Letters (Selected Paper)
FMO Analysis for Intermoleular Interaction Energy between Human Coagulation Factor Xa and Four Ligands
2015 Volume 14 Issue 3 Pages 54-56
Details
2015 Volume 14 Issue 3 Pages 54-56
The role of water in the protein-ligand binding site was investigated from the viewpoint of enthalpic bridging between human coagulation factor Xa (fXa) and four ligands. Ligand-constrained MD simulation was done for identifying hydration sites and representative water molecules were arranged at the sites. The calculated fXa-ligand interaction energy while considering these explicit water molecules as parts of fXa shows excellent correlation to the experimental binding affinity, while the energy with considering them as implicit solvent does not correlate. This result indicates that the representative water molecules at the hydration sites should be explicitly considered when protein-ligand interaction energy is analyzed, and ligand-constrained MD simulation is a powerful tool to identify these hydration sites.
近年,京などに代表される超並列計算機の普及に伴い,コンピュータシミュレーションを利用した創薬の研究が盛んになってきた.タンパク質結晶構造の情報から,薬の候補となり得る化合物(リガンド)を設計するStructure-based drug design (SBDD)も,主要な研究テーマの1つである.SBDDによるリガンド設計では,設計したリガンドとタンパク質との結合活性を如何に評価するかが重要なポイントとなる.ただし結合活性を適切に評価するためには,タンパク質とリガンドとの間に働く相互作用だけでなく,周囲の溶媒の影響を考慮しなければならない [1].今回,我々は,周囲の溶媒をimplicitに考慮する場合とexplicitに考慮する場合のそれぞれについて,タンパク質とリガンドとの間に働く分子間相互作用エネルギーをフラグメント分子軌道(FMO)法 [2]により解析し,結果を結合活性の実測値と比較することで,特定のスポットにexplicitな溶媒を考慮することの重要性を調査した.
タンパク質とリガンド間の相互作用解析には2 layerの2体近似FMO法を用い,リガンドとその近傍6Å内のフラグメントにはRI-MP2/6-31G
(d)を,その他のフラグメントにはHF/3-21Gを用いた.リガンド近傍のカルボキシル基の基底にはdiffuse関数を追加し,RIの外部基底にはcc-pVDZを用いた.fXaのフラグメントはアミノ酸1残基単位とし,Cα炭素とカルボニル炭素間で分割した.Implicitな溶媒効果は2 iterationのPCMにより評価した.タンパク質とリガンドとの間の分子間相互作用エネルギー
| (1) |
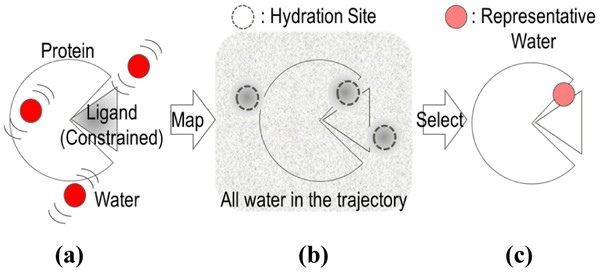
Schematic diagrams to determine representative water molecules using a ligand-constrained MD simulation. (a) Ligand-constrained MD simulation. (b) Hydration sites (sphere, radius: 0.5 Å) whose density of water molecules in the trajectory is relatively high. (c) Representative water molecules selected from the hydration sites to meet distribution conditions.
今回の調査対象には血液凝固因子Xa (fXa) と,4種類のリガンドを用いた.利用した複合体のPDBデータは1NFU (リガンド:RRP),1NFY (リガンド:RTR),1NFX (リガンド:RDR),1NFW (リガンド:RRR)である [5]. MD計算のプログラムにはGromacs v4.6.5を,FMO計算のプログラムにはGAMESS 1 MAY 2013 (R1)を用いた.
解析に用いた4種類のリガンドは全てスルホンアミド構造をリンカー部位に持ち,中性部位がfXaのanionicなS1ポケットに,塩基性部位がfXaのaromaticなS4ポケットに入って結合する.4種類共に分子サイズも結合ポーズもほぼ同様なため,fXaとの結合自由エネルギーの相対値に対するエントロピーの寄与も同等と仮定できる.そのためこれら4種類のリガンドとfXaとの分子間相互作用エネルギーは,実測の結合自由エネルギーと線形の相関を持つことが期待される.しかしimplicitな溶媒効果のみを考慮したFMO計算の結果では,期待された線形の相関からRRRのみ15 kcal/mol
以上外れた(Figure 2 (a)).fXaとリガンドとのbinding
siteには,リガンドのリンカー部位とfXaとの間にhydration siteが存在する.RRRと他のリガンドとの大きな違いは,このhydration
site近傍から,S1ポケットの硫黄原子が離れている点である(Figure 2
(b)).この違いはhydration
siteに含まれる水分子の配向に影響を及ぼし,リガンドとfXaとの間の水素結合の大きさにも影響を生じる可能性が考えらえる.実際にリガンドの重原子拘束MDを利用して水分子の代表構造を特定した結果,リガンド間でhydration
siteの水分子の配向が異なることが分かった(Figure 3 (a), (b), (c)).これらの特定した水分子をfXaの部分構造と見なし,その他の水分子をimplicitな溶媒として考慮したFMO計算を全てのリガンドの場合について実施した結果,RRRだけでなく全てのリガンドで分子間相互作用エネルギーの絶対値が小さくなり,実測の結合自由エネルギーとの相関も

(a) Relationship between experimental binding affinity and protein-ligand intermolecular interaction energy Eint using implicit water. (b) Hydration sites near the linker of each ligand and fXa. The distance between the hydration site and S atom of chlorothiophen (RRR) is larger than that between hydration site and S atom of chlorobenzothiophen (RDR, RTR, RRP).

(a) All water molecules in the ligand-constrained MD trajectory (dots) and hydration sites (spheres) with RRR. (b) Representative water molecules in the hydration site near the linker of RDR and fXa. (c) Representative water molecules in the hydration site near the linker of RRR and fXa. (d) Relationship between experimental binding affinity and protein-ligand intermolecular interaction energy Eint using implicit water with some explicit water molecules, obtained from the ligand-constrained MD, considered part of the protein.