Introduction
Gladiolus (Gladiolus spp.), a bulbous plant with spikes, is cultivated as an ornamental flower. Gladiolus is native to Africa, southern Europe, and western Asia. It is a popular potted and cut flower species because it has various colors and shapes and is easy to cultivate. Gladiolus florets are relatively insensitive to ethylene, with tepal wilting occurring independently of ethylene (Woltering and van Doorn, 1988). However, it was reported that trehalose and cycloheximide extend the vase life of gladiolus florets (Iwaya-Inoue and Takata, 2001; Otsubo and Iwaya-Inoue, 2000; Yamada et al., 2003; Yamane and Ogata, 1995; Yamane et al., 2005). Trehalose is a disaccharide (α-d-glucopyranosyl-α-d-glucopyranosyl) (Sussich et al., 1998) that is present in bacteria, yeast, fungi, insects, and invertebrate animals, but is rarely found in higher plants (Müller et al., 1995). In addition to gladiolus, trehalose can prolong the vase life of other cut-flower species, including tulip and astilbe (Iwaya-Inoue and Takata, 2001; Otsubo and Iwaya-Inoue, 2000; Villanueva et al., 2019; Yamada et al., 2003; Yamane et al., 2005; Yamazaki et al., 2020). Trehalose maintains the turgor pressure in petal tissues and delays senescence (i.e., programmed cell death; PCD) in gladiolus (Iwaya-Inoue and Takata, 2001; Otsubo and Iwaya-Inoue, 2000; Yamada et al., 2003). Cycloheximide inhibits protein synthesis by binding to the E-site of the eukaryotic 60S ribosomal subunit (Garreau de Loubresse et al., 2014; Schneider-Poetsch et al., 2010). Cycloheximide interrupts translation elongation at any time, as its binding to the E site is independent of the occupancy of the E site by de-acylated tRNA (Schneider-Poetsch et al., 2010). Previous research indicated that cycloheximide treatment delays the senescence of some flowers, including gladiolus and daylily (Jones et al., 1994; Lukaszewski and Reid, 1989; Yamane and Ogata, 1995). In an earlier study, cycloheximide inhibited the senescence of gladiolus tepals, but also suppressed flowering when it was applied to pre-flowering florets (Yamane and Ogata, 1995). Therefore, inhibited protein synthesis may contribute to the extension of the vase life of some flowers. However, there are few reports describing expressed genes inhibited by cycloheximide in the petals or tepal senescence.
Chloramphenicol is a protein synthesis inhibitor in prokaryotic cells that adversely affects bacterial growth (Hartke et al., 1998). Chloramphenicol inhibits protein synthesis by binding to the 23S ribosomal subunit, which has peptidyl transferase activity (Thompson et al., 2002). High chloramphenicol concentrations are toxic to mammals, including humans (Holt et al., 1997), but treating cut tulip flowers with chloramphenicol can prolong their vase life (Iwaya-Inoue and Takata, 2001). Unfortunately, because there are few reports regarding chloramphenicol treatments of cut flowers, the utility of chloramphenicol for maintaining the quality of cut flowers is unknown.
Petal senescence is regulated by various enzymes (e.g., proteases, polysaccharide-degrading enzymes, sugar metabolism-related enzymes, and oxidoreductases). The expression of cysteine protease-encoding genes (e.g., senescence-related genes) increases as the petal senescence process progresses in Hemerocallis (Valpuesta et al., 1995), Alstroemeria (Wagstaff et al., 2002) and gladiolus (Arora and Singh, 2004). It was reported that a thiol protease is induced during senescence in pea (Cercós et al., 1999). In Hemerocallis, thiol protease (SEN102 and SEN11) transcripts showed peaks in both petals and sepals, when wilting symptoms were apparent (Guerrero et al., 1998; Valpuesta et al., 1995). Cysteine proteases such as the senescence-associated gene (SAG) 2 and SAG12 are reported to function as developmental markers of senescence in Arabidopsis (Grbić, 2003). In Iris, expression of SAG 39-like is associated with flower senescence (Liu et al., 2022).
Invertase catalyzes sucrose hydrolysis (Kotwal and Shankar, 2009). A previous study on peony revealed that sucrose treatment helped maintain the quality of cut flowers and affected the expression of invertase genes (Xue et al., 2018). Pectinesterase, which is involved in cell wall degradation, promotes the senescence of flowers (van Doorn and Woltering, 2008; van Doorn et al., 2003). This enzyme and polygalacturonase coordinately degrade the glycosidic bonds in the cell wall (Niederhuth et al., 2013).
Peroxidase activity increases during senescence (Abarca et al., 2001; Passardi et al., 2004). Additionally, peroxidase gene expression levels are upregulated in response to wounding stress (Cheong et al., 2002). In senescing gladiolus tepals, peroxidase activity increases, whereas superoxide dismutase activity decreases (Yamane et al., 1999).
Petal senescence and PCD are genetically programmed processes that occur at specific developmental stages (Rogers, 2006, 2013; van Doorn and Woltering, 2008). To identify the regulators of the age-dependent PCD regulatory pathways associated with petal aging, but unrelated to ethylene signaling, ethylene-insensitive flowers, including those of Hemerocallis (Panavas et al., 1999), Iris hollandica (van Doorn et al., 2003), and Alstroemeria (Breeze et al., 2004) plants, were investigated. However, PCD-specific genes influencing petal senescence have not been identified in these species. Conversely, recent studies suggest that the NAC [no apical meristem (NAM), Arabidopsis transcription activation factor (ATAF), and cup-shaped cotyledon (CUC)] transcription factor gene EPHEMERAL1 (EPH1) encodes an important regulator of age-dependent PCD during petal senescence in morning glory (Ipomoea nil) (Shibuya et al., 2014, 2018) and Japanese gentians (Takahashi et al., 2022). The NAC transcription factors were named according to the genes in which their characteristic domain was first detected, namely NAM in petunia and ATAF1-2 and CUC2 (cup-shaped cotyledon 2) in Arabidopsis thaliana (Ooka et al., 2003). The NAC family genes encode plant-specific transcription factors that are involved in developmental processes and stress responses in various tissues (Li et al., 2021; Ni et al., 2017; Olsen et al., 2005; Ooka et al., 2003; Puranik et al., 2012; Yeung et al., 2018). In particular, NAC029 (AtNAP/ANAC029), NAC92 (ORE1/AtNAC2/ANAC092) and NAC083 (VND-INTERACTING2/ANAC083) control leaf senescence (Balazadeh et al., 2010; Guo and Gan, 2006; Kim et al., 2009; Yang et al., 2011; Yeung et al., 2018). NAC transcription factors are also implicated in petal aging (Shibuya et al., 2014, 2018; Trupkin et al., 2019). In tulip, TgNAP (NAC-like, activated by APETALA3/PISTILLATA) positively regulates petal senescence (Meng et al., 2022). WRKY transcription factors are related to various biotic and abiotic stress responses (Woo et al., 2013). Specifically, WRKY22 and WRKY6 regulate leaf senescence (Robatzek and Somssich, 2001; Woo et al., 2013; Zhou et al., 2011).
