研究論文
系列二分決定グラフを用いたタンパク質配列モチーフの多重表現
キーワード:
Sequence binary decision diagram,
Aho-Corasick algorithm,
Multiple representation,
Sequence motif,
Motif search
2020 年 19 巻 1 号 p. 8-17
詳細
2020 年 19 巻 1 号 p. 8-17
本稿では,系列二分決定グラフ(SeqBDD)を用いたタンパク質配列モチーフの多重表現とそのモチーフ検索への応用について述べる.SeqBDDは,複数の文字列のような配列集合の圧縮表現である.本研究では,SeqBDDのための二つのアルゴリズムを開発した.一つ目は,対応するモチーフのアミノ酸配列を表現するSeqBDDを構築するためのもので,二つ目は状態遷移を追加することにより,SeqBDDのための決定性有限オートマトン(DFA)に相当するオートマトンを構築するためのものである.性能評価のために,マトリクスメタロプロテアーゼ(MMP)ファミリーにおいて保存されている三つのドメインを,UniProtKB/Swiss-Prot (Rel. 2017_09)から得られた555,594の全てのアミノ酸配列に対して検索した.PROSITEパターンを使用した同様の検索結果と比較して,本手法は,適合率,再現率,およびF値において良好な結果を示した.
タンパク質は20種類のアミノ酸が直鎖状にペプチド結合して構成され,折りたたまれることで固有の立体構造をとり,機能を発現する.タンパク質のアミノ酸配列中には局所的に保存されている領域が存在し,その領域は配列モチーフと呼ばれている [1].配列モチーフは,機能と関連することも多く,アミノ酸配列中に配列モチーフが存在するか確認することで,機能未知タンパク質の機能推定に活用されている [2,3,4].配列モチーフは,一般に相同性の高いアミノ酸配列や機能を共通して持つタンパク質のアミノ酸配列をマルチプルアラインメントにより整列することで得られる [5].この保存されている領域のアミノ酸配列群をもとに,配列モチーフを表現するモチーフモデルが生成される.
シンプルなモチーフモデルの一つとして,正規表現に基づくPROSITE pattern [6]が広く用いられている.また,出現確率を考慮した定量的なモチーフモデルとして,重み行列によるPROSITE profile [6]や,隠れマルコフモデル(HMM)によるPfam [7]などが提案されている.十分に確立されたモチーフに対して,これらのモチーフモデルは非常に有用である.しかしながら,機能未知のタンパク質も多く存在し,新規モチーフ候補部位の発見に向けて,様々な試みが行われている.そうした中で,従来とは異なる構造特徴表現もまた必要とされている.
特定の機能を有するタンパク質のアミノ酸配列は,一般に相同性が高く,ファミリーを構成する.ファミリーに固有な配列モチーフは,複数種類のモチーフの組み合わせとして存在することも報告されている [8].例えば,マトリクスメタロプロテアーゼ (MMP) ファミリーに属するタンパク質には,活性ドメインを示すZinc proteaseモチーフと,活性を阻害するCysteine switchモチーフが共に存在することが知られている [9].こうした,タンパク質中の配列モチーフの組み合わせに注目した解析も試みられている [10, 11].共起関係にある配列モチーフを表現する場合,PROSITE patternなど既存の方法では,各々のモチーフを一つずつ全て記述する必要がある.
本研究では,配列モチーフ(候補)抽出時のアミノ酸配列群をそのまま表現するモチーフモデルとして,系列二分決定グラフ (sequence binary decision diagram,以下 SeqBDD) [12]の概念を活用し,複数種の配列モチーフ集合を一つのSeqBDDとして圧縮して記述するための新たな方法を提案する.また,配列モチーフ集合のSeqBDD表現を基礎とした配列モチーフ検索のためのアルゴリズムを開発するとともに,PROSITE patternに登録されているMMPタンパク質の共起モチーフ群を例に,その検索精度に関する計算機実験を通じて本提案手法の優位性を示す.
Figure 1に,MMPファミリーに属するタンパク質が持つZinc proteaseモチーフ,Cysteine switchモチーフ,およびHemopexinモチーフの三つの配列モチーフをPROSITE patternにより表現した例を示す.文字列パターンは1残基毎にハイフン (-) で区切られており,P-R-Cのように,それぞれ1文字で表現されるアミノ酸残基は特定の位置に存在することを示している.[GN] のように角括弧でまとめられているものは,出現する残基のうちいずれか一つを示す.xは,任意のアミノ酸残基に対応する.一方,{IL} のような波括弧は,指定されているもの以外のアミノ酸残基を示す.丸括弧で示されるのは,その直前のアミノ酸残基の繰り返しであり,x(2, 3) のように二つの数字の組は繰り返し回数の幅を示す.

