Abstract
Endoplasmic reticulum stress is related to metabolic disorders, including atherosclerosis and type 2 diabetes. It is known that inflammatory adipocytokines and oxidative stress are increased, while anti-inflammatory adipocytokines such as adiponectin are decreased in adipocytes during above conditions. Extracellular-superoxide dismutase is an anti-inflammatory enzyme that protects cells from oxidative stress. Because plasma extracellular-superoxide dismutase levels in type 2 diabetes patients were inversely related to the body mass index and homeostasis model assessment-insulin resistance index, it is speculated that the regulation of extracellular-superoxide dismutase might lead to the suppression of metabolic disorders. Here, we observed the reduction of extracellular-superoxide dismutase and adiponectin in 3T3-L1 adipocytes treated with thapsigargin, an endoplasmic reticulum stress inducer. Interestingly, tunicamycin, another endoplasmic reticulum stress inducer, did not decrease the expression of extracellular-superoxide dismutase in spite of the induction of glucose regulated protein kinase 78 kDa, an endoplasmic reticulum stress marker. Moreover, eukaryotic translation initiation factor 2α signaling cascade plays a pivotal role in the reduction of extracellular-superoxide dismutase in 3T3-L1 adipocytes during endoplasmic reticulum stress conditions.
Introduction
Hypertrophied adipocytes are well known to be implicated in several kinds of disorders, such as atherosclerosis, type 2 diabetes and insulin resistance.(1) Adipocytes suffer chronic inflammation, characterized by the excess production of inflammatory cytokines, including tumor necrosis factor-α and monocyte chemoattractant protein-1 and oxidative stress during hypertrophy.(2) Adiponectin is an anti-inflammatory adipocytokine that plays a pivotal role in the improvement of glucose and lipid metabolism and the prevention of atherosclerosis and inflammation;(3,4) however, during hypertrophy and under hypoxic conditions, the expression of adiponectin is reduced and these events lead to metabolic disturbances.(5,6) On the other hand, to protect the cells from oxidative stress, mammals have several anti-oxidative enzymes such as superoxide dismutase (SOD), catalase and glutathione peroxidase.(7,8) Extracellular-SOD (EC-SOD) is a secretory enzyme, whereas Cu,Zn-SOD and Mn-SOD are intracellular enzymes found predominantly in the cytoplasm and mitochondria, respectively.(9,10) The EC-SOD content is very low in the liver, heart, brain, and other organs, with the exception of the lung and adipose tissue.(11) Moreover, it has been shown that the plasma EC-SOD contents in type 2 diabetes were positively related to the plasma adiponectin level, but inversely related to the body mass index (BMI) and homeostasis model assessment insulin resistance index (HOMA-IR).(12)
Accumulated evidence has revealed that metabolic disorders are closely correlated with endoplasmic reticulum (ER) stress,(5,13) which induces proapoptotic processes. Under ER stress conditions, cells trigger a set of pathways known as the unfolded protein response (UPR), which is mediated by three types of ER-transmembrane proteins: inositol-requiring protein-1 (IRE-1), RNA-dependent protein kinase-like ER kinase (PERK) and activating transcriptional factor 6 (ATF6).(14) Among them, the activation of PERK signaling has been shown to be involved in the reduction of adiponectin.(5) From our previous observation, the expression of EC-SOD was correlated with that of adiponectin during adipocyte differentiation(15) and in hypoxic adipocytes;(16) we therefore hypothesized that the reduction of EC-SOD might be regulated by ER stress mediators similar to adiponectin. In order to address these issues, we studied the regulation of EC-SOD expression by ER stress inducers, thapsigargin and tunicamycin, and examined the involvement of ER stress mediators in these processes.
Materials and Methods
Cell culture
3T3-L1 mouse pre-adipocyte culture and their differentiation into adipocytes were carried out as described in previous reports,(15–17) with minor modification. The effect of ER stress inducers on gene expression levels was investigated using 8-day differentiated adipocytes.
RT-PCR analysis
After the adipocytes had been treated with reagents, the cells were lysed in 1 mL TRIzol® reagent (Invitrogen, Carlsbad, CA). cDNA was prepared and RT-PCR performed using the methods described in our previous report.(16) The primer sequences used in this study are described in Table 1. These PCR products were loaded onto a 2% (w/v) agarose gel for electrophoresis, and densitometric analysis of the PCR products was performed with Multi Gauge ver. 3.0 (Fuji Film, Tokyo, Japan).