In this study, we performed RNA-sequencing (RNA-seq) analysis to clarify the effects of trehalose and cycloheximide on gene expression and to identify senescence-related genes in cut gladiolus tepals. Differentially expressed genes (DEGs) were functionally annotated using the Gene Ontology (GO) (Harris et al., 2004) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto, 2000) databases to characterize the genes associated with floral senescence.
Materials and Methods
Plant materials
Gladiolus (Gladiolus spp.) ‘Fujinoyuki’ plants were grown in an experimental greenhouse at Utsunomiya University (36°32'55" N, 139°54'42" E). Cut inflorescences with 2–3 leaves were harvested when a few tepals appeared on the first florets at the base of the spikes. The cut opened florets were placed in distilled water and then incubated at 20–21°C with 60% relative humidity under cool white fluorescent light (15 μmol·m−2·s−1 photosynthetic photon flux density). The opened florets were collected from the flower stalk and the bracts were removed, after which the florets were placed in a vial containing 10 mL treatment solution. Cut florets treated with 0.1 M trehalose (Tre), 300 μM cycloheximide (CHI), or 50 μM chloramphenicol (CAP) were compared with control cut florets treated with distilled water (Cont). To prevent bacterial contamination, all solutions were supplemented with 200 ppm (v/v) Kathon CG (Dow Chemical Company, Midland, MI, USA). The end of vase life was defined when about half of the tepals wilted or browned.
Total RNA isolation
Total RNA was isolated from the tepals of the cut florets at 0 and 2 days (i.e., 0d and 2d) after the Cont, Tre, and CHI treatments. For each tepal sample, 100 mg tissue was ground to a fine powder in liquid nitrogen. Total RNA was extracted twice using a cetyltrimethylammonium bromide (CTAB) buffer and chloroform:isoamyl alcohol (24:1) as described by Chang et al. (1993) and then further isolated using a Maxwell® RSC Instrument (Promega, Madison, WI, USA) and a Maxwell® RSC Plant RNA Kit (Promega). The isolated total RNA was quantified and examined for protein contamination (A260/A280) and reagent contamination (A260/A230) using a NanoDropTM One spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). RNA integrity was evaluated using a 1.0% agarose gel stained with ethidium bromide.
Library preparation and Illumina sequencing
The RNA integrity number was determined using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and an RNA 6000 Nano kit (Agilent Technologies). The RNA-seq libraries were constructed using a KAPA Stranded mRNA-Seq Kit (Roche Sequencing and Life Science, Indianapolis, IN, USA) according to the manufacturer’s instructions. Double-stranded cDNA fragments were purified using AMPure XP beads (Beckman Coulter, Brea, CA, USA) to eliminate free nucleotides, enzymes, buffer, and RNA and then ligated with adapters (Set A and Set B from TruSeq DNA Sample Prep LT Kits; Illumina, San Diego, CA, USA). The libraries were quantified using a LightCycler® 480 Instrument (Roche, Basel, Switzerland) and a Kapa Library Quantification Kit (Roche Sequencing and Life Science) according to the manufacturer’s instructions. Each library was pooled (4 nM) and quantified again. Finally, the pooled libraries were sent to a sequencing facility for the cluster generation step performed by a MiSeq Sequencer (Illumina). Each paired-end library was prepared according to the instructions provided in the MiSeq Reagent Kit (v3) (150 cycles; Illumina). Three biological replicates (n = 3) of the 0d, 2d_Cont, 2d_Tre, and 2d_CHI samples were sequenced. The data sets are available in the DDBJ Sequence Read Archive (DRA) (accession number DRR459504–DRR459515).
Bioinformatics analysis
The quality of the sequencing reads was assessed using FastQC (v0.11.3). Raw reads were first processed using Trimmomatic-0.39 (Bolger et al., 2014) to remove adapter sequences, low-quality reads (quality scores < 30), and the last (76th) base, as well as reads shorter than 50 bp. High-quality short reads were assembled into unigenes using the Trinity program (v2.8.5), all settings default (Haas et al., 2013). The rRNA sequences among the unigenes were discarded by removing sequences with matches in the SILVA rRNA database using the megablast program (Zhang et al., 2000). Additionally, the high-quality short reads were mapped to the remaining unigenes using Bowtie (Langmead et al., 2009).
To estimate transcript expression levels, the number of uniquely mapped reads for each unigene was determined and normalized to fragments per kilobase per million reads (FPKM) values according to the RSEM method (Li and Dewey, 2011). A pairwise comparison of the FPKM values was performed as follows: 0d vs 2d_Cont, 2d_Cont vs 2d_Tre, and 2d_Cont vs 2d_CHI. The edgeR package (Robinson et al., 2010) was used to calculate p-values and expression-level fold-change (FC) values. The p-value was used to identify genes that were significantly differentially expressed between paired treatments. Significant DEGs were identified on the basis of a false discovery rate (FDR) ≤ 0.05 and |logFC| ≥ 1.
To predict the biological functions of the unigenes, the sequences were annotated according to the Universal Protein Resource KnowledgeBase (UniProtKB)/Swiss-Prot database (NCBI SwissProt) and the NCBI nr/nt database (NCBI nucleotide collection) using the blastx and blastn programs. Additionally, putative coding regions were extracted from the Trinity transcripts using TransDecoder. Then, the coding regions were annotated by GO annotation against the Pfam database, and those data were used for GO enrichment analysis. The results of the GO enrichment analysis are presented in Z-score bar plots for specific GO terms. A higher Z-score indicates the GO term is increasing while a lower Z-score indicates it is decreasing. Heatmaps for the genes annotated with the five senescence-related GO terms were prepared using an online tool (http://shinyheatmap.com/). The upregulated (red) and downregulated (green) expression levels (FPKM values) were indicated by different colors. The enriched KEGG pathways among the unigenes were determined using KofamKOALA (KEGG Orthology And Links Annotation) (Aramaki et al., 2019).
Quantitative gene expression analysis
A Maxwell® RSC Plant RNA Kit (Promega) was used to extract total RNA from 200 mg gladiolus tepals that had been stored at −80°C and lysed in CTAB buffer. The extracted RNA was reverse transcribed to cDNA using a ReverTra Ace® qPCR RT Kit (Toyobo Co. Ltd., Osaka, Japan). The cDNA was stored at −20°C prior to PCR amplification.