PROSITE patterns for the MMP sequence motif
SeqBDDは,系列集合を圧縮して表現する有向非巡回グラフである.SeqBDDの節点は,系列の要素を表現するラベル,要素が存在することを示す1-枝,および要素が存在しないことを示す0-枝で構成される [12].Figure 2に示す節点の例では,ラベルはアスパラギン酸を1文字表記 (D) により表現している.またSeqBDDは,系列が存在することを示す1-終端節点と,系列が存在しないことを示す0-終端節点の二つの終端を持つ.SeqBDDは,内包する系列集合に応じて一つの根節点を持つ.SeqBDDに含まれる系列は,根節点から1-終端節点への経路と対応し,その各経路は,1-枝を通った節点のラベルが示す要素を並べたものにそれぞれ対応する.

The elements of SeqBDD
Figure 3に,配列長3の4種類の異なるアミノ酸配列を用いた,SeqBDDによる系列集合の圧縮表現の例を模式的に示す.このSeqBDDは,アミノ酸配列集合 { DRC, DRY, NRC, NRY } を表現している.

An example of SeqBDD representing a set of amino acid sequences
Figure 4で強調して示す経路は,1-枝を通る節点のラベルD,RおよびCを並べたアミノ酸配列DRCと対応している.一方,Figure 5で強調して示す経路は,Cのラベルを持つ節点は0-枝を通るため,経路上の節点のラベルのうち,Cを除いて並べたアミノ酸配列DRYと対応する.

A path in SeqBDD corresponding to a sequence DRC

A path in SeqBDD corresponding to a sequence DRY
SeqBDDは,系列を表現する複数のSeqBDDの節点を共有することで得られる.例として,アミノ酸配列RC,RYからそれぞれSeqBDDを生成し,各SeqBDDを合成することで得た,アミノ酸配列集合 { RC, RY } を表現する新たなSeqBDDをFigure 6に示す.一つの系列を表現するSeqBDDは,根節点から1-終端節点に向かって,系列の要素を表現するラベルを持つ節点が,1-枝側に並んだ構造をしている.Figure 6のRCを表現するSeqBDDでは,根節点から順に,ラベルR,Cを持つ節点が並んでいる.二つのSeqBDDを合成するには,根節点から再帰的に節点を参照する.以下にその手順を示す.二つのSeqBDDは,それぞれの根節点はラベルRを共有するように新しい根節点のラベルをRとする.次に,それぞれのSeqBDDの1-枝に接続している節点のラベルを参照すると,ラベルはそれぞれC,Yである.このとき,予め定められたラベルの優先順位に従い,節点を0-枝に接続する.本研究では,ラベルの優先順位を辞書順としているため,ラベルCを持つ節点の0-枝に,ラベルYを持つ節点を接続する.最後に,要素が存在することを示す1-終端節点に,それぞれの節点の1-枝を接続し,残った0-枝に0-終端節点を接続することで,目的のSeqBDDを得る.

