Abstract
We have launched a project called "Maizo"-chemistry, which is aimed toward molecular- and
reaction discovery based on big data of quantum mechanical global reaction route mappings.
The global reaction data includes equilibrium structures (EQs), dissociation channels
(DCs), and transition structures (TSs), which are automatically calculated by a global
search on a potential energy surface using the GRRM (global
reaction route
mapping) method. Applications to molecular- and synthesis design
are an important part of the project. Machine learning and visualization techniques as
well as chemoinformatics methods are essential to acquire useful information from the
large reaction data space. We describe here a software system, RMapViewer, which we have
developed to visualize and analyze the GRRM outputs.
1 Introduction
The world's authority for chemical information, the CAS (Chemical Abstract Service)
division of the American Chemical Society announced that more than 100 million organic and
inorganic substances have been registered to their database and the pace of the registration
has accelerated, and approximately 75 million were added over the past 10 years [1]. Automatic global search calculations on
quantum mechanical (QM) potential energy
surfaces by using the GRRM (global reaction
route mapping) methods [2,3,4], however, have demonstrated that far more kinds of
compounds can theoretically exist. Those unknown compounds can potentially be innovative
chemical substances. We therefore have launched a project for the discovery of new molecules
and reactions from the large theoretical data obtained from the QM global reaction route
search. We named the project "Maizo"-chemistry. The term "Maizo [mʌizɔ:]" is Japanese, which
is a prefix for buried precious items [5]. In the
first stage of the project, we have developed a software system, RMapViewer, to visualize
and analyze the global chemical reaction data. In the present article, we will report the
development of the current version 3.0.
2 "Maizo"-chemistry Project
The enumeration of chemical structures is one of the classical topics of chemical
information or chemoinformatics. A topological enumeration, such as MOLGEN [6], is the most conventional method and has been often
used to generate possible chemical structures to diverse a so-called chemical space for drug
discovery or structure elucidation. In the topological methods, a chemical structure is
treated mathematically as a graph, which makes fast processing possible to count isomers.
The topological methods are very useful but not for all cases. For example, one soon
encounters a combinatorial explosion even with rather small molecular size. It will be
difficult to generate chemical structures with all the relevant stereochemistry and the
topological enumeration is only tractable with valence bond theory.
On the other hand, useful methods for chemical structure enumeration along with molecular
orbital theory have been developed, recently. In 2004, Ohno and Maeda published the first
report about the GRRM method, which makes it possible to automatically search global
reaction pathways on a potential energy surface using an anharmonic
downward distortion
following (ADDF) search algorithm [2,3,4]. Using GRRM, one can obtain a global reaction map, which includes equilibrium
structures (EQs), dissociation channels (DCs), and transition structures (TSs), together
with the information about their electronic structures and energy levels along with the QM
theories. Since the enumeration along with the QM theories counts structures beyond the
valence bond theory as well as theoretically rational stereochemistry, it produces far more
structures than the topological enumeration does. For example, in the enumeration for
C6H6, the GRRM exploration has produced more than 5,000 isomers,
while there are just 217 isomers in the topological basis.
The global reaction maps obtained from GRRM can provide useful data for the discovery of
new molecules/reactions, synthetic design, and reaction prediction even by considering the
possibilities of side reactions in the QM level. We focused on the notable characteristics
of the global reaction maps and have launched a project for molecular- and reaction
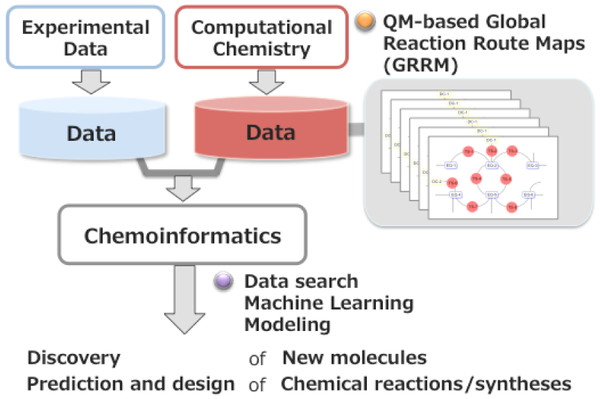
discovery based on the big data of the global reaction route maps. The project scheme is
shown in Figure 1. Applications to molecular- and
synthesis design are an important part of the project. Machine learning and visualization
techniques as well as chemoinformatics methods are essential to acquire useful information
from the large reaction data space. The contents and the type of computational data are
different from those of experimental data. We plan to extract knowledge/models from the data
and to use those knowledge/models independently or complementarily.
