2553026 | + | tasA | major component of biofilm matrix |
| GCGGAATG | 3109582–3109589 | + | bceB | bacitracin ABC transporter (permease) |
| GAGATGAA | 3255545–3255552 | + | comP | two-component sensor kinase |
| AAATGCAT | 3785764–3785771 | + | atpH | ATP synthase (subunit delta) |
a) Position of the direct repeat on the Bacillus subtilis 168 genome (GenBank: CP010052.1). b) cat ORF direction against the genome. c) Gene name in which the IS-cat was inserted.
We then determined which gene was primarily involved in the mini-IS transposition process as a host factor, bearing in mind that the cells in CI medium exhibited the highest TPF value (Fig. 2A). We determined the TPF values arising from the disruptants of each competence-conferring gene, NBS2648 (comKd), NBS2649 (comPd), NBS2650 (comAd), NBS2651 (rokd) and NBS2652 (recAd). As a result, disruption of comP, comK, comA or rok decreased the TPF compared with NBS801 (WT) (P < 0.01, Fig. 2B). Moreover, no Cmr colony was detected from the recA disruptant (Fig. 2B). However, the disruption of recO or recU, both of which, like recA, relate to homologous recombination, resulted in a higher TPF value than in the WT (P < 0.01, Fig. 2C). In B. subtilis, recA is transcribed mainly by the ComK activator (Hamoen et al., 2001), and the decrease in the TPF in competence gene disruptants was presumably attributable to its decline. Considering recO or recU single disruption (Fig. 2C) and the detection of DRs (Table 2), it is conceivable that recA assists mini-IS transposition via a mechanism other than homologous recombination.
Relationship between TPF of mini-IS and RecA active sites
We next attempted to estimate the function of RecA protein in the IS transposition process, using recA point mutants of the jumping cat assay strain. RecA, which has been extensively studied in E. coli, is known to have multiple functions. We compared the B. subtilis and E. coli RecA polypeptide sequences using the EMBOSS Stretcher program (Rice et al., 2000). The results indicated that these sequences are highly conserved in total (identity = 57.8%, similarity = 77.3%), and sequences around the known typical active sites of each domain in E. coli are shown in Fig. 3A. Therefore, we constructed point mutants according to the recA mutations identified in E. coli and determined their TPFs (Fig. 3B).
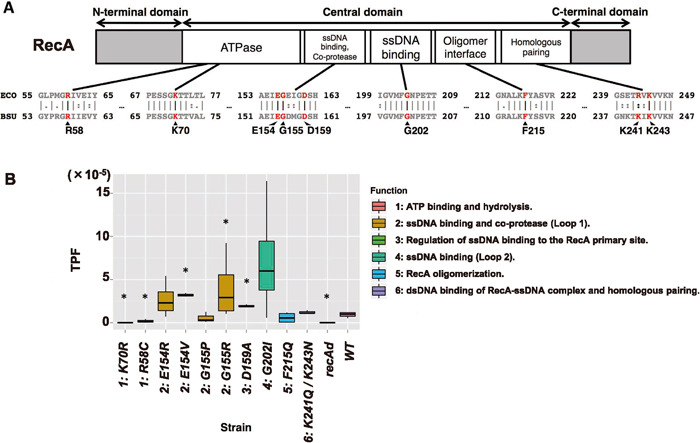
RecA consists of three large domains: the N-terminal domain, the central domain and the C-terminal domain. Its major active sites are within the central domain (Fig. 3A) between amino acids (aa) 53 and 257 in E. coli (Kurumizaka et al., 1999). Strains NBS2656 and NBS2655 contain recA R58C and K70R mutations that are homologous to E. coli R60C and K72R mutations, respectively (Roca and Cox, 1997; Britt et al., 2011), which are in the ATPase active site of E. coli RecA. The TPFs of these two strains were markedly low, as was the TPF for the recA disruptant (recAd), compared with that for the WT (P < 0.05, Fig. 3B). These results suggest that the ATPase activity of RecA is important in the IS transposition process. By contrast, the functions of single-stranded DNA (ssDNA) binding and of homologous DNA exchange with D-loop formation seem to be less important for IS transposition. Four recA mutants—NBS2657 (E154R), 2658 (E154V), 2659 (G155P) and 2660 (G155R)—in which the mutations lie within the predicted loop L1 region containing putative ssDNA-binding activity and co-protease activity, were created based on the E. coli recA mutants E156R, E156V, G157P and G157R, respectively (Nastri and Knight, 1994; Nastri et al., 1997). Mutants NBS2658 (E154V) and 2660 (G155R) exhibited higher TPFs than the WT (3.4-fold and 4.3-fold, respectively, P < 0.05, Fig. 3B). Nastri and Knight (1994) demonstrated that the E. coli E156V mutant does not differ from the WT in terms of DNA damage repair; however, the E. coli RecA G157R mutant exhibits lower DNA repair activity, in spite of the constitutive activation of LexA cleavage (Nastri et al., 1997). NBS2662 (recA G202I) has a mutation that corresponds to E. coli recA G204I; it is located in the loop L2 region, functions as the ssDNA-binding domain, and produces a recombination-defective phenotype (Hortnagel et al., 1999). This mutant exhibited a 7.4-fold higher TPF compared with that of the WT (P = 0.114, Fig. 3B). In E. coli RecA WT, F217 acts as a connector residue for forming the RecA helical nucleoprotein filaments (De Zutter et al., 2001). Understandably, all DNA damage-repairing activities are abolished by the F217Q mutation (Skiba and Knight, 1994). However, the TPF value for NBS2663 carrying the orthologous mutation, F215Q, exhibited no significant difference from that of the