An example of constructing SeqBDD from a set of amino acid sequences
SeqBDDの特徴を以下に述べる.SeqBDDの形状は,内包する系列と節点のラベルの順序により,一意に定まることが知られている [13].SeqBDDは,根節点の0-枝を順に辿ると,グラフが表現する系列集合の先頭のラベルを持つ節点が並ぶ構造である.
Figure 3では,アミノ酸配列集合の先頭の残基DおよびNを持つ節点が並んでいる.これらの先頭の要素の1-枝の先の節点の0-枝を辿ると,2番目の要素を持つ節点が同様に並ぶ構造をしている.Figure 3では,各アミノ酸配列が共有する残基Rを持つ節点のみが存在している.このように,内包する系列の先頭の要素から順番に共有する構造となっている.また,1-終端節点に接続される節点は,最大でも要素の種類数,アミノ酸残基の場合は高々20個であり,末尾の要素についても共有される.SeqBDDで文字列集合を表現する場合には,それぞれ接頭辞および接尾辞が共有されると考えられる.さらに,Table 1に示すような,SeqBDDで表現する系列集合に対する集合演算のアルゴリズムが知られている [14].
| Operation | Result |
| Empty set (0-terminal node) | |
| A set of empty series (1-terminal node) | |
| Intersection of P and Q | |
| Union of P and Q | |
| Difference of P and Q | |
| A set of series with x at the beginning of P | |
| A set of series without x at the beginning of P | |
| A set with x appended to the beginning of P | |
| Root node label of P | |
| Number of elements of P |
本研究では,配列モチーフの記述表現として,SeqBDDを用いる.SeqBDDを用いることで,配列モチーフに対応するアミノ酸配列を圧縮して,網羅的に含むことができる.Figure 3の例では,合計12残基からなるアミノ酸配列を,5節点と二つの終端で表現している.SeqBDDでは,条件に一致するアミノ酸配列を明示的に表現するため,正規表現と比較して精度の向上が期待できる.また,配列モチーフの組み合わせが存在するとき,Table 1に示す集合演算を用いることで,複数のモデルを一つに集約することができる.すなわち,SeqBDDを用いたモチーフモデルでは,一つのモデルで複数の配列モチーフ群を記述できる.
なお,任意のアミノ酸xが位置する部分は,SeqBDDでは20種類のアミノ酸と0-枝の組み合わせで表現できる.ただし,例えば x (10,20) のように配列的なギャップ領域と考えられるものは,むしろ,その前後の保存領域の二つのパターンが共起するモチーフとして表現する.これにより,シャッフリングのように進化の過程で配列的には大きな変異があるものの,空間的な関係を保っているような機能部位の構造特徴も扱うことができると考える.
2.4 配列モチーフ検索配列モチーフ検索は文字列処理により行われるため,例えば正規表現を用いたモチーフ表現モデルでは,文字列パターンから決定性有限オートマトン (deterministic finite automaton,DFA) を構築することにより検索を行う [15].正規表現は,一般に一致する複数の文字列を表現するため,DFAを用いることで単純な網羅的文字列マッチング (以下,単純文字列マッチング) よりも効率的に検索可能である.SeqBDDを用いたモチーフモデルの場合,単純文字列マッチングでは,検索対象のアミノ酸配列を1残基ずつずらしてグラフ中の経路と比較することで検索を行う.しかしこの方法では,アミノ酸配列長をn,根節点と1-終端節点の間の最長経路長をhとするとき,最悪時間計算量はO(nh)となる.
SeqBDDと同様に文字列集合を表現するデータ構造に,トライ [16]と呼ばれる木構造がある.このトライを基にして,文字列検索のためのDFAを構築するアルゴリズム (Aho-Corasick法 [17]) が知られている.Figure 7に,Aho-Corasick法により得られた,アミノ酸配列集合 { DR, DRY, R, RY, YDR, YDRY } により配列モチーフ検索を行うDFAの例を示す.ここで,状態を丸,特に受理状態を二重丸で表し,一致時の遷移をラベル付き有向辺,不一致時の状態遷移をラベルなし有向辺でそれぞれ示す.本研究では,このAho-Corasick法による複数文字列検索と同様に,SeqBDD上に探索失敗時の状態遷移を追加することで,SeqBDDをDFAとして配列モチーフ検索に応用することを試みた.

An example of DFA for a set of sequence { DR, DRY, R, RY, YDR, YDRY } obtained by Aho-Corasick algorithm
Aho-Corasick法では,文字の不一致時の状態遷移をfailure関数で表現する.Figure 7の例では,Figure 8に示すfailure関数を用いる.このfailure関数をSeqBDDにおいて再現することで,トライの場合と同様に効率的に文字列検索が可能になる.しかし,Figure 9に示すように,SeqBDDはトライと異なり,一つの節点が複数の文字列の中で使われる.特に,Figure 9のアミノ酸配列集合 { D, YD } の状態を表現するRのラベルを持つ節点は,その節点の1-枝が接続している,Yのラベルを持つ節点の0枝が1-終端節点に接続されているため,DFAにおける受理状態に相当する.このような各節点の文字列,すなわち状態を区別することで,SeqBDDにおいてもfailure関数による状態遷移を実現できる.本研究では,failure関数構築時にはスタック,検索時にはキューを用いて複数の文字列を区別する.