Table 1
Primers used for amplification of RT-PCR analysis
| Genes |
Primer sequences |
| EC-SOD |
F; 5'-AGGTGGATGCTGCCGAGAT-3' |
|
R; 5'-TCCAGACTGAAATAGGCCTCAAG-3' |
| Cu,Zn-SOD |
F; 5'-GTGTCAGGACAGATTACAGG-3' |
|
R; 5'-TTCTCGTGGACCACCATAGT-3' |
| Mn-SOD |
F; 5'-ACAATCTGAACGTCACCGAG-3' |
|
R; 5'-AGTGGGTCCTGATTAGATCA-3' |
| Adiponectin |
F; 5'-CAGGATGCTACTGTTGCAAGC-3' |
|
R; 5'-TGCAGTCAGTTGGTATCATGG-3' |
| GRP78 |
F; 5'-TTTCTGCCATGGTTCTCACT-3' |
|
R; 5'-CCCAGATGAGTATCTCCATT-3' |
| C/EBP-a |
F; 5'-AGCCAAGAAGTCGGTGGACAAG-3' |
|
R; 5'-TAGAGATCCAGCGACCCGAAAC-3' |
| PPAR-g |
F; 5'-GAGCTGACCCAATGGTTGCTG-3' |
|
R; 5'-CCAAACCTGATGGCATTGTGAGAC-3' |
| MyD116 |
F; 5'-GAGAAGAGGGAGTGGCTGAGC-3' |
|
R; 5'-AGCATTCCGACAAGGGTGACC-3' |
| XBP-1 |
F; 5'-AGTTAAGAACACGCTTGGGAAT-3' |
|
R; 5'-AAGATGTTCTGGGGAGGTGA-3' |
| GAPDH |
F; 5'-ACCACAGTCCATGCCATCAC-3' |
|
R; 5'-TCCACCACCTGTTGCTGTA-3' |
Whole cell extracts were prepared in lysis buffer as described previously.(18) Extracts containing 20 µg protein were separated by SDS-PAGE on 10 or 12% (w/v) polyacrylamide gels. After being transferred electrophoretically onto PVDF membranes, nonspecific binding sites were blocked with PBS containing 1% bovine serum albumin. Subsequently, the membranes were incubated with the respective specific primary antibodies [1:1,000 dilution for phospho-eukaryotic translation initiation factor 2α (eIF2α) from Cell signaling Technology, Danvers, MA, #9721; 1:1,000 dilution for eIF2α, Cell Signaling Technology, #9722; 1:1,000 dilution for ATF6 from IMGENEX, San Diego, CA, #IMG-273; 1:1,000 for actin from Millipore Co., Billerica, MA, #MAB1501], 1:1,000 dilution biotin-conjugated goat anti-rabbit or -mouse antibody from Zymed Laboratories, San Francisco, CA, and 1:5,000 dilution ABC reagents (Vector Laboratories, Inc., Burlingame, CA). The bands were detected using ImmunoStar®LD (Wako Pure Chem. Ind. Ltd., Osaka, Japan) or SuperSignal® West Pico (Thermo Scientific, Rockford, IL), and imaged using an LAS-3000 UV mini (Fuji Film, Tokyo).
Statistical analysis
Data are expressed as the means ± SD of three independent experiments. Statistical evaluation of the data was performed using ANOVA followed by post-hoc Bonferroni tests. A p value less than 0.05 was considered significant.
