速報
カチオン性イリジウム触媒を用いた均一系触媒反応における相対論効果
キーワード:
C-H Bond activation,
Transition metal catalyst,
Relativistic effect,
Oxidative addition,
Iridium
2019 年 18 巻 3 号 p. 136-138
詳細
2019 年 18 巻 3 号 p. 136-138
A cationic Ir complex was reported to show specific catalytic activity in C-H bond activation reaction of benzanilides. The present study examined reaction energy profiles of the C-H bond activation with Ir and Rh catalysts based on non-relativistic and relativistic quantum chemical calculations. We found that the relativistic effect is essential to demonstrate the difference in the catalytic activity. In particular, the activation of the d orbital of Ir, which is caused by the s- and p- orbital contraction followed by the self-consistent d-orbital expansion, leads to stabilization of the transition state and product of the C-H bond activation.
アルカン,芳香族化合物,アルキル鎖などにおける炭素-水素 (C-H) 結合は通常不活性であり,直接切断したり官能基化したりすることは困難である [1].村井らが有機金属錯体を触媒としてC-H結合活性化反応に成功して以来,様々な研究が行われてきた [1, 2].C-H結合活性化反応は炭素鎖伸長反応の工程数を低減し,原子効率が向上するため,環境負荷の少ない有機合成化学手法として注目されている.最近柴田らはカチオン性Ir触媒を用いたベンズアニリド類のC-H結合活性化反応を報告した [3].特に,この反応は同族のカチオン性Rh触媒では進行しないという興味深い知見を得ている [4].本研究では,カチオン性Ir触媒とカチオン性Rh触媒の反応性の違いを理論的に解明することを目指した.非相対論的 (NR) および相対論的 (Rel) 量子化学計算に基づき解析を行った.
対象とした反応はScheme 1に示す重水添加反応である.この結果より,Ir錯体により,ベンズアニリドのカルボニル,アミノ基の両オルト位のC-H結合が開裂していることがわかる.
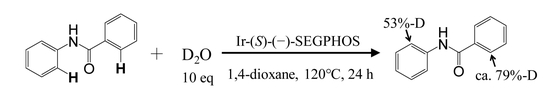
Deuterium addition reaction of N-phenylbenzamide.
反応における中間体および遷移状態に対して,ωB97X-D汎関数 [5] を用いた密度汎関数理論計算により構造最適化・振動数計算を行った.Irは[Kr]4d104f14,Rhは[Ar]3d10の内殻電子に対して,非相対論的および相対論的なStuttgart-Dresden (SDD) [6, 7] 擬ポテンシャル (PP) を用いた.対応する価電子軌道の基底関数として,非相対論的PPには(18s7p6d)/[4s2p2d],相対論的PPには(20s9p8d)/[4s2p2d]を用いた.その他の元素は全電子基底6-31G (d,p)を用いた.振動数計算では温度,圧力をそれぞれ393.15 K, 1 atmとした.溶媒効果 (1,4-ジオキサン;比誘電率2.2099) はSolvation Model Density [8] により取り入れた.計算に用いたプログラムはGaussian09 [9] である.
Figure 1に得られた中間体および遷移状態を示す.(a),(b)はそれぞれN-フェニルベンズアミドのアミノ基,カルボニル基のオルト位において酸化的付加が起こる場合の構造である.
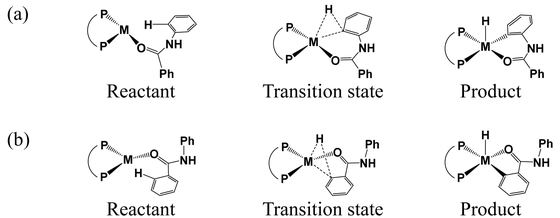
Schematic illustrations of intermediates and transition states (M = Ir or Rh). Activated C-H bonds are (a) ortho position of the amino group and (b) that of the carbonyl group, respectively.