An example of a failure function for Figure 7

SeqBDD and trie for an identical set of sequence
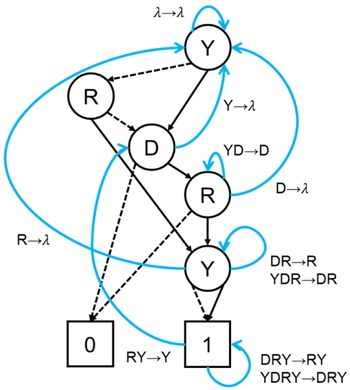
An example of SeqBDD by adding state transitionin Figure 9 (a)
まず,任意のSeqBDDにおけるfailure関数を求めるアルゴリズムを示す.Ahoらは,現在の節点を入力,遷移先の節点を出力とするfailure関数を用いたが,提案手法ではこれを拡張する.SeqBDDを用いて文字列を検索するために,現在の状態を節点とスタックにより表現する.そのため,本研究におけるfailure関数は,現在の節点とその文字列を示すスタックを入力,遷移先の節点と文字列の先頭から除去する文字数を出力とする.このfailure関数で表現する状態の遷移先は,その状態と対応する文字列の先頭の1文字を除いた最長接尾辞と対応する状態であるため,遷移先の状態は0-枝を優先して探索することで求めることができる.SeqBDDにおいてfailure関数を求めるアルゴリズムを Algorithm1に示す.ここでは,節点nodeのラベルをnode.label,0-枝をnode.branch0,および1-枝をnode.branch1と表すものとする.また,0-終端節点を0,1-終端節点を1でそれぞれ示す.Algorithm 1におけるtrance関数では,各節点における遷移先をnext_state関数により求め,各節点の0-枝,1-枝の順にpre-orderで根節点から探索を行う.next_state関数では,再帰的に最長接尾辞に対応する節点を探索するnext_node関数を用いて,次の節点を得る.目的のfailure関数は,make_failure関数を呼び出すことで取得できる.

次に,SeqBDDと得られたfailure関数による状態遷移を組み合わせて文字列検索を行うアルゴリズムをAlgorithm 2に示す.Algorithm 2におけるis_terminal関数では,現在の状態が持つ文字列を出力すべきかを,現在の状態が終端であるかどうか,すなわち0-枝を辿った先に1-終端節点が接続されているかどうかで判断している.fail_state関数では,failure関数の出力を用いて次の状態,すなわち次の節点とキューおよび検索対象の文字列の位置を得る.search関数では,これらの関数を利用し,各節点の文字と比較しながら探索する.

Figure 10に,Figure 9 (a) のSeqBDDにfailure関数による状態遷移を追加したグラフの例を示す.このグラフを用いることで,単純文字列マッチングよりも効率的に計算量O(n + h)で配列モチーフ検索することができる.
SeqBDDを用いたモチーフモデル (以下,提案手法) とPROSITE pattern (以下,従来法) による検索精度の比較を,適合率,再現率,およびF値によって行った.検索対象として,UniProtKB/Swiss-Prot (Release 2017_09,以下Swiss-Prot) [18]に登録されている全アミノ酸配列データ555,594配列を用いた.PROSITEに記載されている真陽性配列,偽陽性配列,および偽陰性配列の情報を参照して,精度を計算した.モチーフモデルで表現するデータとして,MMPファミリーにおいて共起する配列モチーフであるHemopexinモチーフ,Zinc proteaseモチーフ,およびCysteine switchモチーフを用いた.PROSITEによると,配列モチーフを持つタンパク質は,それぞれ98件,1,643件および137件である.従来法には,それぞれの配列モチーフに対応するPROSITE pattern: PS00024,PS00142,およびPS00546を用いる (Figure 1).