Results
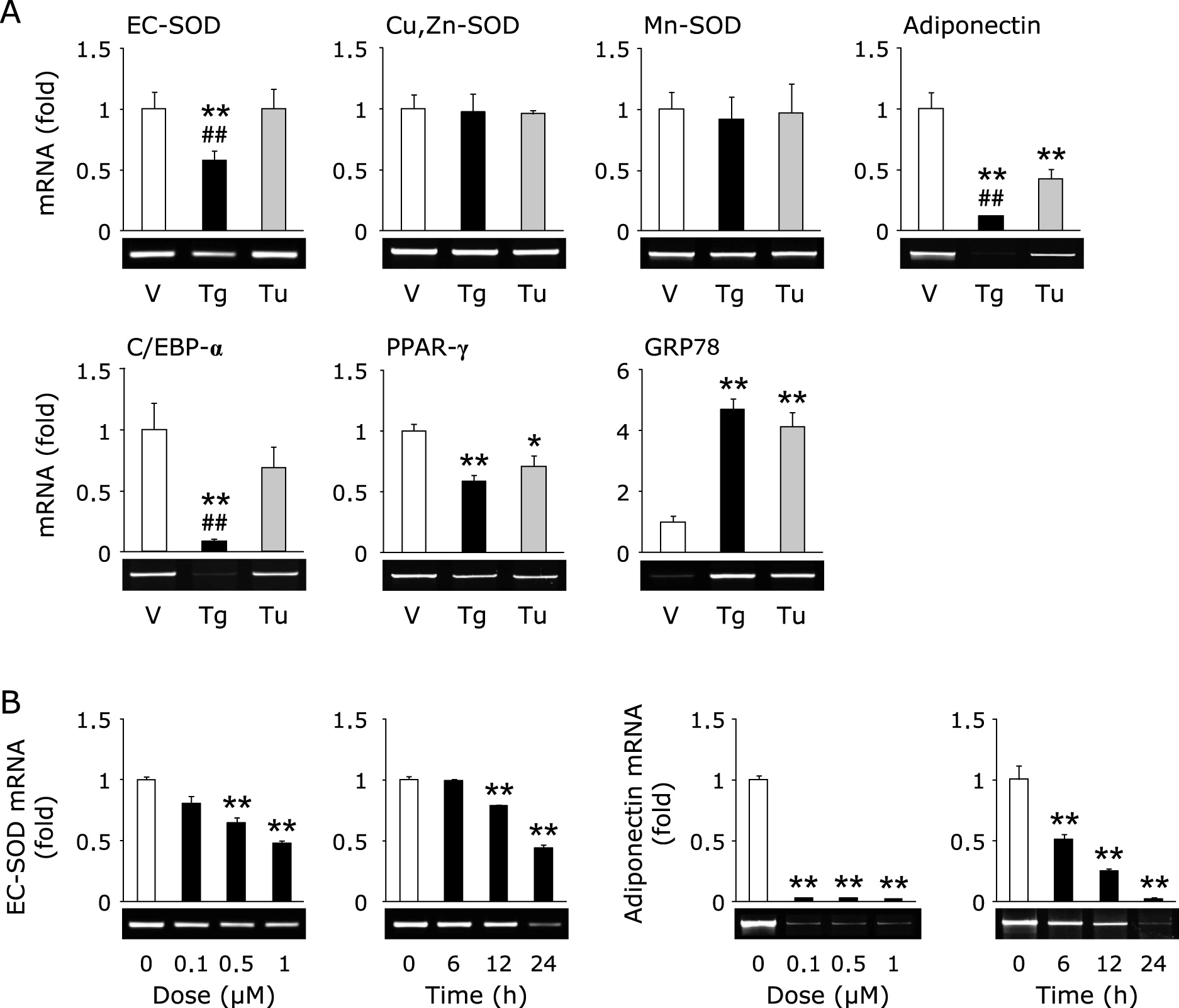
Effect of ER stress inducer, thapsigargin and tunicamycin, on the expression of EC-SOD
Recently, we reported that an ER stress inducer, thapsigargin, decreases the expression of EC-SOD in kidney tubule epithelial cells.(19) As shown in Fig. 1, treatment with thapsigargin decreased the expression of EC-SOD and adiponectin in adipocytes, whereas the expression of Cu,Zn-SOD and Mn-SOD was not changed. On the other hand, treatment with tunicamycin, another ER stress inducer, did not affect the expression of EC-SOD in spite of the equal induction of glucose-regulated protein 78 kDa (GRP78), an ER stress marker (Fig. 1A). Moreover, the reduction of adiponectin, CCAAT/enhancer-binding protein-α (C/EBP-α) and peroxisome proliferator-activated receptor-γ (PPAR-γ) by tunicamycin treatment was also less than that by thapsigargin treatment (Fig. 1A). Further, we confirmed the dose- and time-dependent reduction of EC-SOD and adiponectin by thapsigargin treatment (Fig. 1B).