For the quantitative real-time PCR (qPCR) analysis, cDNA samples were diluted to 20 ng·μL−1 in 100 μL TE (Tris-EDTA) buffer. The concentrations were determined using a NanoDropTM One spectrophotometer (Thermo Fisher Scientific). The qPCR analysis was conducted using a KAPA SYBR Fast qPCR Kit (Roche Diagnostics, Mannheim, Germany) and the LightCycler 480 system (Roche Diagnostics). Actin (139 bp) was selected as the reference gene for the analysis of NAC048 (126 bp), NAC68 (119 bp), NAC073 (110 bp), WRKY6 (101 bp), WRKY11 (141 bp), and WRKY24 (150 bp) (Table S1). Abbreviations of NAC transcription factors followed UniProt (https://www.uniprot.org). The qPCR program was as follows: 95°C for 3 min; 45 cycles of 95°C for 3 s and 60°C for 20 s. This was followed by a melting curve analysis.
Construction and analysis of a phylogenetic tree comprising NAC proteins
A phylogenetic tree was constructed based on the sequence alignments of the NAC family proteins detected in this study. The senescence-related NAC proteins included EPHEMERAL1 (EPH1) (Shibuya et al., 2014, 2018), NAC029, NAC083, and NAC92. The following representative NAC proteins from seven groups were also included in the phylogenetic tree: NAM/NAC1 (group I), OsNAC7/NAC076 (group II), ANAC011 (group III), AtNAC3/NAC59 (group IV), OsNAC8/NAC074 (group VI), OsNAC3/NAC067 (group VII), and NAC2/NAC063 (group VIII) except ONAC022 (group V) (Ooka et al., 2003; Li et al., 2021).
Results and Discussion
Effects of trehalose or protein synthesis inhibitor treatments on ornamental quality
The Tre and CHI treatments significantly extended the vase life of gladiolus florets by approximately 1 day, i.e., 30% of Cont (Fig. 1). The Tre treatment affected the fresh weight less than the other treatments. The fresh weight after 1 day was significantly lower for the Tre-treated samples than for the other samples (Fig. S1). The wilting of the samples also differed among the treatments (Fig. S2). Specifically, the Cont and Tre treatments caused the whole sample to wilt, whereas the CHI treatment resulted in senescence symptoms, including browning at the flower edge, but the floral shape was maintained. In response to the CAP treatment, the outer tepal drooped within 2–3 days, but many of the inner tepals, stamens, and pistils were relatively unaffected.
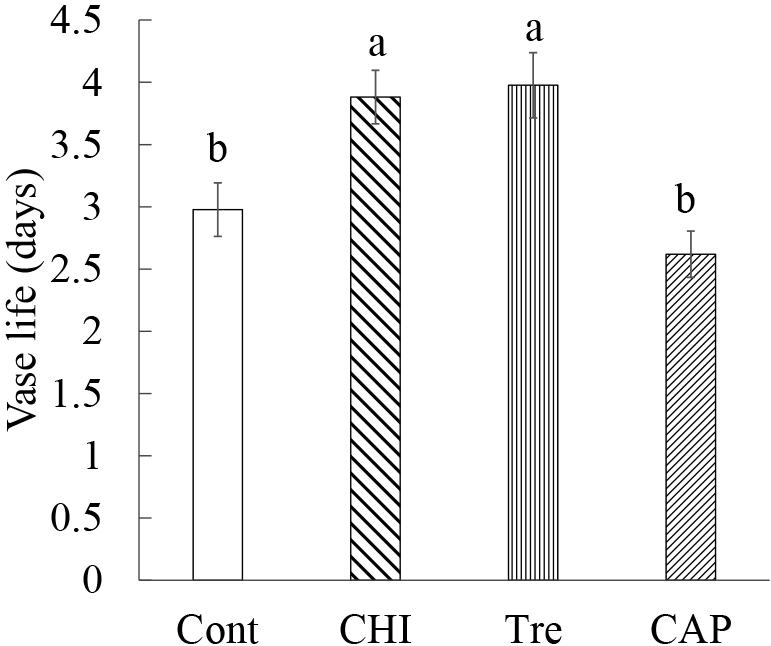
The increase in the fresh weight after 1 day was smaller for the Tre treatment than for the other treatments (Fig. S1), suggesting the Tre treatment did not promote tissue development and water absorption. However, the decrease in the fresh weight after 2 days tended to be more gradual in the Tre treatment than in the other treatments. Previous studies indicated that trehalose treatments are conducive to stabilizing the water content and turgor pressure in gladiolus and tulip tepals (Iwaya-Inoue and Takata, 2001; Otsubo and Iwaya-Inoue, 2000). Another study confirmed that trehalose suppresses nuclear fragmentation in gladiolus petal cells and inhibits PCD (Yamada et al., 2003). Therefore, the more gradual decrease in the fresh weight after the Tre treatment than after the other treatments likely indicates a loss of water from the tepals and a PCD-related decrease in the number of cells was suppressed.
Following the CHI treatment, browning and water-soaked flowers were detected before wilting (Fig. S2). Petals wilt because of damage to biological membranes and decreases in the turgor pressure of petal cells during senescence (Zhang et al., 2021). Therefore, we speculated that the browning and water-soaked flowers before wilting reflected the phytotoxic effects of cycloheximide rather than tepal senescence.
The extension of the vase life with the Tre and CHI treatments was consistent with the results of previous studies (Iwaya-Inoue and Takata, 2001; Jones et al., 1994; Otsubo and Iwaya-Inoue, 2000; Yamane and Ogata, 1995; Yamane et al., 2005). The CAP treatment is reported to help preserve the quality of cut tulip flowers (Iwaya-Inoue and Takata, 2001), but did not effectively preserve the ornamental value of gladiolus florets. However, the quality of the inner tepals was higher in the CAP treatment than in the Cont. Because outer tepals are at a more advanced developmental stage than inner tepals, it is possible that the timing of the CAP treatment was inappropriate for the onset of aging, but this possibility will need to be experimentally verified.
De novo transcriptome assembly and DEG analysis
We obtained 81,136 unique sequences, with an average sequence length of approximately 946.67 bp and a total sequence length of 70,809,222 bp (Table 1). The N50 value was 1,540 bp.
We compared the gene expression levels among the 0d, 2d_Cont, 2d_CHI, and 2d_Tre florets to identify DEGs. Using FDR ≤ 0.05 and |logFC| ≥ 1 as the thresholds, 2,892, 4,670, and 57 significant DEGs were detected for the 0d vs 2d_Cont, 2d_Cont vs 2d_CHI, and 2d_Cont vs 2d_Tre comparisons, respectively. Table S2 presents the number of DEGs that were expressed at higher or lower levels in the 2d_Cont florets for each comparison. These results suggest that the Tre treatment had almost no effect on gene expression.