MMP sequence motifs extracted by MEME
提案手法では,モチーフモデルを構築するために,配列モチーフに相当するアミノ酸配列を収集する必要がある.しかし,PROSITEからは,偽陰性に分類されるタンパク質の配列モチーフに相当するアミノ酸配列を得ることができない.そのため,真陽性および偽陰性に分類されるタンパク質を対象に,配列モチーフ抽出ツールMEME [19]を用いた配列モチーフ抽出により得られたアミノ酸配列を用いる.MEMEによる配列モチーフ抽出では,配列モチーフ長をPROSITE patternの最大長に固定し,PROSITE patternに対応する部位のうち最もE値が低いものを使用する.今回,提案手法で表現する配列モチーフを,sequence logo形式でFigure 11に示す.
実験では,提案手法については,Denzumiらにより提案されているアルゴリズム [12],および2.5節に示すアルゴリズムをC++により実装したものを,Pythonより呼び出して使用した.従来法については,PROSITE patternを正規表現に変換した上で,Pythonに標準で含まれている正規表現モジュールを用いて検索を行った.
3.2 結果と考察従来法については,PROSITE patternを変換した三つの正規表現パターンを用いて配列モチーフ検索を行う.このとき,一つのタンパク質に複数ヒットする場合は,重複を除き1件とカウントする.従来法による検索結果をTable 2に示す.Table 2の適合率と再現率を配列モチーフごとに比較すると,Hemopexinモチーフについては適合率,Zinc proteaseモチーフおよびCysteine switchモチーフについては再現率が低いことが確認できる.これは,配列モチーフに相当するアミノ酸配列を,簡潔な文字列パターンで表現することの困難さを示している.
次に,提案手法による配列モチーフ検索を行う.例えば,Hemopexinモチーフを有するタンパク質について,98件のモチーフ対応部位から重複を除いた75件,総残基数1125の配列集合を,接点数482のSeqBDDにより漏れなく表現することができる.三つの配列モチーフを表現するそれぞれのSeqBDDを,Table 1の和集合演算により一つのSeqBDD (重複を除き計821件,総残基数8473,接点数2438)に集約した後に状態遷移を追加し,アミノ酸配列に対して検索を行った.配列モチーフ検索で得られたアミノ酸配列は,Hemopexinモチーフ,Zinc proteaseモチーフ,およびCysteine switchモチーフをそれぞれ表現するSeqBDDの経路と比較することで分類した.提案手法による配列モチーフ検索の結果をTable 3に示す.Table 3の再現率が全て1であることから,SeqBDDを用いることで,配列モチーフに相当するアミノ酸配列を漏れなく表現できることが確認できる.更に,Hemopexinモチーフについては,偽陽性配列が存在しなかったことから,適合率についても1となっている.その他の二つの配列モチーフについては,偽陽性配列を含んでいることから,今回の条件では配列モチーフを識別できない配列パターンを含んでいることがわかる.
| False negative sequence | False positive sequence | True positive sequence | Precision | Recall | F-measure | |
| Hemopexin | 23 | 50 | 75 | 0.600 | 0.765 | 0.673 |
| Zinc protease | 485 | 212 | 1,168 | 0.846 | 0.707 | 0.770 |
| Cysteine switch | 65 | 2 | 72 | 0.973 | 0.526 | 0.682 |
| False negative sequence | False positive sequence | True positive sequence | Precision | Recall | F-measure | |
| Hemopexin | 0 | 0 | 98 | 1.000 | 1.000 | 1.000 |
| Zinc protease | 0 | 30 | 1,643 | 0.982 | 1.000 | 0.991 |
| Cysteine switch | 0 | 19 | 137 | 0.878 | 1.000 | 0.935 |
| Original sequence | Accession number | ID | Proposed sequence |
| PICGNGMV | Q10743 | ADA10_RAT | MRCRLVDA |
| O35598 | ADA10_MOUSE | GGCADHSV | |
| O14672 | ADA10_HUMAN | GGCADHSV | |
| Q10741 | ADA10_BOVIN | GGCADHSV | |
| KRCGNGMV | Q9JI76 | ADA21_MOUSE | MLCSLTEK |
| KRCGNGVV | Q9UKJ8 | ADA21_HUMAN | MRCGLTEK |
| DKCGVCGG | Q9P2N4 | ATS9_HUMAN | HACDTSEH |
| P59384 | ATS15_MOUSE | SRCGVASG | |