WT, although it was 0.6-fold lower in the mutant (P = 0.609, Fig. 3B). Both the R243 and K245 residues in E. coli RecA bind to donor double-stranded DNA (dsDNA) and enable it to interact with the RecA-ssDNA presynaptic nucleoprotein filament during homologous recombination (Kurumizaka et al., 1999; Lee and Wang, 2009). The R243Q/K245N double mutant RecA fails to form the D-loop, and causes defects in homologous recombination (Kurumizaka et al., 1999). This double mutant in E. coli RecA was mimicked in B. subtilis NBS2664 (K241Q/K243N). However, this strain exhibited almost the same TPF as the WT (1.3-fold higher, P = 0.202, Fig. 3B). According to the above study of the corresponding E. coli mutant, the function of the domain including these residues (R243 and K245) is searching homologous dsDNA strands as part of the ssDNA-RecA complex, and is distinct from dsDNA binding by free RecA itself. The result with NBS2664 (K241Q/K243N) suggests that the process of searching complementary dsDNA by ssDNA-RecA complex is not essential for mini-IS transposition. Recently, detailed analyses in E. coli have revealed that residue D161 in the loop L1 region controls the affinity of RecA for ssDNA or dsDNA, and the RecA D161A mutant binds to dsDNA rather than ssDNA (Shinohara et al., 2015). Bacillus subtilis NBS2661 having the orthologous mutation, D159A, exhibited a higher TPF than the WT (2.2-fold, P < 0.05, Fig. 3B). Altogether, these results suggest that the ATPase activity of RecA is an essential feature for mini-IS transposition, but ssDNA-binding and homologous recombination activities are not required.
muB is not derived from the recA gene cluster, but may be an ancestor of RecA-like recombinase
As mentioned above, recA, at least as a component of the homologous recombination machinery, may be nonessential for IS transposition. To elucidate its role, we focused on the muB gene of the enterobacteriophage Mu (Adzuma and Mizuuchi, 1988), which is a replicative transposition-type bacteriophage that was reported by Taylor (1963). It possesses the genes muA and muB: the former encodes a DDE-type, mutator-like transposase, and the latter encodes a DNA-binding protein that determines the location of the Mu genome on its host genome by transposition (Morgan et al., 2002; Harshey, 2012). Based on information from the NCBI Conserved Domain Database, both MuB and RecA belong to the P-loop dNTPase superfamily; both proteins form filaments on DNA, and hydrolyze ATP before detachment from the substrate DNA (Greene and Mizuuchi, 2002). To confirm the phylogenetic position of muB in the RecA-like superfamily of genes, we collected RecA-like recombinase gene sequences and muB sequences from 406 taxa for which the entire genomes had been sequenced; this group of taxa contains archaea, bacteria, eukaryotes and viruses. In the RecA-like recombinase group, protein sequence similarities among RecA, Rad51, RadA and UvsX are known (Bianco et al., 1998; Haldenby et al., 2009); therefore, we included these gene types in the analysis. Two types of recA gene, derived from mitochondria and chloroplasts in Streptophyta (plants), and from mitochondrial recA in Dictyostelium spp. (Mycetozoa) (Hasegawa et al., 2004), were added, in addition to their rad51 sequences. A phylogenetic tree was constructed from these nucleotide sequences, as described in Materials and Methods.
Figure 4 shows the bootstrap consensus, maximum likelihood phylogenetic tree, constructed by increasing the node order (in which operational taxonomic units [OTUs] were collapsed by their classes). The result demonstrates that each of the five genes, muB, radA, recA, rad51 and uvsX, clearly forms a clade. Of particular note, the muB clade neighboring uvsX was not nested within the other RecA-like recombinase clades (> 99%). This reveals that muB is not derived from a preexisting RecA-like recombinase. The rad51 clade, constituted entirely of eukaryotes, was separated from the nearby archaeal radA clade (> 98%). The recA clade mostly contained bacterial OTUs; however, they exhibited patchy distribution, except for those of mitochondria and the chloroplasts in Streptophyta. This tendency was also seen in the radA clade (Fig. 4). The sequence in Halosimplex carlsbadense 2-9-1 (NZ_AOIU01000018.1: 12005–13468 bp) was separated from the other clades, which were annotated as “recombinase RecA”. However, it seems that H. carlsbadense’s “kaiC” should belong to the RecA protein family, because two duplicated ATPase domains for kaiC have been found (Haldenby et al., 2009). On the other hand, radA in H. carlsbadense 2-9-1 was correctly classified into the halobacterial clade. The uncollapsed phylogenetic tree is available in Supplementary Fig. S1.