GO enrichment analysis of DEGs
The significant DEGs in each treatment were subjected to GO enrichment analysis and assigned to biological process (BP), molecular function (MF), and cellular component (CC) categories. In the MF category, the most enriched GO terms among the DEGs for the 0d vs 2d_Cont comparison were pectinesterase activity, enzyme inhibitor activity, polygalacturonase activity, hydrolase activity, hydrolyzing O-glycosyl compounds, cysteine-type endopeptidase activity, and structural constituent of ribosome (Fig. 2A). The significantly enriched GO terms assigned to the DEGs for the 2d_CHI vs 2d_Cont comparison included pectinesterase activity, oxidoreductase activity, structural constituent of ribosome, and sequence-specific DNA binding transcription factor activity (Fig. 2B). The GO terms were enriched in 2d_Cont except sequence-specific DNA binding transcription factor activity (Fig. 2B). There were no enriched GO terms among the DEGs for the 2d_Tre vs 2d_Cont comparison.
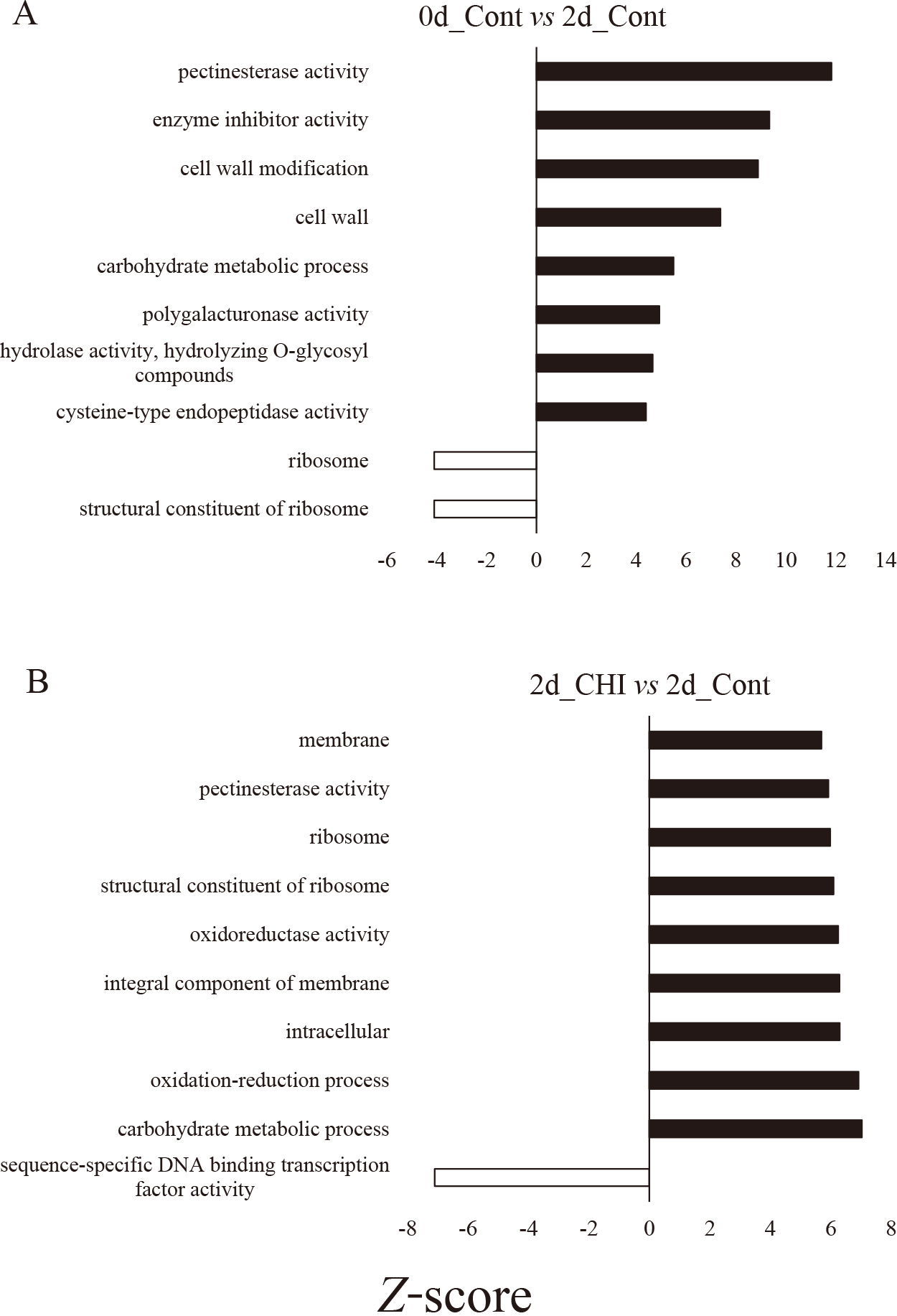
In the BP category, cell wall modification and carbohydrate metabolic process were the enriched GO terms among the DEGs for the 0d_Cont vs 2d_Cont comparison (Fig. 2A), whereas carbohydrate metabolic process and oxidation-reduction process were the most enriched GO terms assigned to the DEGs for the 2d_CHI vs 2d_Cont comparison (Fig. 2B).
In the CC category, cell wall and ribosome were the significantly enriched GO terms among the DEGs for the 0d vs 2d_Cont comparison (Fig. 2A), while intracellular, integral component of membrane, ribosome, and membrane were among the most enriched GO terms assigned to the DEGs for the 2d_CHI vs 2d_Cont comparison (Fig. 2B).
Heatmaps were created on the basis of the FPKM values for the genes annotated with the following five senescence-associated GO terms: cysteine-type endopeptidase activity (Fig. S3A), pectinesterase activity (Fig. S3B), polygalacturonase activity (Fig. S3C), cell wall (Fig. S3D), and cell wall modification (Fig. S3E).
For the GO term cysteine-type endopeptidase activity, the z-score was significantly higher for 2d_Cont than for 0d (Fig. 2A). In the corresponding heatmap, the expression level of most of the genes tended to be higher for 2d_Cont than for 0d (Fig. S3A). Cysteine protease gene expression levels were reported to increase in the senescing tepals of Alstroemeria (Wagstaff et al., 2002), Hemerocallis (Valpuesta et al., 1995) and gladiolus (Arora and Singh, 2004). Our results reconfirmed that the expression of cysteine protease genes is upregulated in senescing gladiolus tepals.