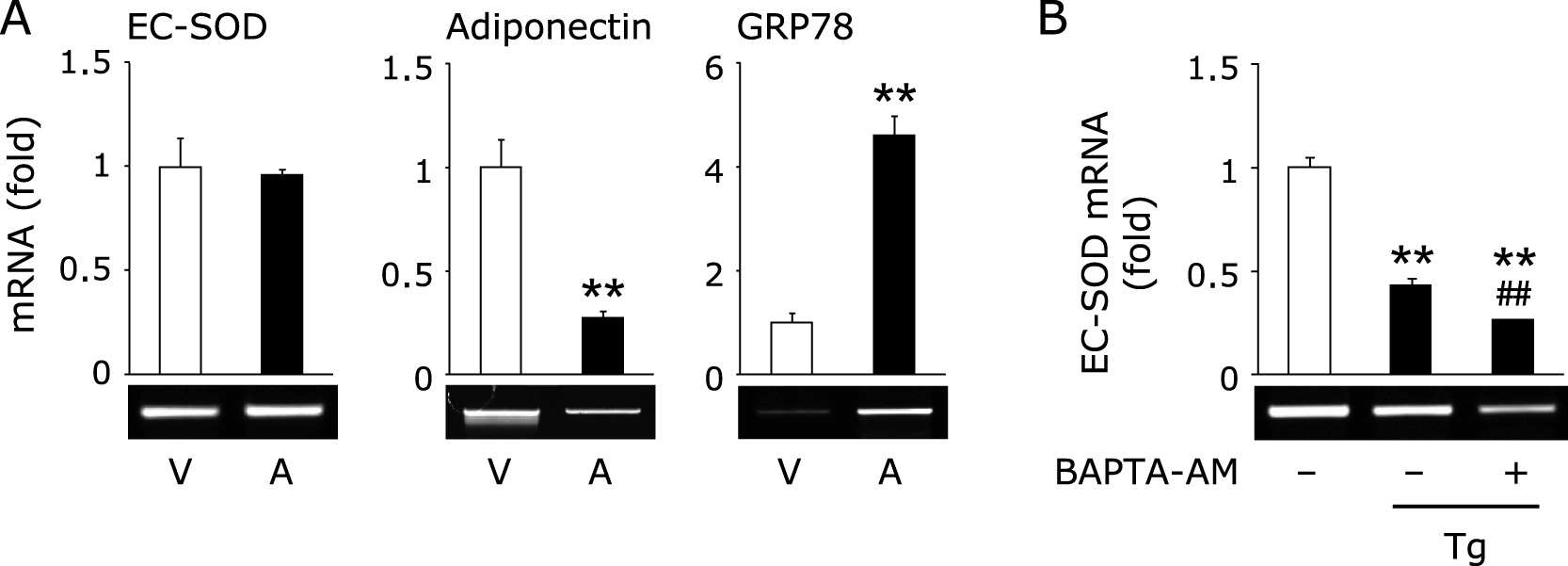
Involvement of calcium-derived signaling in the thapsigargin-triggered reduction of EC-SOD
Because it is well known that thapsigargin induces ER stress by disturbing intracellular calcium homeostasis.(20) As shown in Fig. 2A, treatment with A23187, a calcium ionophore, significantly induced GRP78 and decreased adiponectin expression; however, EC-SOD was not affected by this treatment. Moreover, pretreatment with BAPTA-AM, an intracellular calcium chelator, did not suppress the thapsigargin-triggered reduction of EC-SOD (Fig. 2B).