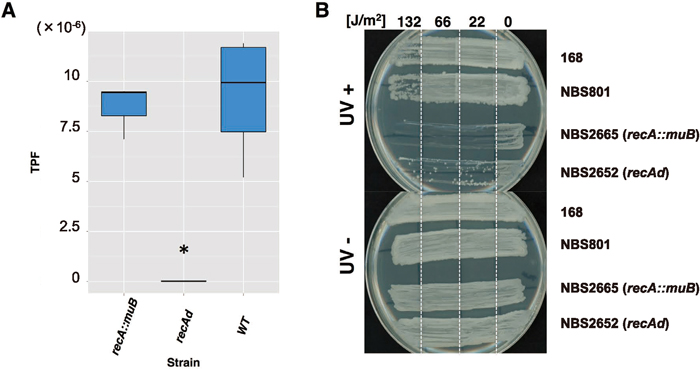
Using the jumping cat assay, we determined whether muB was capable of complementing the IS transposition-defective phenotype of the recA deletant. Surprisingly, the TPF of NBS2665 (recA::muB) was almost identical to that of the WT (0.93-fold, P = 0.40, Fig. 5A). However, NBS2665 was sensitive to UV irradiation, as seen in the recA mutant (Fig. 5B), suggesting that the muB complementation (or compensation) of recA function is restricted to IS transposition. These results demonstrate that recA is related to muB both genetically and phylogenetically, and the relationship is mediated by the mutator-like transposase.
DISCUSSION
When applied to the insertion sequence IS256Bsu1, the newly developed jumping cat assay system revealed high TPFs and enabled us to analyze quantitatively various mutants of B. subtilis 168. In agreement with our previous report (Takahashi et al., 2007b), the TPF was high in the competence induction medium (Fig. 2A). This result prompted us to investigate several competence-related genes, which revealed a definite effect of recA (Fig. 2B). This was reasonable, since the expression of recA is induced in CI medium and before sporulation (Lovett et al., 1989; Dubnau, 1991; Grossman, 1995). The major conclusions in this investigation are: (i) recA is required for the transposition of IS256Bsu1; (ii) the ATP-binding/hydrolyzing activity of RecA appears to be involved in this activity; and (iii) the MuB protein belonging to the P-loop dNTPase superfamily, to which RecA also belongs, can compensate for RecA deficiency in terms of the ability to support the transposition of IS256Bsu1.
The E. coli RecA protein has homologous recombination activity and plays a key role in DNA repair. The DNA repair pathway, which is also referred to as the SOS response, involves RecA and several other factors including the proteins LexA and RecX; similar systems are highly conserved across species (Shibata et al., 1979; Little et al., 1980; Wojciechowski et al., 1991; Drees et al., 2004; Gruenig et al., 2010; van der Veen et al., 2010). RecA forms a complex with ATP and ssDNA to form a helical filament that binds to dsDNA, and can therefore be used to search for a homologous genomic region (Shibata et al., 1979; Drees et al., 2004). The ssDNA in a cell is usually coated with single-stranded binding proteins (SSBs), and RecA is loaded onto the ssDNA after the removal of the SSBs by the RecFOR complex (Sakai and Cox, 2009). In B. subtilis, RecA is also essential for DNA damage repair and homologous recombination, although RecO and RecU are mandatory for plasmid transformation (Kidane et al., 2009). However, our construction of various recA mutants based on homology with E. coli recA revealed that the amino acids and domains encoded by the two genes were highly conserved when the genes were compared, especially in regions where clear functions were examined and mutants were isolated (Fig. 3A). Therefore, we assumed that B. subtilis recA mutants would exhibit the same phenotype as their counterparts in E. coli. Accordingly, strains with mutations in the ATPase domain completely lost transposition activity, whereas mutations in the other domains had a limited effect, suggesting that the ssDNA-binding and homologous recombination activities are not required for IS transposition (Fig. 3B). The D159A mutant showed a slightly higher TPF than the WT, however, which indicated that RecA’s affinity for dsDNA influenced mini-IS mobility (Fig. 3B).
