
Current issue
Special Issue: Selected Papers from the Annual Autumn Meeting 2024
Displaying 1-9 of 9 articles from this issue
- |<
- <
- 1
- >
- >|
Foreword
-
2025 Volume 24 Issue 1 Pages A1-A2
Published: 2025
Released on J-STAGE: March 14, 2025
Download PDF (382K) Full view HTML
Commentary
-
2025 Volume 24 Issue 1 Pages A3-A11
Published: 2025
Released on J-STAGE: March 14, 2025
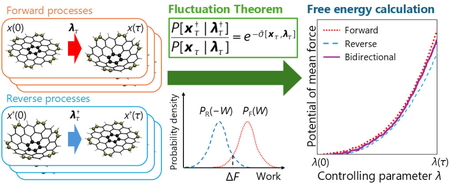 Download PDF (1229K) Full view HTML
Download PDF (1229K) Full view HTML
Software News and Review
-
2025 Volume 24 Issue 1 Pages A12-A17
Published: 2025
Released on J-STAGE: March 28, 2025
Advance online publication: February 28, 2025Download PDF (536K) Full view HTML
Letters (SCCJ Annual Meeting 2024 Autumn Poster Award Articles)
-
2025 Volume 24 Issue 1 Pages 1-4
Published: 2025
Released on J-STAGE: March 14, 2025
 Download PDF (1120K) Full view HTML
Download PDF (1120K) Full view HTML -
2025 Volume 24 Issue 1 Pages 5-9
Published: 2025
Released on J-STAGE: March 29, 2025
 Download PDF (1084K) Full view HTML
Download PDF (1084K) Full view HTML
Letters (Selected Papers)
-
2025 Volume 24 Issue 1 Pages 10-13
Published: 2025
Released on J-STAGE: March 29, 2025
 Download PDF (1719K) Full view HTML
Download PDF (1719K) Full view HTML -
2025 Volume 24 Issue 1 Pages 14-16
Published: 2025
Released on J-STAGE: March 31, 2025
Download PDF (2090K) Full view HTML -
2025 Volume 24 Issue 1 Pages 17-19
Published: 2025
Released on J-STAGE: April 01, 2025
 Download PDF (1310K) Full view HTML
Download PDF (1310K) Full view HTML -
2025 Volume 24 Issue 1 Pages 20-26
Published: 2025
Released on J-STAGE: April 12, 2025
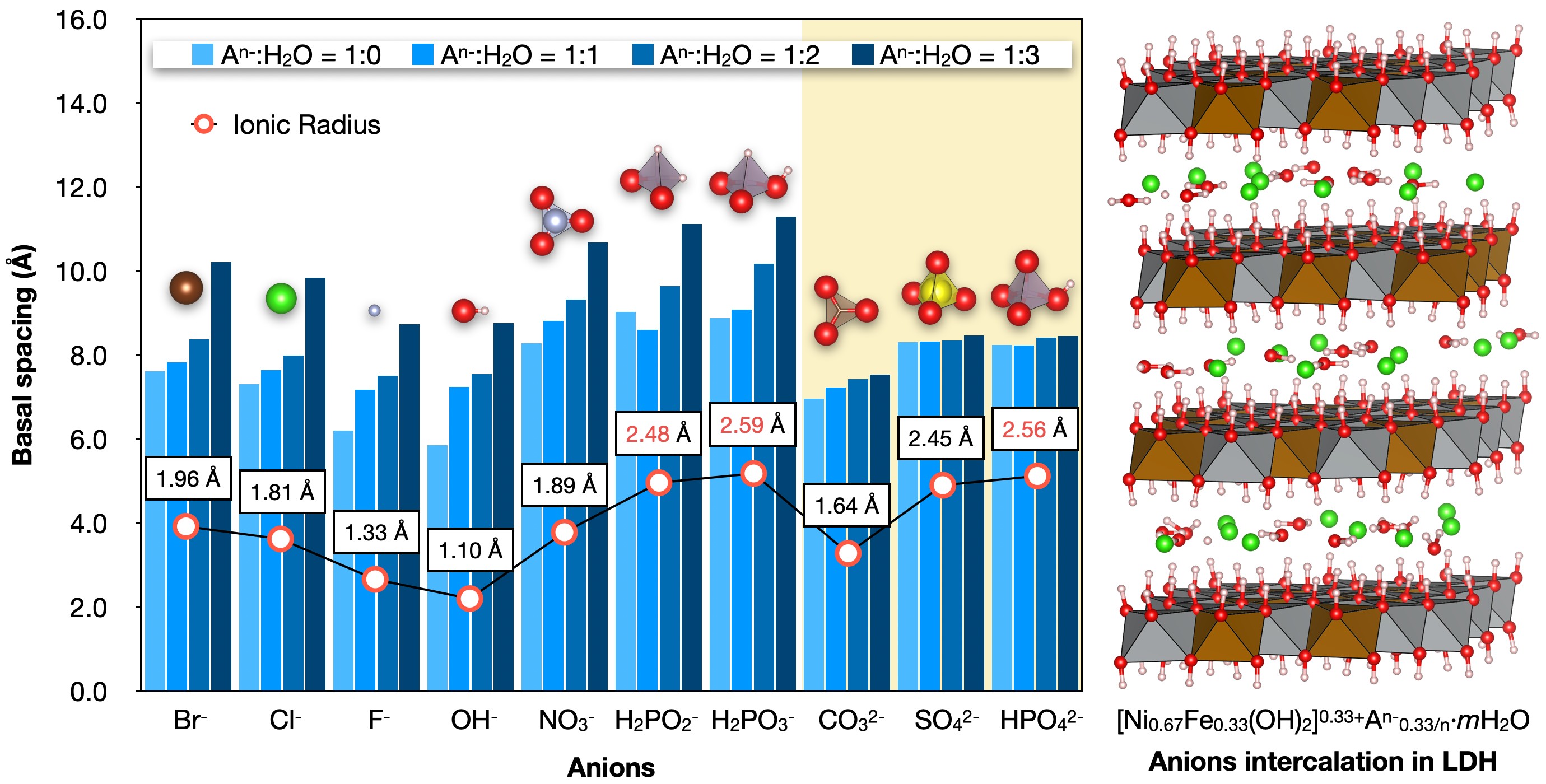 Download PDF (1813K) Full view HTML
Download PDF (1813K) Full view HTML
- |<
- <
- 1
- >
- >|