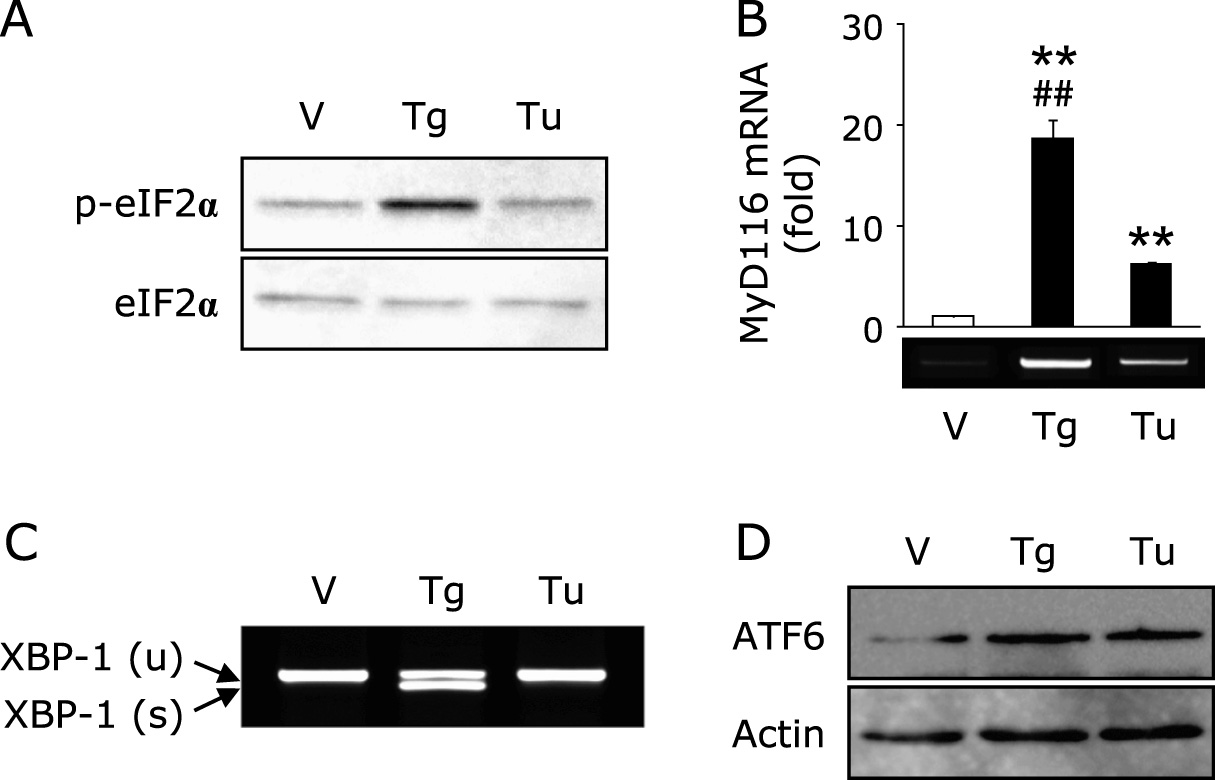
Involvement of ER stress mediators in the thapsigargin-triggered reduction of EC-SOD
It is well known that ER stress sets up three types of signaling, such as PERK-derived eukaryotic translation initiation factor 2α (eIF2α), IRE-1-derived X-box binding protein-1 (XBP-1) and ATF6.(14) As shown in Fig. 3A, treatment with thapsigargin phosphorylated eIF2α at 1 h; however, treatment with tunicamycin did not phosphorylate that modulator. Moreover, treatment with thapsigargin significantly increased the expression of myeloid differentiation primary response gene 116 (MyD116), a downstream target of eIF2α, compared to tunicamycin treatment (Fig. 3B). We also detected the spliced form of XBP-1, an active form of XBP-1, only in thapsigargin treatment (Fig. 3C). On the other hand, the nuclear translocation of ATF6 was promoted equally by thapsigargin and tunicamycin treatment (Fig. 3D). It has been recognized that salubrinal activates eIF2α signaling by inhibiting dephosphorylation of this mediator.(21) Moreover, Cnop et al.(22) reported that treatment with salubrinal potentiates palmitate-induced XBP-1 splicing. In this study, pretreatment with salubrinal promoted the reduction of thapsigargin-trigged reduction of EC-SOD (Fig. 4A). Moreover, co-treatment with salubrinal and tunicamycin decreased the expression of EC-SOD, suggesting that the downstream of eIF2α plays a pivotal role in that reduction. On the other hand, pretreatment with salubrinal did not activate XBP-1 in either thapsigargin- or tunicamycin-treated adipocytes (Fig. 4B).