The 0d vs 2d_Cont comparison revealed that the Z-scores for cell wall-related GO terms, such as pectinesterase activity, polygalacturonase activity, cell wall, and cell wall modification, were higher for 2d_Cont (Fig. 2A). In the heatmaps for all four of these GO terms, the expression levels of many genes were higher for 2d_Cont than for 0d (Fig. S3B–E). The 2d_CHI vs 2d_Cont comparison indicated the expression levels of the genes annotated with the GO term of cell wall (Z-score: 5.6) and cell wall modifications (Z-score: 4.0) were significantly higher for 2d_Cont (data not shown). Petal wilting is associated with cell wall changes (De Vetten and Huber, 1990; Eason, 2006; van Doorn and Woltering, 2008). These findings are suggestive of a relationship between the senescence of gladiolus florets and cell wall modifications. Future studies will need to characterize the cell wall changes during the senescence of gladiolus florets.
KEGG pathway analysis of DEGs
Among the KO (KEGG Orthology)-annotated DEGs, those with |logFC| ≥ 1 were mapped. The 0d vs 2d_Cont and 2d_Cont vs 2d_CHI comparisons detected 2,646 and 11,290 KO-annotated DEGs, respectively (Table S3). The KEGG Mapper Reconstruct mapping results (green: relatively high expression levels for 0d or 2d_CHI; pink: relatively high expression levels for 2d_Cont) are presented in Figures S4–S7. The 0d vs 2d_Cont and 2d_CHI vs 2d_Cont comparisons indicated the expression levels of the genes in the galactose metabolic pathway related to sucrose synthesis were higher for 2d_Cont (Figs. S4 and S5). In daylily and gladiolus, sucrose is synthesized in senescing petals and then translocated to other tissues (Bielski, 1995; Yamane et al., 1993). Therefore, the sucrose synthetic pathway may be activated by the senescence-related translocation. Of the DEGs associated with starch and sucrose metabolism revealed by the 2d_CHI vs 2d_Cont comparison, the expression levels of the genes such as beta-glucosidase (EC 3.2.1.21) and alpha-amylase (EC 3.2.1.1) were higher for 2d_Cont than for 2d_CHI (Figs. S6 and S7). It was reported that the expression of a gene related to cell wall degradation increased during senescence (van Doorn and Woltering, 2008; van Doorn et al., 2003). This also suggests that cycloheximide delays the glycometabolism and cell wall degradation associated with senescence.
Senescence-related genes
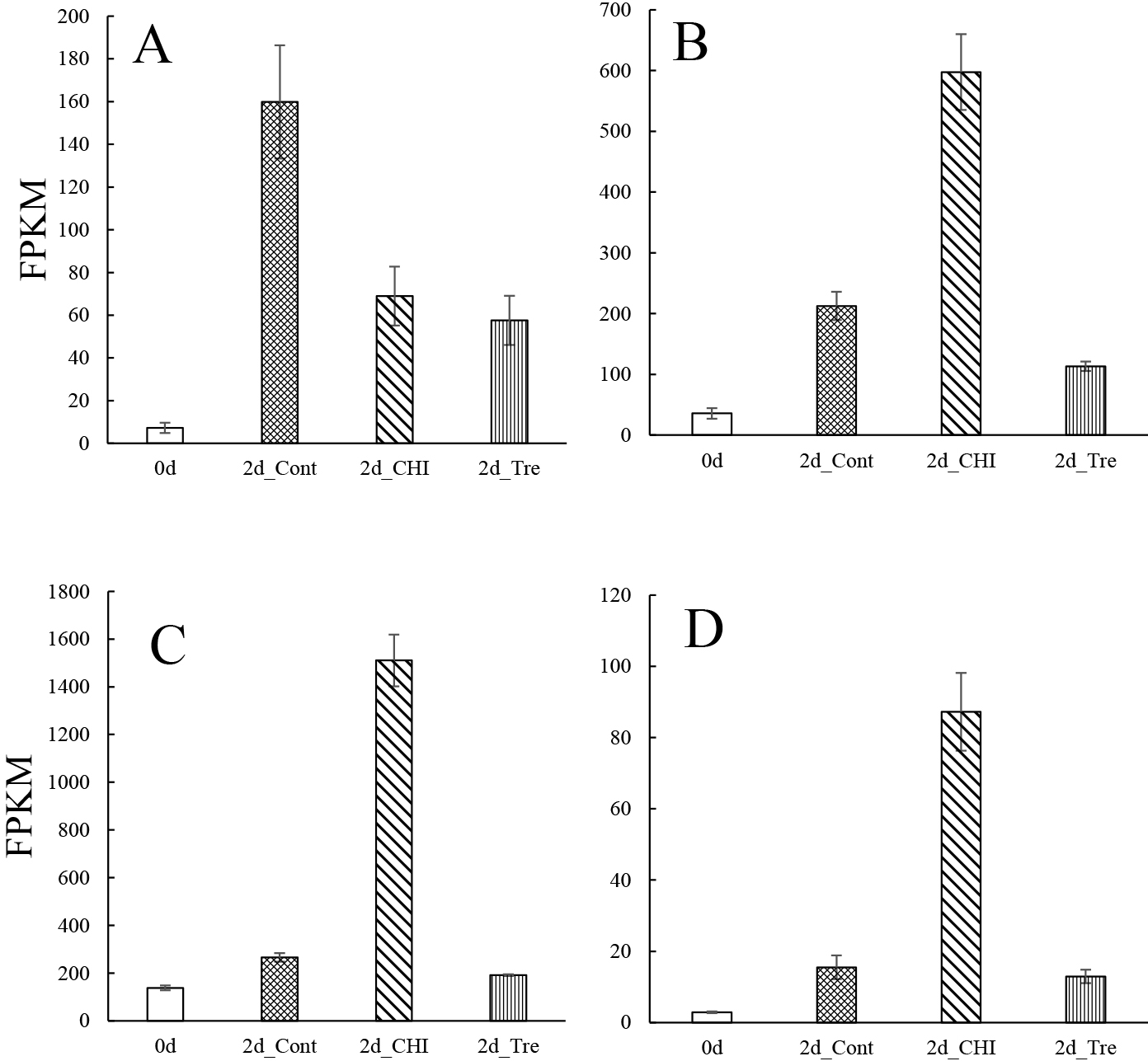
On the basis of previous reports, we analyzed the DEGs encoding cysteine proteases (Fig. 3), invertases (Fig. S8), peroxidases (Fig. S9), pectinesterases (Fig. 4), NAC transcription factors (Figs. 5 and S10), and WRKY (Figs. 6 and S11) transcription factors in terms of their FPKM values.