We then focused on the muB gene of bacteriophage Mu (Adzuma and Mizuuchi, 1988), which, like RecA, encodes a DNA-binding protein belonging to the P-loop dNTPase superfamily. The main role of MuB is to place the MuA (transposase)-DNA complex into the target site of the genome via filamentation and then to hydrolyze ATP when MuB detaches from dsDNA (Kruklitis et al., 1996; Levchenko et al., 1997; Greene and Mizuuchi, 2004). RecA also binds to dsDNA, and detaches from it by hydrolyzing ATP (Muller et al., 1990; Conover et al., 2011; Shinohara et al., 2015). The amino acid sequences of RecA and MuB are moderately conserved but RecA has additional sequences that are missing in MuB. There is a 12-aa insertion in RecA between the two domains corresponding to the N-terminal appendage and α/β domain of MuB, and also a 16-aa insertion in the homologous loop L1 region. Mutations at basic residues in the L1 region of MuB cause MuA to lose responsive ATPase activity (Mizuno et al., 2013). According to their corresponding residues in the higher structure of MuB, R58 of B. subtilis RecA resides in the N-terminal appendage and E154, G155, D159 and G202 are within the α/β domain of the AAA+ module. In particular, G202 of B. subtilis RecA is in the corresponding Walker B motif within the α/β domain of MuB, while K70, F215, K241 and K243 of RecA have no homologous counterpart in MuB although the flanking regions are conserved. The N-terminal appendage of MuB has nothing to do with ATP hydrolysis, interaction with MuA or DNA binding (Mizuno et al., 2013). On the other hand, as the K72R mutant of E. coli exhibited (Britt et al., 2011), the N-terminal side of the central domain of RecA, which governs ATPase activity, is responsible for ATP hydrolysis. The finding that our mutational analysis in this region, K70R, could not lead to detectable mini-IS transposition (Fig. 3B) indicates that the residues responsible for ATP hydrolysis are different between RecA and MuB.
Pairwise-alignment analysis of MuB (NP_050608.1) and RecA in B. subtilis (WP_003245789.1) by EMBOSS Stretcher (Rice et al., 2000) revealed 17.7% identity and 34.5% similarity. These scores are somewhat higher than those for the comparison of RecA with Rad51 in Homo sapiens (NP_002866.2) (identity = 13.1%, similarity = 29.5%), although Rad51 is known to be a eukaryotic homolog of RecA. This strongly suggests that not only RecA and Rad51 (Chintapalli et al., 2013) but also MuB have evolved from a common ancestral sequence. Overall, the most important point revealed by the phylogenetic analysis is that muB-homologous genes formed a clade, which was not nested into the other recA-like gene clades (Fig. 4). This result indicated that homologous recombinases have common ancestral sequences with bacteriophage Mu and T4-like viruses and may thus be derived from these bacteriophages.
The similarities between RecA and MuB are paralleled by similarities between IS256Bsu1 and MuA. IS256Bsu1 is a member of the IS256 family, whose transposase also belongs to the DDE-type, prokaryotic mutator-like transposase 1 (p-MULT1) group (Guérillot et al., 2014). DDE/D-type transposases in eukaryotes have evolved from a common ancestral gene (Yuan and Wessler, 2011), and it has been demonstrated that one of these transposase members in Zea mays has a similar sequence to the IS256 family (Eisen et al., 1994). This relationship between prokaryotic and eukaryotic DDE/D-type transposases was also confirmed by secondary structure predictions (Guérillot et al., 2014). Furthermore, MuA has been designated as the representative DDE-type transposase (Montaño et al., 2012).
It should be noted that muB was able to complement the function of recA for mini-IS transposition (Fig. 5A). However, we also demonstrated that muB does not confer the ability to survive under UV light (Fig. 5B). Therefore, what is the common function of recA and muB? The requirement for the ATP hydrolysis activity of RecA in IS256Bsu1 transposition may be similar to that of MuB in MuA transposition. According to the model of Mizuno et al. (2013), the MuB filament on dsDNA is partially decomposed by MuB-ATP hydrolysis enhanced by MuA. The interaction of MuA and MuB nicks the terminus of the Mu genome and initiates transposition onto dsDNA exposed by MuA. Similar to this model, the function of RecA in IS256Bsu1 transposition may be to recruit the transposase-mini-IS complex to the target region and, after release from DNA by accompanying ATP hydrolysis, to make nicks at the terminus of mini-IS and promote transposition. A recent study revealed that the reactant DNA is determined by the D161 residue on the loop L1 region of RecA in E. coli (Shinohara et al., 2015). The amino acid residue of MuB corresponding to this D161 is in the loop L1 region of the α/β domain. RecA has a 16-aa insertion in the loop L1 region, as described above, and this slight difference may confer on RecA two specific modes of ssDNA and dsDNA binding. Besides the D159A mutant of RecA, E154V, G155R and G202I mutants exhibited higher TPFs than WT (Fig. 3B). These residues are related to the function of ssDNA binding, and, therefore, when these RecA mutants are reduced in ssDNA binding activity the likelihood of dsDNA binding may simultaneously increase. The requirement of dsDNA binding activity in mini-IS transposition can be explained by this hypothesis. The high TPF in the recO mutant is also interpreted in the same way, that is, RecO recruits RecA onto SSB-coated ssDNA (Manfredi et al., 2008) and thus the recO mutant raises the likelihood of RecA binding to dsDNA, resulting in the promotion of mini-IS transposition. On the other hand, the effect of recU is unclear. RecU traps incorporated ssDNA with RecA, and the recU mutant may have a similar effect to RecO. In any case, the recU mutant decreases transformation efficiency (Kidane et al., 2009) and may influence the homologous recombination process involving RecA binding to dsDNA.
