
最新号
日本コンピュータ化学会2024 秋季年会精選論文特集号
選択された号の論文の9件中1~9を表示しています
- |<
- <
- 1
- >
- >|
巻頭言
-
2025 年 24 巻 1 号 p. A1-A2
発行日: 2025年
公開日: 2025/03/14
PDF形式でダウンロード (382K) HTML形式で全画面表示
解説
-
2025 年 24 巻 1 号 p. A3-A11
発行日: 2025年
公開日: 2025/03/14
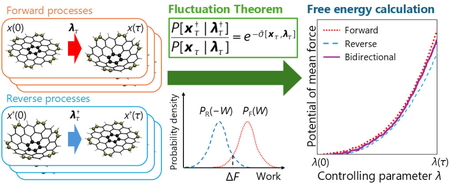 PDF形式でダウンロード (1229K) HTML形式で全画面表示
PDF形式でダウンロード (1229K) HTML形式で全画面表示
ソフトウェアニュース・レビュウ
-
2025 年 24 巻 1 号 p. A12-A17
発行日: 2025年
公開日: 2025/03/28
[早期公開] 公開日: 2025/02/28PDF形式でダウンロード (536K) HTML形式で全画面表示
速報 (SCCJ Annual Meeting 2024 Autumn Poster Award Articles)
-
2025 年 24 巻 1 号 p. 1-4
発行日: 2025年
公開日: 2025/03/14
 PDF形式でダウンロード (1120K) HTML形式で全画面表示
PDF形式でダウンロード (1120K) HTML形式で全画面表示 -
2025 年 24 巻 1 号 p. 5-9
発行日: 2025年
公開日: 2025/03/29
 PDF形式でダウンロード (1084K) HTML形式で全画面表示
PDF形式でダウンロード (1084K) HTML形式で全画面表示
速報 (Selected Papers)
-
2025 年 24 巻 1 号 p. 10-13
発行日: 2025年
公開日: 2025/03/29
 PDF形式でダウンロード (1719K) HTML形式で全画面表示
PDF形式でダウンロード (1719K) HTML形式で全画面表示 -
2025 年 24 巻 1 号 p. 14-16
発行日: 2025年
公開日: 2025/03/31
PDF形式でダウンロード (2090K) HTML形式で全画面表示 -
2025 年 24 巻 1 号 p. 17-19
発行日: 2025年
公開日: 2025/04/01
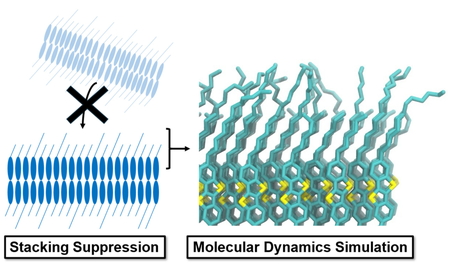 PDF形式でダウンロード (1310K) HTML形式で全画面表示
PDF形式でダウンロード (1310K) HTML形式で全画面表示 -
2025 年 24 巻 1 号 p. 20-26
発行日: 2025年
公開日: 2025/04/12
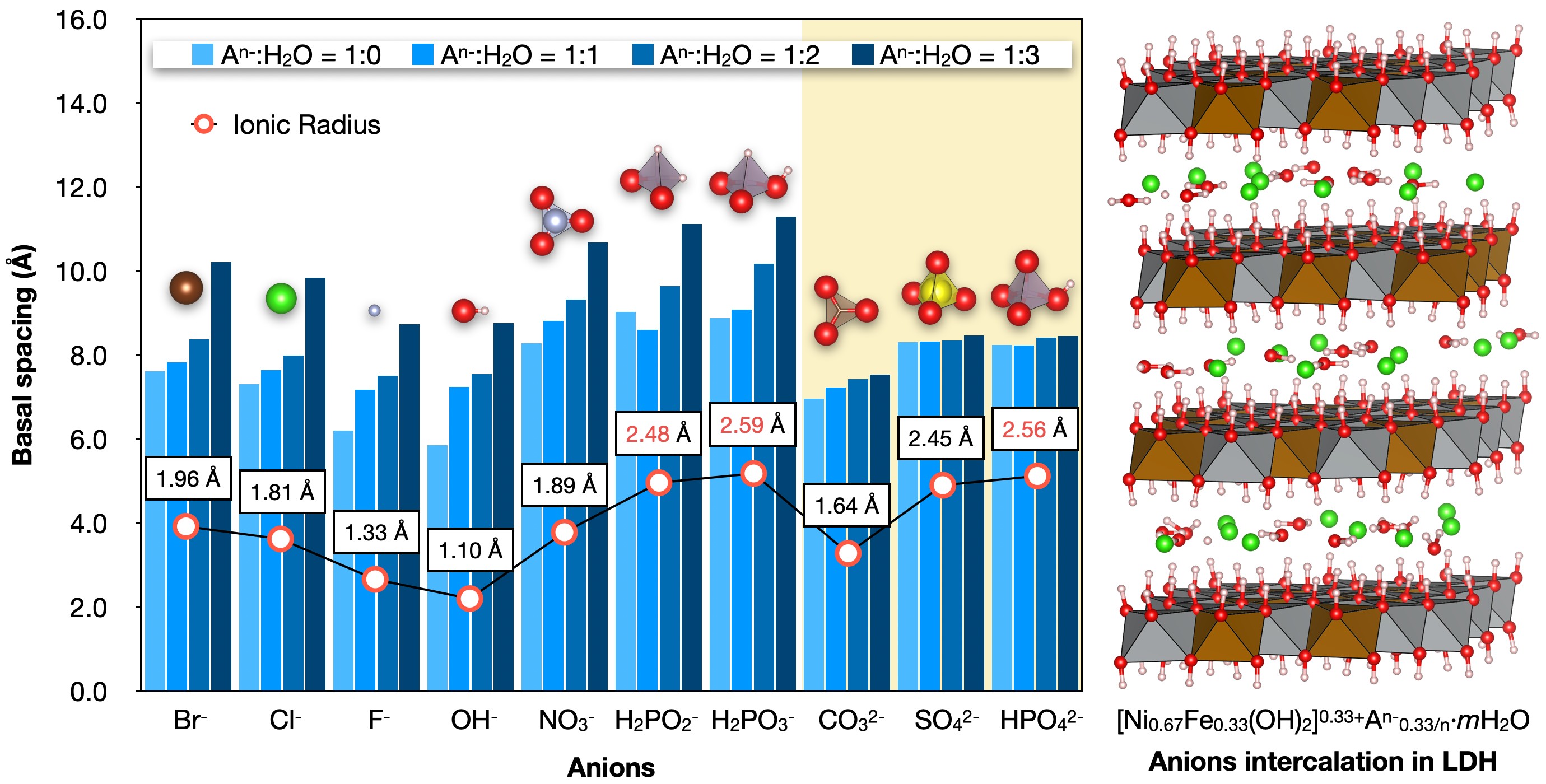 PDF形式でダウンロード (1813K) HTML形式で全画面表示
PDF形式でダウンロード (1813K) HTML形式で全画面表示
- |<
- <
- 1
- >
- >|