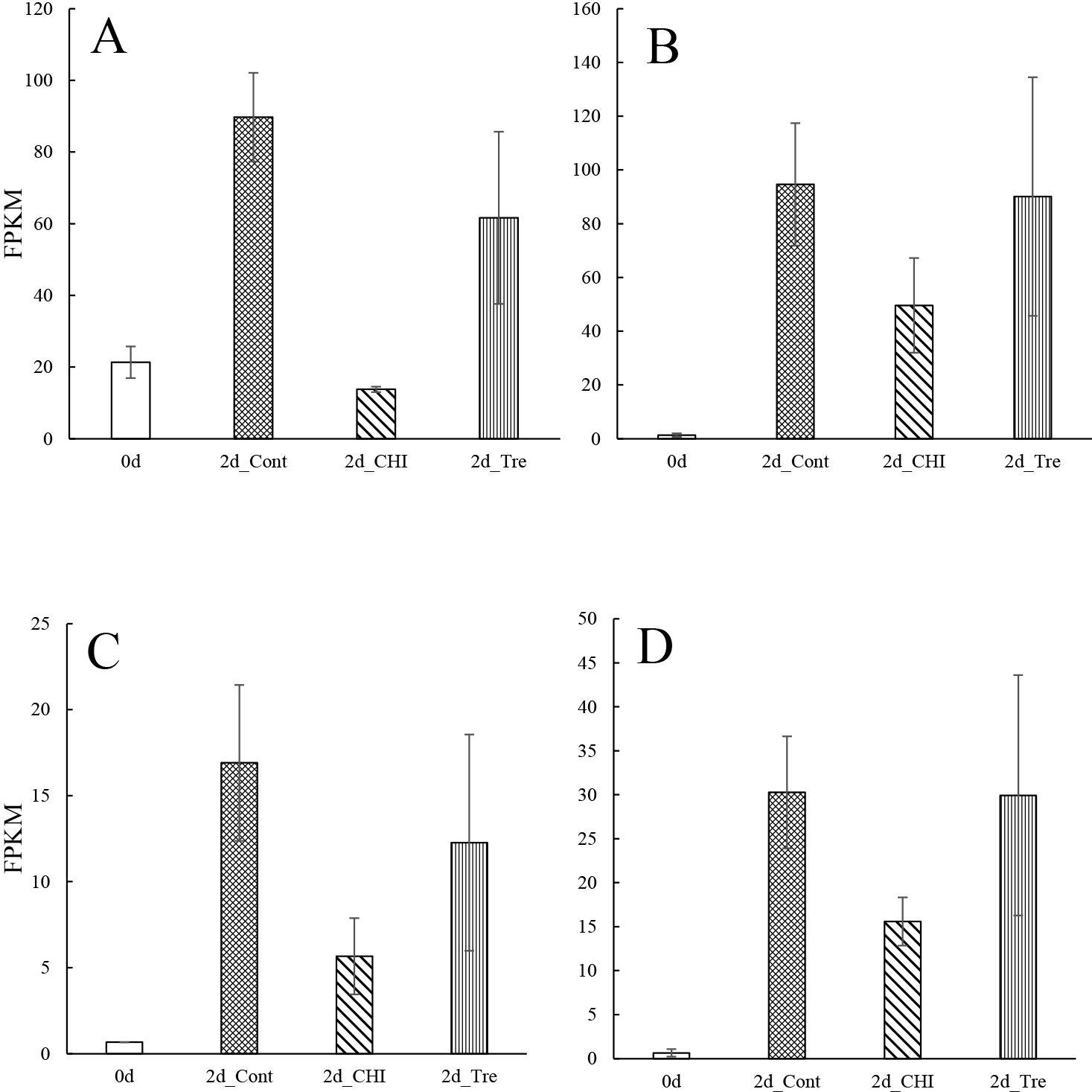
Earlier research confirmed cysteine protease activities increase during petal senescence (Grbić, 2003; Jones et al., 1995; Valpuesta et al., 1995; Wagstaff et al., 2002). The protein content decreases as gladiolus tepals wither after the full bloom stage (Yamane et al., 1991). In the current study, the thiol protease SEN102 and senescence-specific cysteine protease SAG39 had the highest FPKM values in 2d_Cont and were suppressed by CHI and Tre (Fig. 3), which is consistent with previously reported results for SEN102 in Hemerocallis (Guerrero et al., 1998; Valpuesta et al., 1995) and SAG 39-like in Iris (Liu et al., 2022), suggesting that SEN102 and SAG39 are associated with tepal senescence in gladiolus.
Invertase is an enzyme that catalyzes the hydrolysis of sucrose (Kotwal and Shankar, 2009). Acid-invertase activities and the osmotic potential affect the growth of gladiolus tepals (Yamane et al., 1991). In the present study, the expression of three invertase genes decreased in three DEGs (Fig. S8A–C), was maintained in three DEGs (Fig. S8D–F) and increased in two DEGs (Fig. S8G, H) during senescence. Only the gene encoding beta-fructofuranosidase, insoluble isoenzyme 4, was significantly differentially expressed between 2d_Cont and the other treatment groups (Fig. S8H). Treating gladiolus with sucrose did not significantly delay senescence (Otsubo and Iwaya-Inoue, 2000). Therefore, invertase may not strongly influence gladiolus tepal senescence.
Peroxidase activities increase as senescence progresses (Abarca et al., 2001; Passardi et al., 2004). Although there is a significant increase in peroxidase activities at the onset of senescence, cycloheximide inhibits this increase in gladiolus tepals (Yamane et al., 1999). Our analysis of the expression of 10 peroxidase genes (Fig. S9A–J) indicated that the FPKM values for the peroxidase N, peroxidase 15, and peroxidase P7 genes were more than two times higher for 2d_Cont than for the treatment group with the second highest FPKM values (Fig. S9A, C, E). Based on the data presented in Figure 1, these three peroxidase genes are likely involved in senescence.
Pectinesterase mediates cell wall degradation and the activities of this enzyme are reported to increase during senescence (van Doorn and Woltering, 2008; van Doorn et al., 2003). Consistent with these earlier findings, the FPKM values for the most pectinesterase genes were more than two times greater for 2d_Cont than for the other treatment groups in this study (Figs. 4 and S3B). Thus, pectinesterases probably contribute to the wilting of gladiolus tepals through cell wall degradation together with polygalactulonase (Fig. S3C).