ACKNOWLEDGMENTS
We are grateful to Takehiko Shibata Ph.D. for graciously providing information on recA mutants, and to Masaya Fujita Ph.D. for kindly providing plasmid pDR111 that contains Phyper-spank. We also thank Fujio Kawamura Ph.D., Mariko Shimizu-Kadota Ph.D., Mari Tokuyama, Ryudo Obayashi Ph.D., and Tomoko Araya-Kojima for helpful comments on this research. This work was supported by a Grant-in-Aid for Scientific Research (B) from the Japan Society for the Promotion of Science (to H.Y. (16H04903)), and the Research Fund for the Advancement of the Graduate School, Tokyo University of Agriculture (to M.A).
REFERENCES
- Adzuma, K., and Mizuuchi, K. (1988) Target immunity of Mu transposition reflects a differential distribution of Mu B protein. Cell 53, 257–266.
- Anagnostopoulos, C., and Spizizen, J. (1961) Requirements for transformation in Bacillus subtilis. J. Bacteriol. 81, 741–746.
- Barbe, V., Cruveiller, S., Kunst, F., Lenoble, P., Meurice, G., Sekowska, A., Vallenet, D., Wang, T., Moszer, I., Médigue, C., et al. (2009) From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology 155, 1758–1775.
- Bianco, P. R., Tracy, R. B., and Kowalczykowski, S. C. (1998) DNA strand exchange proteins: a biochemical and physical comparison. Front. Biosci. 3, D570–D603.
- Britt, R. L., Chitteni-Pattu, S., Page, A. N., and Cox, M. M. (2011) RecA K72R filament formation defects reveal an oligomeric RecA species involved in filament extension. J. Biol. Chem. 286, 7830–7840.
- Britton, R. A., Eichenberger, P., Gonzalez-Pastor, J. E., Fawcett, P., Monson, R., Losick, R., and Grossman, A. D. (2002) Genome-wide analysis of the stationary-phase sigma factor (sigma-H) regulon of Bacillus subtilis. J. Bacteriol. 184, 4881–4890.
- Chang, J. M., Di Tommaso, P., and Notredame, C. (2014) TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol. Biol. Evol. 31, 1625–1637.
- Chintapalli, S. V., Bhardwaj, G., Babu, J., Hadjiyianni, L., Hong, Y., Todd, G. K., Boosalis, C. A., Zhang, Z., Zhou, X., Ma, H., et al. (2013) Reevaluation of the evolutionary events within recA/RAD51 phylogeny. BMC Genomics 14, 240.
- Conover, A. J., Danilowicz, C., Gunaratne, R., Coljee, V. W., Kleckner, N., and Prentiss, M. (2011) Changes in the tension in dsDNA alter the conformation of RecA bound to dsDNA-RecA filaments. Nucleic Acids Res. 39, 8833–8843.
- De Zutter, J. K., Forget, A. L., Logan, K. M., and Knight, K. L. (2001) Phe217 regulates the transfer of allosteric information across the subunit interface of the RecA protein filament. Structure 9, 47–55.
- Drees, J. C., Lusetti, S. L., Chitteni-Pattu, S., Inman, R. B., and Cox, M. M. (2004) A RecA filament capping mechanism for RecX protein. Mol. Cell 15, 789–798.
- Dubnau, D. (1991) Genetic competence in Bacillus subtilis. Microbiological Reviews 55, 395–424.
- Dubnau, D. (1999) DNA uptake in bacteria. Annu. Rev. Microbiol. 53, 217–244.
- Eisen, J. A., Benito, M. I., and Walbot, V. (1994) Sequence similarity of putative transposases links the maize Mutator autonomous element and a group of bacterial insertion sequences. Nucleic Acids Res. 22, 2634–2636.
- Felsenstein, J. (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791.
- Grant, S. G. N., Jessee, J., Bloom, F. R., and Hanahan, D. (1990) Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 87, 4645–4649.
- Greene, E. C., and Mizuuchi, K. (2002) Target immunity during Mu DNA transposition: transpososome assembly and DNA looping enhance MuA-mediated disassembly of the MuB target complex. Mol. Cell 10, 1367–1378.
- Greene, E. C., and Mizuuchi, K. (2004) Visualizing the assembly and disassembly mechanisms of the MuB transposition targeting complex. J. Biol. Chem. 279, 16736–16743.
- Grossman, A. D. (1995) Genetic networks controlling the initiation of sporulation and the development of genetic competence in Bacillus subtilis. Annu. Rev. Genet. 29, 477–508.