3 RMapViewer 3.0
We assume that there are two ways of molecular/reaction discovery by using our system. One
is a human-dominated way with the help of a computer, and the other is a computer-driven
way. In the first stage of the project, we have developed a software system for the former
way, RMapViewer, to visualize and analyze the global reaction route maps (R-maps). An
example output display of RMapViewer is shown in Figure
2. In the current version of RMapViewer3.0, basic functions to analyze the R-maps
have been implemented, including functions for visualization of R-maps (Figure 2–a,b), searching all possible paths between two molecules,
corresponding to a reactant and a product, sorting the paths in order of TS energies and/or
the number of reaction steps (Figure 2–c), and
displaying a movie along a reaction coordinate. In the current version, we use the JMol
tools [7] to visualize molecular models (Figure 2–d). Using the path search function (Figure 2–a,b), one can get
minimum energy paths
(MEPs) from reactant (s) to product (s).

In July 2014, we released RMapViewer as freeware, which is delivered from the Sourceforge
service [8]. The current version as well as the
detailed usage is available with some sample input files in the RMapViewer format. We will
soon release also a file converter module from the GRRM outputs to the RMapViewer inputs
from the same web page.
We have been calculating the global reaction route maps for several organic molecules
including C, O, N, S, H, Cl, Br, F using the GRRM system mainly at rather moderate levels of
theory, such as RHF/6–31+G (d,p) and RHF/6–31+G (d). We plan to add calculations at higher
theory level with larger basis sets in future. Comparing between R-maps obtained at
different levels/basis sets would be also useful for the evaluation of the QM methods.
Because the shape of a potential energy surface can change by a theory and/or a basis set,
the number of EQ, TS, and DC structures can be changed depending on the theory/basis set
used in the calculations.
In the context of the "Maizo"-chemistry project, some of the authors have found a new
carbon family consisting of a prism carbon unit [9,10,11]. We also have performed conformational analyses for monosaccharides and found
several MEPs between representative conformers [12].
4 Conclusions
We have given a brief introduction about a new project called "Maizo"-chemistry. Since
there was no method to explore such a global range of QM potential energy surfaces before
GRRM, it is expected that the large part of the global reaction route maps we have been
exploring is totally new data that had not existed anywhere in the world. In the project, we
plan to do mining precious findings of chemistry, which are buried in the large QM data
space, by using advanced information science techniques. The global reaction data will help
researchers to get an idea of molecular modeling, including in drug discovery and material
design research.
The authors were supported by a Grant from the Data Centric Science Research Commons
Project of the Research Organization of Information and Systems (ROIS), Japan. H.S. and T.U.
were supported by a Grant-in-Aid for Challenging Exploratory Research (Grant No. 25540017)
from the Japan Society for Promotion of Science.
References and Notes
- 1 On June 29th, 2015, the
Chemical Abstracts Service (CAS) announced that they registered the 100 millionth chemical
substance in the CAS REGISTRY:
https://www.cas.org/news/media-releases/100-millionth-substance. (accessed August 4th
2015)
- 2 K. Ohno, S. Maeda, Chem.
Phys. Lett., 384, 277 (2004).
- 3 S. Maeda, K. Ohno, J. Phys.
Chem. A, 109, 5742 (2005).
- 4 K. Ohno, S. Maeda, J. Phys.
Chem. A, 110, 8933 (2006).
- 5 "Maizo" [mʌizɔ:] is a term
used for buried precious items, such as cultural properties, fossil, a treasure trove, and
deposits of precious metals, oil, or coal. For example, "Maizo"-gold is a usage of this
word. We employed this word as part of the name of our project, since this nicely
expresses the concept toward finding out new chemistry buried in the QM-based big data
space.
- 6 http://molgen.de. (accessed
August 4th 2015)
- 7 http://jmol.sourceforge.net
(accessed August 4th 2015)
- 8
http://sourceforge.net/projects/rmapviewer/ (accessed August 4th 2015)
- 9 K. Ohno, H. Satoh, T.
Iwamoto, Chem. Lett., 44, 712 (2015).
- 10 K. Ohno, H. Satoh, T.
Iwamoto, Chem. Phys. Lett., 633, 120 (2015).
- 11 K. Ohno, H. Tokoyama, H.
Yamakado, Chem. Phys. Lett., 635, 180 (2015).
- 12 To be
submitted.