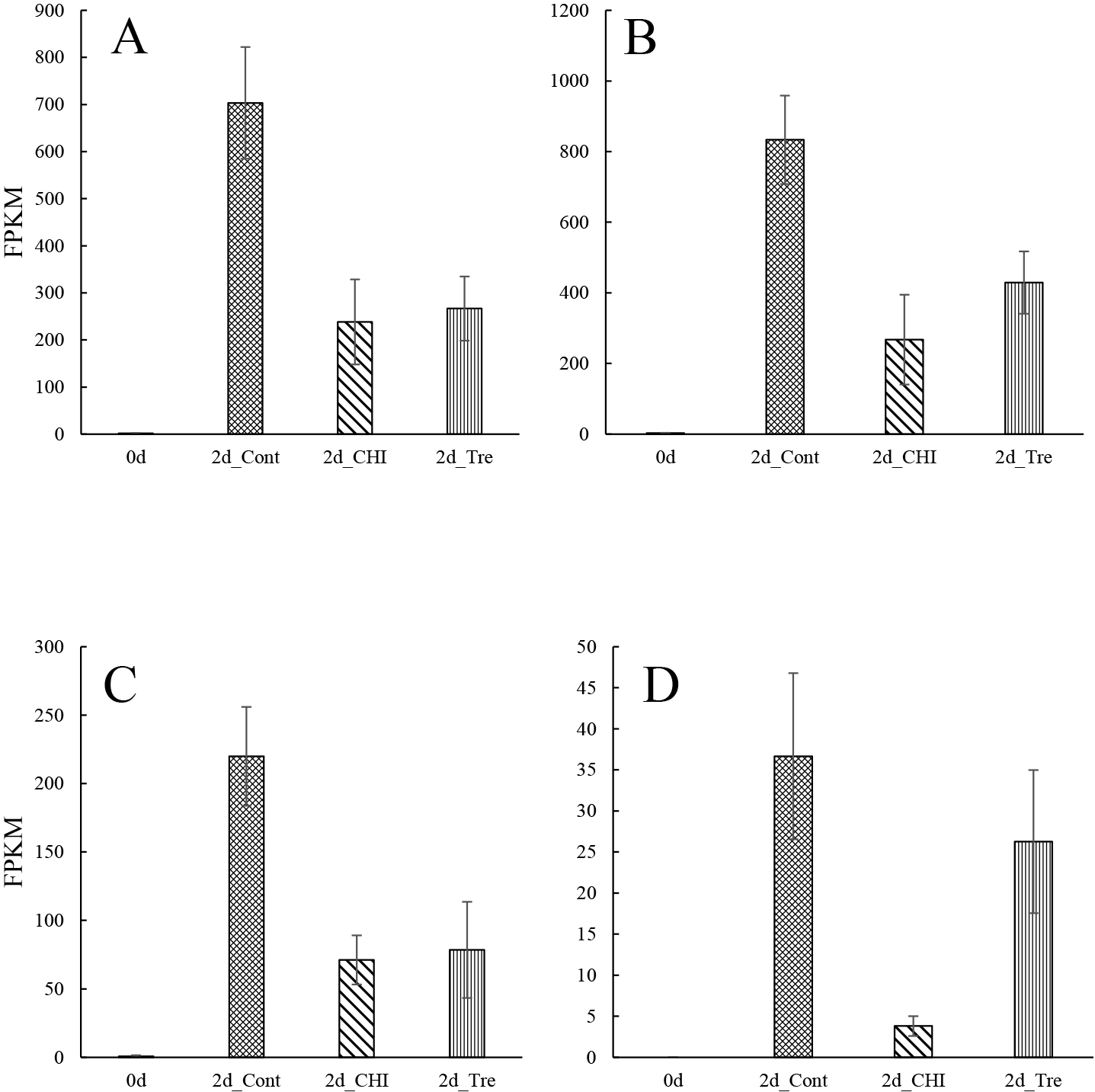
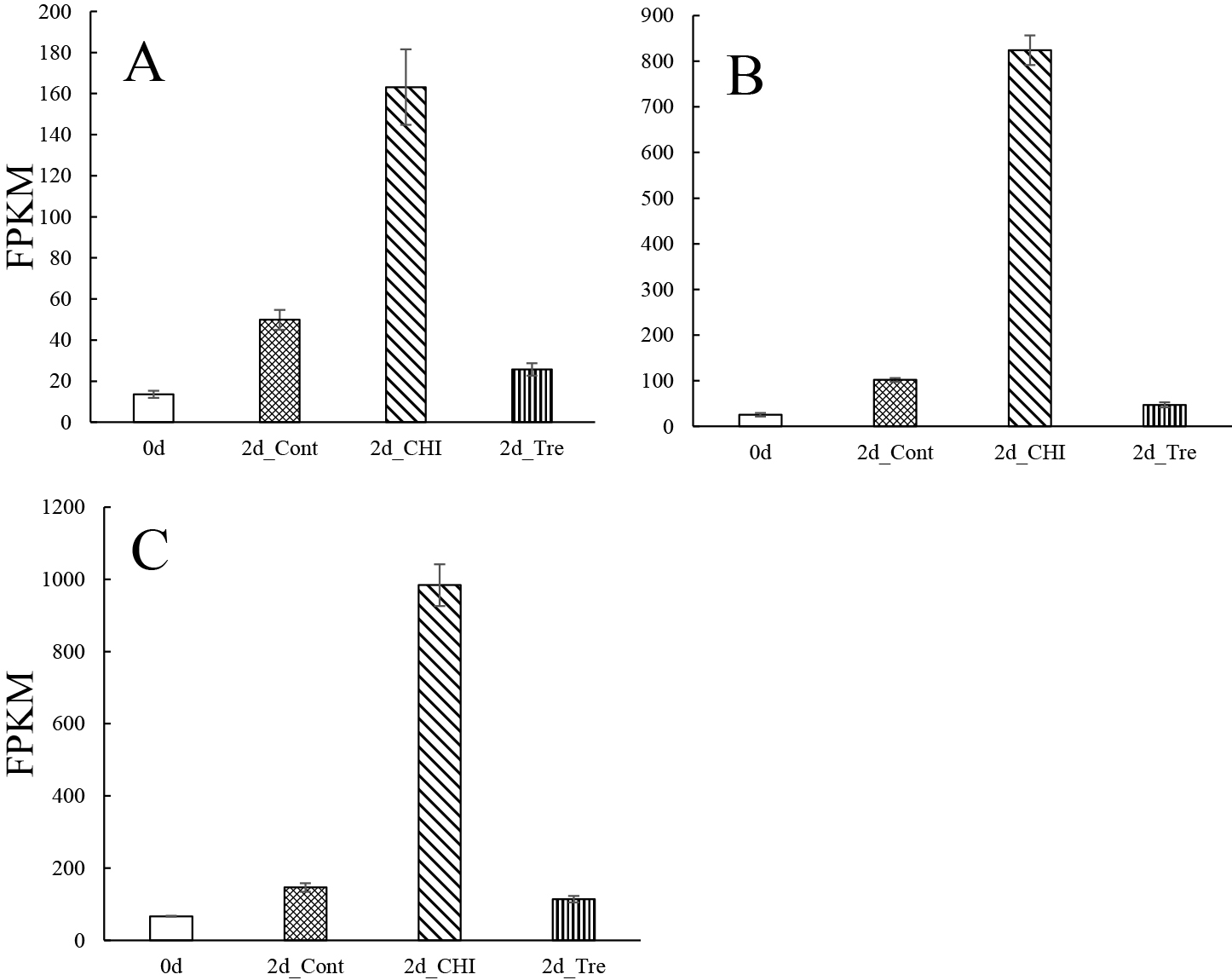
NAC transcription factors control stress-induced PCD during leaf and petal senescence in an age-dependent manner (Balazadeh et al., 2010; Guo and Gan, 2006; Kim et al., 2009; Meng et al., 2022; Shibuya et al., 2014; Takahashi et al., 2022; Trupkin et al., 2019; Yang et al., 2011; Yeung et al., 2018). The FPKM values for the NAC transcription factor genes in this study are presented in Figures 5A–D and S10A–N. The FPKM value for the NAC transcription factor 47 gene (NAC047) was highest for 2d_Cont (Fig. 5A) and was more than double that for the other 2d treatment groups. In Arabidopsis, NAC047 is involved in the leaf senescence process (Yan et al., 2019), suggesting that NAC047 encodes a transcriptional regulator that modulates the senescence of gladiolus tepals.