- Gruenig, M. C., Stohl, E. A., Chitteni-Pattu, S., Seifert, H. S., and Cox, M. M. (2010) Less is more: Neisseria gonorrhoeae RecX protein stimulates recombination by inhibiting RecA. J. Biol. Chem. 285, 37188–37197.
- Guérillot, R., Siguier, P., Gourbeyre, E., Chandler, M., and Glaser, P. (2014) The diversity of prokaryotic DDE transposases of the mutator superfamily, insertion specificity, and association with conjugation machineries. Genome Biol. Evol. 6, 260–272.
- Haldenby, S., White, M. F., and Allers, T. (2009) RecA family proteins in archaea: RadA and its cousins. Biochem. Soc. Trans. 37, 102–107.
- Hamoen, L. W., Haijema, B., Bijlsma, J. J., Venema, G., and Lovett, C. M. (2001) The Bacillus subtilis competence transcription factor, ComK, overrides LexA-imposed transcriptional inhibition without physically displacing LexA. J. Biol. Chem. 276, 42901–42907.
- Harshey, R. M. (2012) The Mu story: how a maverick phage moved the field forward. Mob. DNA 3, 21.
- Hasegawa, Y., Wakabayashi, M., Nakamura, S., Kodaira, K., Shinohara, H., and Yasukawa, H. (2004) A homolog of Escherichia coli RecA in mitochondria of the cellular slime mold Dictyostelium discoideum. DNA Repair 3, 515–525.
- Higuchi, R. (1989) Using PCR to Engineer DNA. In PCR Technology – Principles and Applications for DNA Amplification. (ed.: Erlich, H.A.), pp. 61–70. Stockton Press, New York.
- Horinouchi, S., and Weisblum, B. (1982) Nucleotide sequence and functional map of pC194, a plasmid that specifies inducible chloramphenicol resistance. J. Bacteriol. 150, 815–825.
- Hörtnagel, K., Voloshin, O. N., Kinal, H. H., Ma, N., Schaffer-Judge, C., and Camerini-Otero, R. D. (1999) Saturation mutagenesis of the E. coli RecA loop L2 homologous DNA pairing region reveals residues essential for recombination and recombinational repair. J. Mol. Biol. 286, 1097–1106.
- Itaya, M., and Matsui, K. (1999) Conversion of Bacillus subtilis 168: Natto producing Bacillus subtilis with mosaic genomes. Biosci. Biotechnol. Biochem. 63, 2034–2037.
- Itaya, M., Yamaguchi, I., Kobayashi, K., Endo, T., and Tanaka, T. (1990) The blasticidin S resistance gene (bsr) selectable in a single copy state in the Bacillus subtilis chromosome. J. Biochem. 107, 799–801.
- Jain, C., and Kleckner, N. (1993) Preferential cis action of IS10 transposase depends upon its mode of synthesis. Mol. Microbiol. 9, 249–260.
- Jang, S., Sandler, S. J., and Harshey, R. M. (2012) Mu insertions are repaired by the double-strand break repair pathway of Escherichia coli. PLoS Genet. 8, e1002642.
- Johnson, R. C., and Reznikoff, W. S. (1984) Copy number control of Tn5 transposition. Genetics 107, 9–18.
- Kamada, M., Hase, S., Sato, K., Toyoda, A., Fujiyama, A., and Sakakibara, Y. (2014) Whole genome complete resequencing of Bacillus subtilis natto by combining long reads with high-quality short reads. PLoS ONE 9, e109999.
- Kamada, M., Hase, S., Fujii, K., Miyake, M., Sato, K., Kimura, K., and Sakakibara, Y. (2015) Whole-genome sequencing and comparative genome analysis of Bacillus subtilis strains isolated from non-salted fermented soybean foods. PLoS One 10, e0141369.
- Katoh, K., and Standley, D. M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780.
- Kidane, D., Carrasco, B., Manfredi, C., Rothmaier, K., Ayora, S., Tadesse, S., Alonso, J. C., and Graumann, P. L. (2009) Evidence for different pathways during horizontal gene transfer in competent Bacillus subtilis cells. PLoS Genet. 5, e1000630.
- Kimura, K., and Itoh, Y. (2007) Determination and characterization of IS4Bsu1-insertion loci and identification of a new insertion sequence element of the IS256 family in a natto starter. Biosci. Biotechnol. Biochem. 71, 2458–2464.
- Kleckner, N., Chalmers, R. M., Kwon, D., Sakai, J., and Bolland, S. (1996) Tn10 and IS10 transposition and chromosome rearrangements: mechanism and regulation in vivo and in vitro. Curr. Top Microbiol. Immunol. 204, 49–82.
- Kruklitis, R., Welty, D. J., and Nakai, H. (1996) ClpX protein of Escherichia coli activates bacteriophage Mu transposase in the strand transfer complex for initiation of Mu DNA synthesis. EMBO J. 9, 935–944.