Discussion
ER stress is closely related to several pathological disorders, such as metabolic disorders(13) and atherosclerosis.(23) It is well known that adipocytes during hypertrophy suffer chronic ER stress conditions.(5) During these conditions, the homeostasis of adipocytokines is disturbed, and these events may lead to and/or exacerbate metabolic disorders. Among the adipocytokines, adiponectin has anti-inflammatory properties and excess production of adiponectin effectively improves glucose and lipid metabolism and insulin resistance.(24) It has been reported that ER stress decreases the expression of adiponectin and transcriptional factors including C/EBP-α and PPAR-γ.(15) In this study, we confirmed the reduction of adiponectin and these transcriptional factors by thapsigargin treatment (Fig. 1A).
Similar to inflammatory cytokines, the excess production of ROS is involved in the exacerbation of metabolic disorders. EC-SOD is a major SOD isozyme which is relatively highly expressed in adipocytes. The plasma EC-SOD contents in type 2 diabetic patients are significantly and inversely related to BMI and HOMA-IR.(12) In this study, we confirmed the reduction of EC-SOD in thapsigargin-treated cells (Fig. 1). On the other hand, treatment with tunicamycin did not decrease the expression of EC-SOD (Fig. 1A), indicating that the reduction of EC-SOD was not directly regulated by ER stress, but other mechanisms.
Intracellular calcium ion may play an important role in the reduction of EC-SOD during ER stress conditions, because it is well recognized that thapsigargin, but not tunicamycin, disturbs intracellular calcium homeostasis.(20) However, we did not observe the reduction of EC-SOD by A23187 and the suppression of thapsigargin-triggered EC-SOD reduction by pretreatment with BAPTA-AM (Fig. 2), indicating that the expression of EC-SOD is not regulated by intracellular calcium signaling. It is well recognized that ER stress sets up three types of signaling, such as PERK-derived eIF2α, IRE-1-derived XBP-1 and ATF6.(14) In this study, we observed the phosphorylation of eIF2α and induction of spliced XBP-1 only in thapsigargin-treated adipocytes, whereas the induction of ATF6 was observed both in thapsigargin- and tunicamycin-treated adipocytes (Fig. 3D). We also determined that co-treatment with salubrinal and thapsigargin or tunicamycin decreased the expression of EC-SOD (Fig. 4A), suggesting the involvement of eIF2α signaling in the reduction of EC-SOD under our experimental conditions. Recently, it has been reported that salubrinal not only inhibits the dephosphorylation of eIF2α but also potentiates palmitate-induced XBP-1 splicing;(22) however, pretreatment with salubrinal did not promote or induce spliced XBP-1 in thapsigargin- and tunicamycin-treated adipocytes (Fig. 4B). These results suggest that XBP-1-derived signaling is not involved in the reduction of EC-SOD under our experimental conditions. However, additional studies will be necessary to determine the exact molecular mechanism governing the regulation of EC-SOD.
Oxidative stress via the activation of NAD(P)H oxidase (NOX) augmented in obese adipose tissue and infiltrated and activated macrophages leads to establish a second vicious paracrine loop.(25) On the other hand, it has been reported that exogenous addition of EC-SOD and over-expression of EC-SOD prevented vascular endothelial cell-mediated oxidative modification of low-density lipoprotein.(26,27) Overall, it is speculated that the reduction of EC-SOD by thapsigargin leads to a decrease in the resistance to oxidative and other inflammatory stress. Further, these findings contribute to the control of metabolic disorder exacerbation and knowledge about cytotoxicity induced by oxidative stress under ER stress conditions.
Abbreviations
ATF6; activating transcriptional factor 6,
BMI; body mass index,
C/EBP-α; CCAAT/enhancer-binding protein-α,
EC-SOD; extracellular-superoxide dismutase,
eIF2α; eukaryotic translation initiation factor 2α,
ER; endoplasmic reticulum,
GRP78; glucose-regulated protein 78 kDa,
HOMA-IR; homeostasis model assessment insulin resistance index,
IRE-1; inositol-requiring protein-1,
MyD116; myeloid differentiation primary response gene 116,
PERK; RNA-dependent protein kinase-like ER kinase,
PPAR-γ; peroxisome proliferator-activated receptor-γ,
UPR; unfolded protein response,
XBP-1; X-box binding protein-1
Conflict of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was supported in part by a Grant-in-aid for Scientific Research from the Japan Society for the Promotion for Science (TK, No. 21790153 and TA, No. 21590169).
References
- 1 Eriksson JW. Metabolic stress in insulin’s target cells leads to ROS accumulation—a hypothetical common pathway causing insulin resistance. FEBS Lett 2007; 581: 3734–3742.