| Q8TE58 | ATS15_HUMAN | SRCGVASG | |
| Q8WXS8 | ATS14_HUMAN | KACGGGIQ |
二つの手法の検索精度についての検証を,Table 2とTable 3の比較により行う.Cysteine switchモチーフの適合率を除き,提案手法の方が適合率,再現率,およびF値について良いことが示される.これは,従来法による検索では偽陰性となる配列を,提案手法では検索することができ,従来法の検索で偽陽性となる配列は検索しないことにより,検索精度の向上が達成されたと考えられる.一方,Cysteine switchモチーフの検索精度では,適合率について提案手法の精度が劣っている.これらの配列について検証するため,Swiss-Protのエントリ情報をもとに調査したところ,配列モチーフではないアミノ酸配列を含んでいることが判明した (Table 4).ここで,Table 4のIDがADA10_RAT,ATS14_HUMANであるタンパク質については,Swiss-ProtにCysteine switchモチーフの情報が存在しなかった.そのため,真陽性配列により最も類似性スコアの高いアミノ酸配列,すなわち配列モチーフに相当すると期待されるアミノ酸配列を,BLASTを用いて検索した.提案手法の含まれるアミノ酸配列をTable 4に従い修正して,Cysteine switchモチーフについて再検索した結果をTable 5に示す.Table 5より,Cysteine switchモチーフの適合率についても,精度が改善されたことが確認できる.しかし,修正後の結果であるTable 5には,偽陽性に分類されているタンパク質が1件存在する.このタンパク質 (MMP24_RAT) について調査するために,Swiss-Protのエントリ情報 (Q99PW6) を確認したところ,このタンパク質はCysteine switchモチーフを持つことを確認した.このことから,このタンパク質については,検索精度の計算に用いた分類が間違っていたと考えられる.
| False negative sequence | False positive sequence | True positive sequence | Precision | Recall | F-measure | |
| Cysteine switch | 0 | 1 | 137 | 0.993 | 1.000 | 0.996 |
| False negative sequence | False positive sequence | True positive sequence | Precision | Recall | F-measure | |
| SeqBDD | 0 | 1 | 80 | 0.988 | 1.000 | 0.994 |
| PROSITE pattern | 27 | 0 | 53 | 1.000 | 0.663 | 0.797 |
最後に,Hemopexinモチーフ,Zinc proteaseモチーフ,およびCysteine switchモチーフの三つの配列モチーフを持つタンパク質,すなわちMMPファミリーとしての特徴を持つタンパク質についての検索結果をTable 6に示す.この表は,Table 2およびTable 3から計算できる.提案手法では,個々の配列モチーフにおける検索精度が良いことから期待されるように,これらの全ての配列モチーフを持つタンパク質の検索についても,検索精度が良いことが確認できる.その一方で,Table 6における提案手法の適合率は従来法の適合率よりも低くなっている.この原因を調査するために,偽陽性に分類されたタンパク質1件のIDを確認したところ,このタンパク質は,分類の間違いだと考えられるMMP24_RATであることが判明した.すなわち,Table 6における提案手法の適合率についても,従来法の適合率と同じく1となる.このことから,提案手法は,従来法と比較して,適合率,再現率およびF値が全て改善されていることが示された.
本研究では,系列二分決定グラフを用いた配列モチーフ群を表現するモチーフモデルの提案を行った.提案手法では,配列モチーフに相当するアミノ酸配列を漏れなく表現できる.また,提案手法による配列モチーフ検索を行うために,Aho-Corasick法のアイディアを基にした文字列検索アルゴリズムを導入した.文字列検索アルゴリズムでは,系列二分決定グラフとトライのラベルを,根節点からトレースすることで文字列を得られるという特徴を活用して,Aho-Corasick法と同様の状態遷移を系列二分決定グラフ上に実現した.このアルゴリズムと集合演算を用いることで,複数の配列モチーフを一つのモデルで表現し,そのまま配列モチーフ検索を行うことができる.計算機実験では,マトリクスメタロプロテアーゼ・ファミリーに属するタンパク質が持つ三つの配列モチーフを用いて,系列二分決定グラフとPROSITE patternの検索精度を,適合率,再現率,およびF値により比較した.その結果,全てのケースにおいて提案手法の方が検索精度が良いことを示した.また,共起するモチーフ群を同時探索できることから,これらの組み合わせで構成される新規モチーフの発見につながるものと考える.これは,条件に一致する系列集合を網羅的に圧縮して表現できる,系列二分決定グラフの特徴によるものである.
本研究の一部は,栢森情報科学振興財団の研究助成(K30研ⅩⅩⅢ第536号)を受けて実施したものであることを明記して謝意を表す.