In addition, compared with the other treatment groups, the FPKM values for NAC transcription factor 48 (NAC048), NAC transcription factor 73 (NAC073), and NAC transcription factor 68 (NAC68) genes were highest for 2d_CHI (Fig. 5B–D). Expression of the transcription factors including NAC048, NAC68 and NAC073 were validated by RT-qPCR, and they were upregulated by CHI in accordance with FPKM (Fig. S12). NAC048 may function in vascular meristem activity (Yang et al., 2020). NAC68 (NAC with transmembrane motif1) is activated by proteolytic cleavage through regulated intramembrane proteolysis and is involved in cytokinin signaling during cell cycling (Kim et al., 2006). NAC073 is involved in the regulation of secondary cell wall biosynthesis in Arabidopsis (Zhong et al., 2008). Previous studies did not reveal the involvement of these three NAC transcription factors in flower senescence.
WRKY transcription factors have been implicated in a variety of biotic and abiotic stress responses (Woo et al., 2013). Most of the FPKM values for the WRKY transcription factor genes in the present study were highest in response to the CHI treatment (Figs. 6A–C and S11A, B, D). Notably, WRKY transcription factor 11 (WRKY11), WRKY transcription factor 6 (WRKY6), and WRKY transcription factor 24 (WRKY24) gene expression levels were very high (Fig. 6). Expression of transcription factors including WRKY 6, 11 and 24 were validated by RT-qPCR, and they were upregulated by CHI in accordance with FPKM (Fig. S13). WRKY6 helps control leaf senescence (Robatzek and Somssich, 2001; Woo et al., 2013; Zhou et al., 2011). Earlier investigations demonstrated that WRKY11 is involved in non-biotic stress and defense responses (Lee et al., 2018), and WRKY24 suppresses abscisic acid and gibberellin signaling (Zhang et al., 2009). On the other hand, WRKY75 was significantly increased at 2d_Cont, although it was not affected by CHI (Fig. S11B). Since WRKY75 is reported to positively regulate leaf senescence in an age-dependent manner in Arabidopsis (Guo et al., 2017; Yan et al., 2019), WRKY75 could be involved in the normal aging of gladiolus tepals.
Effects of CHI treatment on the expression of transcriptional factor genes
In morning glory petals, EPH1 is a key NAC transcription factor that regulates PCD during petal senescence. In an earlier study, suppressed EPH1 expression resulted in a decrease in the expression of a vacuolar protease gene and SAG12 (Shibuya et al., 2014). Although we did not directly examine protein synthesis in this study, protein biosynthesis in the tepal cells was likely almost completely inhibited because of the high cycloheximide concentration in the CHI treatment (Schneider-Poetsch et al., 2010). We speculate that the genes with increased expression levels (e.g., accumulation of mRNA) even after the CHI treatment may be regulated and induced independently of the genes encoding other proteins. Similar to EPH1, they may be involved in triggering senescence. In this context, the upregulated NAC transcription factor genes (NAC048, NAC68, and NAC073) and WRKY transcription factor genes (WRKY6, WRKY11, and WRKY24) are candidate genes that are actively induced in gladiolus tepals during senescence.
It is also possible that the expression of transcription factor genes increased following the exposure to stress simply as a side effect of the CHI treatment. According to the GO analysis, the genes with upregulated expression levels at 2 days after the CHI treatment were associated with DNA binding transcription factor activity (e.g., NAC and WRKY genes) (Fig. 2B). Recent research confirmed WRKY11 is involved in abiotic stress and defense responses (Lee et al., 2018). However, mRNA degradation and translation are closely related and the degradation of individual mRNAs may be triggered or antagonized by translational impairment (Roy and Jacobson, 2013). Moreover, CHI delays the degradation of eukaryotic mRNA and indirectly enhances mRNA stability (Beelman and Parker, 1994). Therefore, the possibility that mRNA accumulated because of the CHI treatment should be considered.
Phylogenetic relationships among NAC proteins
For the differentially expressed NAC transcription factor genes in this study, a phylogenetic tree was constructed to explore their relationships with NAC proteins associated with senescence (Fig. S14). In gladiolus, NAC021 belongs to group I, NAC002 and NAC048 belong to group IV, and NAC68 belongs to group VII. The NAC079 gene is closely related to EPH1 (Shibuya et al., 2014, 2018) and NAC92 (Balazadeh et al., 2010; Kim et al., 2009; Yeung et al., 2018), which induce petal and leaf senescence. Additionally, NAC047 and NAC083 in gladiolus are in the same clade as NAC029 (Guo and Gan, 2006) and NAC083 (Yang et al., 2011), which are also associated with senescence. Because genes clustered in the same subgroup may have similar functions (Li et al., 2021), future investigations should clarify the functions of NAC047, NAC079, and NAC083.
Conclusion
In this study, we used RNA-seq data to comprehensively analyze the expression of specific genes during tepal senescence. In addition, we functionally characterized several genes and clarified their relationships with senescence on the basis of the published literature and the results of vase life analysis. However, the relationship between gene expression and protein biosynthesis in response to the application of cycloheximide remains unclear. Because we examined the senescence of cut florets in vials, our findings may not necessarily be applicable to the senescence of florets on cut spikes. Therefore, florets on cut spikes should be analyzed to clarify the involvement of genes, including transcription factor genes, in gladiolus senescence. In conclusion, this study revealed several candidate genes that could contribute to the senescence of gladiolus tepals, but additional research involving intact florets and proteomic analyses is required.