- Kumar, S., Stecher, G., and Tamura, K. (2016) MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874.
- Kunst, F., Ogasawara, N., Moszer, I., Albertini, A. M., Alloni, G., Azevedo, V., Bertero, M. G., Bessieres, P., Bolotin, A., Borchertet, S., et al. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256.
- Kurumizaka, H., Ikawa, S., Sarai, A., and Shibata, T. (1999) The mutant RecA proteins, RecAR243Q and RecAK245N, exhibit defective DNA binding in homologous pairing. Arch. Biochem. Biophys. 365, 83–91.
- Lavoie, B. D., and Chaconas, G. (1996) Transposition of phage Mu DNA. Curr. Top Microbiol. Immunol. 204, 83–102.
- Lazazzera, B. A. (2000) Quorum sensing and starvation: signals for entry into stationary phase. Curr. Opin. Microbiol. 3, 177–182.
- Lee, C. D., and Wang, T. F. (2009) The N-terminal domain of Escherichia coli RecA have multiple functions in promoting homologous recombination. J. Biomed. Sci. 16, 37.
- Leighton, T. J., and Doi, R. H. (1971) The stability of messenger ribonucleic acid during sporulation in Bacillus subtilis. J. Biol. Chem. 246, 3189–3195.
- Levchenko, I., Yamauchi, M., and Baker, T. A. (1997) ClpX and MuB interact with overlapping regions of Mu transposase: implications for control of the transposition pathway. Genes Dev. 11, 1561–1572.
- Little, J. W., Edmiston, S. H., Pacelli, L. Z., and Mount, D. W. (1980) Cleavage of the Escherichia coli lexA protein by the recA protease. Proc. Natl. Acad. Sci. USA 77, 3225–3229.
- Liu, J., and Zuber, P. (1998) A molecular switch controlling competence and motility: competence regulatory factors ComS, MecA, and ComK control ςD-dependent gene expression in Bacillus subtilis. J. Bacteriol. 180, 4243–4251.
- Loessner, I., Dietrich, K., Dittrich, D., Hacker, J., and Ziebuhr, W. (2002) Transposase-dependent formation of circular IS256 derivatives in Staphylococcus epidermidis and Staphylococcus aureus. J. Bacteriol. 184, 4709–4714.
- Lovett, C. M. Jr., Love, P. E., and Yasbin, R. E. (1989) Competence-specific induction of the Bacillus subtilis RecA protein analog: evidence for dual regulation of a recombination protein. J. Bacteriol. 171, 2318–2322.
- Maamar, H., and Dubnau, D. (2005) Bistability in the Bacillus subtilis K-state (competence) system requires a positive feedback loop. Mol. Microbiol. 56, 615–624.
- Mahillon, J., and Chandler, M. (1998) Insertion sequences. Microbiol. Mol. Biol. Rev. 62, 725–774.
- Manfredi, C., Carrasco, B., Ayora, S., and Alonso, J. C. (2008) Bacillus subtilis RecO nucleates RecA onto SsbA-coated single-stranded DNA. J Biol Chem. 283 24837–24847.
- Mizuno, N., Dramićanin, M., Mizuuchi, M., Adam, J., Wang, Y., Han, Y. W., Yang, W., Steven, A. C., Mizuuchi, K., and Ramón-Maiques, S. (2013) MuB is an AAA+ ATPase that forms helical filaments to control target selection for DNA transposition. Proc. Natl. Acad. Sci. USA 110, E2441–2450.
- Montaño, S. P., Pigli, Y. Z., and Rice, P. A. (2012) The Mu transpososome structure sheds light on DDE recombinase evolution. Nature 491, 413–417.
- Morgan, G. J., Hatfull, G. F., Casjens, S., and Hendrix, R. W. (2002) Bacteriophage Mu genome sequence: analysis and comparison with Mu-like prophages in Haemophilus, Neisseria and Deinococcus. J. Mol. Biol. 317, 337–359.
- Muller, B., Koller, T., and Stasiak, A. (1990) Characterization of the DNA binding activity of stable RecA-DNA complexes: interactions between the two DNA binding sites within RecA helical filaments. J. Mol. Biol. 212, 97–112.
- Mustard, J. A., and Little, J. W. (2000) Analysis of Escherichia coli RecA interactions with LexA, λCI, and UmuD by site-directed mutagenesis of recA. J. Bacteriol. 182, 1659–1670.
- Nagai, T., Tran, L. S., Inatsu, Y., and Itoh, Y. (2000) A new IS4 family insertion sequence, IS4Bsu1, responsible for genetic instability of poly-γ-glutamic acid production in Bacillus subtilis. J Bacteriol. 182, 2387–2392.
- Nastri, H. G., Guzzo, A., Lange, C. S., Walker, G. C., and Knight, K. L. (1997) Mutational analysis of the RecA protein L1 region identifies this area as a probable part of the co-protease substrate binding site. Mol. Microbiol. 25, 967–978.