- 2 Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab 2007; 293: E1118–E1128.
- 3 Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999; 257: 79–83.
- 4 Ouchi N, Kihara S, Arita Y, et al. Novel modulator for endothelial adhesion molecules: adipocyte-derived plasma protein adiponectin. Circulation 1999; 100: 2473–2476.
- 5 Hosogai N, Fukuhara A, Oshima K, et al. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 2007; 56: 901–911.
- 6 Schuster SJ, Badiavas EV, Costa-Giomi P, Weinmann R, Erslev AJ, Caro J. Stimulation of erythropoietin gene transcription during hypoxia and cobalt exposure. Blood 1989; 73: 13–16.
- 7 Amstad P, Cerutti P. Genetic modulation of the cellular antioxidant defense capacity. Environ Health Perspect 1990; 88: 77–82.
- 8 Cerutti P, Ghosh R, Oya Y, Amstad P. The role of the cellular antioxidant defense in oxidant carcinogenesis. Environ Health Perspect 1994; 102 (Suppl 10): 123–129.
- 9 Faraci FM. Vascular protection. Stroke 2003; 34: 327–329.
- 10 Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol 2004; 24: 1367–1373.
- 11 Marklund SL. Extracellular superoxide dismutase in human tissues and human cell lines. J Clin Invest 1984; 74: 1398–1403.
- 12 Adachi T, Inoue M, Hara H, Maehata E, Suzuki S. Relationship of plasma extracellular-superoxide dismutase level with insulin resistance in type 2 diabetic patients. J Endocrinol 2004; 181: 413–417.
- 13 Ozcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004; 306: 457–461.
- 14 Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000; 101: 451–454.
- 15 Adachi T, Toishi T, Wu H, Kamiya T, Hara H. Expression of extracellular superoxide dismutase during adipose differentiation in 3T3-L1 cells. Redox Rep 2009; 14: 34–40.
- 16 Kamiya T, Hara H, Inagaki N, Adachi T. The effect of hypoxia mimetic cobalt chloride on the expression of EC-SOD in 3T3-L1 adipocytes. Redox Rep 2010; 15: 131–137.
- 17 Mori T, Sakaue H, Iguchi H, et al. Role of Krüppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem 2005; 280: 12867–12875.
- 18 Kamiya T, Hara H, Yamada H, Imai H, Inagaki N, Adachi T. Cobalt chloride decreases EC-SOD expression through intracellular ROS generation and p38-MAPK pathways in COS7 cells. Free Radic Res 2008; 42: 949–956.
- 19 Kamiya T, Obara A, Hara H, Inagaki N, Adachi T. ER stress inducer, thapsigargin, decreases extracellular-superoxide dismutase through MEK/ERK signalling cascades in COS7 cells. Free Radic Res 2011; 45: 692–698.
- 20 Werno C, Zhou J, Brune B. A23187, ionomycin and thapsigargin upregulate mRNA of HIF-1α via endoplasmic reticulum stress rather than a rise in intracellular calcium. J Cell Physiol 2008; 215: 708–714.
- 21 Boyce M, Bryant KF, Jousse C, et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005; 307: 935–939.
- 22 Cnop M, Ladriere L, Herkerman P, et al. Selective inhibition of eukaryotic translation initiation factor 2α dephosphorylation potentiates fatty acid-induced endoplasmic reticulum stress and causes pancreatic β-cell dysfunction and apoptosis. J Biol Chem 2007; 282: 3989–3997.
- 23 Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med 2010; 16: 396–399.
- 24 Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002; 8: 1288–1295.
- 25 Reilly PM, Schiller HJ, Bulkley GB. Pharmacologic approach to tissue injury mediated by free radicals and other reacitive oxygen metabolites. Am J Surg 1991; 161: 488–503.
- 26 Takatsu H, Tasaki H, Kim HN, et al. Overexpression of EC-SOD suppresses endothelial-cell-mediated LDL oxidation. Biochem Biophys Res Commun 2001; 285: 84–91.
- 27 Laukkanen MO, Lehtolainen P, Turunen P, et al. Rabbit extracellular superoxide dismutase: expression and effect on LDL oxidation. Gene 2000; 254: 173–179.