Figure 2は,反応物を基準とした遷移状態および生成物のGibbsエネルギーダイアグラムである.相対論計算の結果では活性化エネルギーはIr触媒の方がRh触媒よりも低く,その差はアミノ基側で10.7 kcal/mol,カルボニル基側で11.3 kcal/molである.また,反応エネルギーはIr触媒ではアミノ基側で−0.41 kcal/mol,カルボニル基側で−6.20 kcal/molであり,負の値となった.一方,Rh触媒ではアミノ基側で17.68 kcal/mol,カルボニル基側で14.86 kcal/molであり正の値となった.この結果は,酸化的付加がIr触媒存在下では自発的に進行し,Rh触媒存在下では進行しないことを示唆している.この結果は,実験事実と対応している.非相対論計算ではIr触媒とRh触媒は同様の傾向を示している.活性化エネルギーは約25 ∼ 30 kcal/molであり,反応エネルギーは正の値である.またIr,Rhともに相対論計算よりもエネルギー障壁が高くなっている.このことは,Ir触媒の活性は相対論効果に由来していることを示している.

Calculated Gibbs energy diagrams of the oxidative addition along with the C-H bond cleavage.
Figure 3,4はそれぞれカルボニル基側の反応物,生成物における,反応中心 (中心金属,開裂するC-H) のPDOSである.−7 eV 以下は占有軌道,−2 eV 以上は非占有軌道である.反応物では約−9,−8 eV のピークは中心金属の占有d軌道に,約−13 eV のピークC-H結合性軌道に,約+3.6 eV のピークはC-H反結合性軌道にそれぞれ相当する.Ir触媒では相対論効果を考慮することにより,占有d軌道の準位が上昇している.これはd軌道の自己無撞着的膨張に対応する.一方,C-H反結合性軌道の変化は小さい.Scheme 1に示す反応ではIr占有軌道からC-H空軌道へ電子の逆供与が起こっていると考えられる.これらのことから電子を供与する軌道と供与される非占有軌道とのエネルギー差が低下し,相互作用しやすくなっていると考えられる.一方,Rh触媒では相対論計算と非相対論計算の差は小さい.
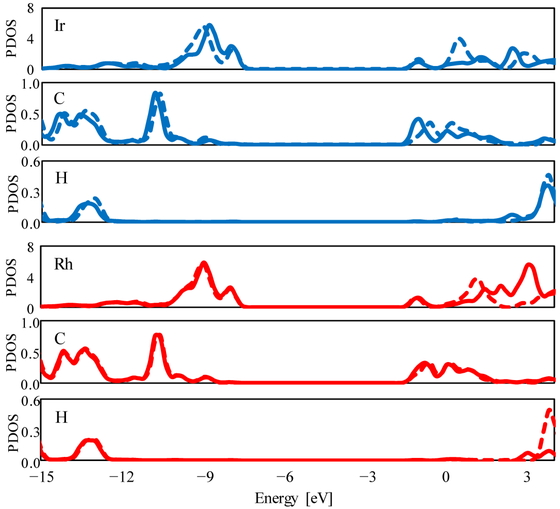
PDOS of Ir/Rh, H, and C atoms in reactant. Solid and dashed lines represent Rel and NR results, respectively.

PDOS of Ir/Rh, H, and C atoms in product. Solid and dashed lines represent Rel and NR results, respectively.
生成物では約−13 eV のピークは金属-水素結合性軌道に,約−11 eV のピークは金属-炭素結合性軌道に相当する.対応する反結合性軌道は非占有軌道に非局在化している.Ir触媒では相対論を考慮することにより,Ir-H結合およびIr-C結合の準位が低下している.Rh触媒ではこのような効果は見られない.生成した結合が安定化したことがIr触媒における劇的な反応エネルギーの低下をもたらしたと考えられる.また反応物に対して生成物のエネルギーが低下することに伴って早い遷移状態になっている.このことが活性化エネルギーの低下を引き起こしている.
本研究では,カチオン性Ir触媒を用いたベンズアニリド類のC-H結合活性化反応において,相対論効果が重要な役割を果たすことを明らかにした.つまり,d軌道の自己無撞着的膨張がd軌道の活性化をもたらし,その結果,C-H結合活性化反応によって生じるIr-H結合を安定化する.Ir (原子番号77) と同じ9族であるが第5周期のRh (原子番号45) ではこのような相対論効果は見られず,結果としてRh触媒とIr触媒の反応性に大きな違いが生じることが明らかとなった.
本研究で行った量子化学計算の一部は,自然科学研究機構 (NINS)・計算科学研究センター (RCCS) の計算機を利用した.