- Nastri, H. G., and Knight, K. L. (1994) Identification of residues in the L1 region of the RecA protein which are important to recombination or coprotease activities. J. Biol. Chem. 269, 26311–26322.
- Nei, M., and Kumar, S. (2000) Molecular Evolution and Phylogenetics. Oxford University Press, New York.
- Neubert, K., and Brunner, E. (2007) A studentized permutation test for the non-parametric Behrens – Fisher problem. Comput. Stat. Data Anal. 51, 5192–5204.
- Nishito, Y., Osana, Y., Hachiya, T., Popendorf, K., Toyoda, A., Fujiyama, A., Itaya, M., and Sakakibara, Y. (2010) Whole genome assembly of a natto production strain Bacillus subtilis natto from very short read data. BMC Genomics 11, 243.
- Polard, P. I., Prère, M. F., Fayet, O., and Chandler, M. (1992) Transposase-induced excision and circularization of the bacterial insertion sequence IS911. EMBO J. 11, 5079–5090.
- Rice, P., Longden, I., and Bleasby, A. (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277.
- Roca, A. I., and Cox, M. M. (1997) RecA protein: structure, function, and role in recombinational DNA repair. Prog. Nucleic Acid Res. Mol. Biol. 56, 129–223.
- Saito, T., Chibazakura, T., Takahashi, K., Yoshikawa, H., and Sekine, Y. (2010) Measurements of transposition frequency of insertion sequence IS1 by GFP hop-on assay. J. Gen. Appl. Microbiol. 56, 187–192.
- Sakai, A., and Cox, M. M. (2009) RecFOR and RecOR as distinct RecA loading pathways. J. Biol. Chem. 284, 3264–3272.
- Sekine, Y., and Ohtsubo, E. (1989) Frameshifting is required for production of the transposase encoded by insertion sequence 1. Proc. Natl. Acad. Sci. USA 86, 4609–4613.
- Shibata, T., DasGupta, C., Cunningham, R. P., and Radding, C. M. (1979) Purified Escherichia coli recA protein catalyzes homologous pairing of superhelical DNA and single-stranded fragments. Proc. Natl. Acad. Sci. USA 76, 1638–1642.
- Shiga, Y., Sekine, Y., Kano, Y., and Ohtsubo, E. (2001) Involvement of H-NS in transpositional recombination mediated by IS1. J. Bacteriol. 183, 2476–2484.
- Shinohara, T., Ikawa, S., Iwasaki, W., Hiraki, T., Hikima, T., Mikawa, T., Arai, N., Kamiya, N., and Shibata, T. (2015) Loop L1 governs the DNA-binding specificity and order for RecA-catalyzed reactions in homologous recombination and DNA repair. Nucleic Acids Res. 43, 973–986.
- Sierro, N., Makita, Y., de Hoon, M., and Nakai, K. (2008) DBTBS: a database of transcriptional regulation in Bacillus subtilis containing upstream intergenic conservation information. Nucleic Acids Res. 36 (Database issue), D93–D96.
- Skiba, M. C., and Knight, K L. (1994) Functionally important residues at a subunit interface site in the RecA protein from Escherichia coli. J. Biol. Chem. 269, 3823–3828.
- Takahashi, K., Chibazakura, T., Sekine, Y., and Yoshikawa, H. (2007a) Development of a new “GFP hop-on assay” system for insertion sequence transposition in Bacillus subtilis 168 using IS4Bsu1 from B. subtilis (natto). Biochem. Biophys. Res. Commun. 355, 426–430.
- Takahashi, K., Sekine, Y., Chibazakura, T., and Yoshikawa, H. (2007b) Development of an intermolecular transposition assay system in Bacillus subtilis 168 using IS4Bsu1 from Bacillus subtilis (natto). Microbiology 153, 2553–2559.
- Taylor, A. L. (1963) Bacteriophage-induced mutation in Escherichia coli. Proc. Natl. Acad. Sci. USA 50, 1043–1051.
- Tran, L. S. P., Nagai, T., and Itoh, Y. (2000) Divergent structure of the ComQXPA quorum-sensing components: molecular basis of strain-specific communication mechanism in Bacillus subtilis. Mol. Microbiol. 37, 1159–1171.
- van der Veen, S., van Schalkwijk, S., Molenaar, D., de Vos, W. M., Abee, T., and Wells-Bennik, M. H. J. (2010) The SOS response of Listeria monocytogenes is involved in stress resistance and mutagenesis. Microbiology 156, 374–384.
- Wojciechowski, M. F., Peterson, K. R., and Love, P. E. (1991) Regulation of the SOS response in Bacillus subtilis: evidence for a LexA repressor homolog. J. Bacteriol. 173, 6489–6498.
- Yuan, Y. W., and Wessler, S. R. (2011) The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. USA 108, 7884–7889.